

Abstract
Genetic and pathologic studies have associated angiotensin-converting enzyme (ACE) with Alzheimer disease. Previously, we and others have reported that ACE degrades in vitro the amyloid β-protein (Aβ), a putative upstream initiator of Alzheimer disease. These studies support the hypothesis that deficiency in ACE-mediated Aβ proteolysis could increase Alzheimer disease risk, and raise the question of whether ACE inhibitors, a commonly prescribed class of anti-hypertensive medications, can elevate Aβ levels in vivo. To test this hypothesis, we administered the ACE inhibitor captopril to two lines of APP transgenic mice harboring either low levels of Aβ or high levels of Aβ with associated plaque deposition. In both models, we show that captopril does not affect cerebral Aβ levels in either soluble or insoluble pools. Further, we find no change in plaque deposition or in peripheral Aβ levels. Data from these Alzheimer models suggest that captopril and similar ACE inhibitors do not cause Aβ accumulation in vivo.
Keywords: Alzheimer disease, amyloid β-protein, β-amyloid precursor protein, angiotensin-converting enzyme, Aβ degradation
Introduction
Alzheimer disease (AD) is characterized by the progressive accumulation and deposition into plaques of the pathogenic amyloid β-protein (Aβ) in various brain regions, leading to impairments in memory, language, cognition and behavior. Brain levels of soluble Aβ species correlate with the presence and extent of AD-type dementia (Lue et al., 1999). In familial cases of AD, mutations in genes encoding the β-amyloid precursor protein (APP) or the presenilins cause accelerated accumulation and toxicity of Aβ, resulting in an early onset and aggressive form of the disease. Typical, late-onset AD cases are of unknown etiology. However, several studies highlight decreased Aβ degradation (Caccamo et al., 2005) or impaired Aβ transport (Shibata et al., 2000) in aging and in AD, leading to the hypothesis that deficits in Aβ clearance spur disease progression. One mechanism of Aβ clearance is the direct degradation of the peptide by proteases residing within the brain. Several Aβ-degrading enzymes have been shown, by knockout and overexpression studies, to regulate the levels of cerebral Aβ. These include neprilysin (NEP) (Iwata et al., 2001), insulin-degrading enzyme (Farris et al., 2003; Miller et al., 2003) and the endothelin-converting enzymes (Eckman et al., 2003). The serine protease plasmin has been shown to promote Aβ clearance from the brain (Melchor et al., 2003), though genetic deficiency does not change steady-state murine Aβ metabolism in vivo (Tucker et al., 2004). As more Aβ-degrading proteases are identified -- recently including matrix metalloproteinase-9 (Yan et al., 2006) and the mitochondrial presequence peptidase (Falkevall et al., 2006) -- novel pathways to manipulate Aβ clearance are being proposed. Supporting a potential therapeutic role for Aβ proteolysis in AD, overexpressing Aβ-degrading proteases has been found to modulate the progression of Aβ deposition and neuropathology in a mouse model of AD (Leissring et al., 2003).
Angiotensin-converting enzyme (ACE) is a zinc metalloprotease containing two homologous proteolytically active domains. A major function of ACE is to regulate blood pressure by cleaving angiotensin I to angiotensin II and inactivating bradykinin (Coates, 2003). Small molecule competitive inhibitors of ACE have been developed to prevent vasopeptide proteolysis, which has proven to be a successful strategy for the management of hypertension. A link between ACE and AD was first hypothesized by human genetic studies, which have found both single nucleotide polymorphisms (Katzov et al., 2004) and an intronic insertion (I)/deletion (D) polymorphism within the ACE gene that associate with AD (Lehmann et al., 2005). The I allele has been associated with an increased risk for AD, but not for vascular dementia (Kolsch et al., 2005). The D allele is associated with reduced risk for AD, and carriers of this protective allele have elevated levels of the ACE protein (Rigat et al., 1990). Some post-mortem studies have reported elevated ACE expression in AD brain (Savaskan et al., 2001), suggesting a role for ACE in disease response. ACE has been shown to degrade synthetic Aβ in vitro (Hu et al., 2001; Oba et al., 2005) and to regulate naturally produced Aβ in cell culture models (Hemming and Selkoe, 2005). These genetic, pathological, biochemical and cell biological studies all support the hypothesis that ACE is linked to AD by regulating Aβ metabolism. Further, if ACE inhibition decreases Aβ catabolism, ACE inhibitors could be an avoidable risk factor for AD.
In our previous studies, we demonstrated that cellular overexpression of ACE promotes the clearance of naturally produced Aβ40 and Aβ42. This Aβ-degrading activity was found to occur at each of the two ACE active sites. Inhibition of ACE activity by a widely prescribed ACE inhibitor prevented ACE-mediated Aβ clearance and resulted in accumulation of the Aβ peptide in cell culture. In the present study, we explore the ability of ACE to regulate Aβ metabolism in vivo by treating APP transgenic mice with an orally administered ACE inhibitor. In young APP transgenic mice harboring predominantly soluble Aβ species, prolonged ACE inhibition caused no change in cerebral Aβ concentrations. In aged APP transgenic mice having extensive Aβ deposits, prolonged ACE inhibition did not affect cerebral or peripheral levels of Aβ, or plaque deposition, suggesting that ACE inhibitors do not significantly compromise Aβ metabolism in vivo.
Materials and Methods
Mice and Drug Treatments
The 3xTg-AD line of transgenic mice (Oddo et al., 2003), expressing PS1M146V, APPSwe and tauP301L transgenes, were aged to ~3 months before experimental treatment. The J20 line of APPSwe/Ind heterozygous transgenic mice (C57BL/6 × DBA2 background) (Mucke et al., 2000) were aged 16–17 months before treatment. Captopril (Sigma) was prepared at 2 g/l, unless otherwise specified, in drinking water and the pH adjusted to 7.0. Losartan was prepared by pulverizing Cozaar® (Merck & Co., INC.) tablets and dissolving the resulting powder in drinking water to a final concentration of 0.6 g/l. This losartan regimen has been shown to therapeutically lower blood pressure and protect against left ventricular remodeling following myocardial infarction in the mouse, to a similar extent as ACE inhibition (Patten et al., 2003). To determine the effects of varying doses of the ACE inhibitor on central nervous system and peripheral ACE enzymatic activity, age-matched C57BL/6 wild-type mice were treated for 10 days at variable concentrations of captopril. Prolonged drug treatments consisted of treating APP transgenic mice with vehicle (water), captopril, or losartan for 28 days. Drinking water with drug was changed three times per week, and was prepared fresh from powder every time. All animal procedures were approved by the Harvard Standing Committee for Animal Use.
ACE Activity Assay
ACE enzymatic activity was determined using the substrate hippuryl-L-histidyl-L-leucine (Hip-His-Leu; Sigma) as previously described (Hemming and Selkoe, 2005; Santos et al., 1985). Tissue was homogenized in 6 volumes (wt:vol) of 50 mM Tris (pH 7.4) and debris removed by centrifuging 1,000 × g for 10 min. To assay ACE activity, 10 µg of protein homogenate were incubated with 1 mM Hip-His-Leu in 0.4 M sodium borate buffer (pH 8.3) with 0.3 M NaCl in a volume of 35 µl. Samples were developed first by the addition of 150 µl of 0.34 M NaOH, followed by a 10 min incubation at 25°C with 20 µl of 20 mg/ml o-phthaldialdehyde (Sigma). This reaction was stopped by acidification with 50 µl of 3 N HCl. The fluorescence of o-phthaldialdehyde-modified His-Leu was measured in a 96-well plate format using a Victor2 multilabel plate reader (excitation, 355; emission, 535) (PerkinElmer Life Sciences). ACE activity, measured in arbitrary units (a.u.), is defined as the fluorescent signal inhibitable by 1 µM captopril.
NEP Activity Assay
NEP enzymatic activity was determined using the substrate 3-dansyl-D-Ala-Gly-p-(nitro)-Phe-Gly (DAGNPG; Sigma) (Florentin et al., 1984), which is principally degraded by NEP, and to a smaller extent by ACE. Tissue was homogenized in 6 volumes (wt:vol) of 50 mM Tris (pH 7.4) and debris removed by centrifuging 1,000 × g for 10 min. One hundred µg of tissue homogenate were incubated with 50 µM DAGNPG and 1 µM captopril in a volume of 200 µl at 37°C. Reactions were stopped by heating samples to 100°C for 5 min, then spinning 5,000 g × 5 min to remove the denatured protein. The supernatant was diluted into 400 µl of 50 mM Tris (pH 7.4) and fluorescence determined using a Victor2 multilabel plate reader (excitation, 342; emission, 562). NEP activity, measured in arbitrary units, is defined as the fluorescent signal above that present in NEP knockout mouse (Iwata et al., 2001) brain or kidney.
Tissue Aβ Extraction
Fresh-frozen mouse brain was serially homogenized into aqueous, detergent soluble, and guanidine HCl soluble fractions. First, brains were homogenized in 4 volumes (wt:vol) of TBS Extraction Buffer (140 mM NaCl, 3 mM KCl, 25 mM Tris (pH 7.4), 5 mM EDTA, 2 mM 1,10-phenanthroline and protease inhibitor cocktail (Roche)). Homogenate was spun 100,000 g × 1 hr, and the supernatant saved as the aqueous fraction. The insoluble pellet was resuspended and briefly sonicated in TBS Extraction Buffer containing 1% Nonidet P-40 (NP-40), homogenized, and spun at 100,000 g × 1 hr. The resulting supernatant was saved as the NP-40 soluble fraction. The resulting pellet was homogenized in 6.25 M guanidine HCl in 50 mM Tris (pH 8.0), incubated for 2 hrs at 25°C, and spun at 20,800 g × 20 min at 4°C. The resulting supernatant was saved as the guanidine HCl soluble fraction. Fresh-frozen mouse kidney was homogenized in TBS Extraction Buffer containing 1% NP-40 and spun 100,000 g × 1 hr, and the soluble fraction saved for Aβ quantifications.
Enzyme-linked Immunosorbant Assay (ELISA)
ELISAs for Aβ were performed as previously described (Hemming and Selkoe, 2005; Johnson-Wood et al., 1997) with few modifications. 96-well ELISA plates (Costar) were coated with 3.5 µg/ml of the capture antibody. Aβ1-Total, Aβ1–40 and Aβ1–42 were measured by capturing with antibodies specific to residues 13–28 of Aβ (266), or to the Aβ C-terminal 40 (2G3) or 42 (21F12) residues, respectively. Captured Aβ was detected with biotinylated 3D6, specific to residues 1–5 of the Aβ N-terminus (all antibodies gift of Elan Pharmaceuticals, San Francisco, CA). ELISAs were developed by incubating the Aβ-bound biotinylated 3D6 with Avidin-horseradish peroxidase (Vector Labs), followed by a 1 hr incubation with QuantaBlu Fluorogenic Peroxidase Substrate (Pierce), and the resulting fluorescence determined (excitation, 340; emission, 400). Plate washing after antibody and enzyme binding steps was performed three times for 1 min with Tris-buffered saline, 0.05% Tween 20. Kidney homogenates were found to produce a modest, non-specific ELISA signal in wild-type mice; this value was subtracted from APP transgenic mouse kidney readings to calculate Aβ levels.
Immunohistochemistry and Image Analysis
Ten µm thick sagittal brain sections were deparaffinized and hydrated through a series of graded alcohol steps and washed in phosphate buffered saline. Endogenous peroxidase activity was quenched with 0.6% hydrogen peroxide in methanol for 15 min. Sections were blocked in goat serum for 25 min, then incubated with the anti-Aβ antibody R1282 (1:1000 dilution) (Seabrook et al., 2006) for 1 hr. After washing, an anti-rabbit biotinylated secondary antibody was applied for 30 min, washed, and developed using the Avidin/biotin/HRP method (ABC Elite Kit, Vector Labs) and DAB chromogenic reaction (Liquid DAB, BioGenex). Images from stained sections were captured and quantified from 4 sections per brain region of each mouse using a 2× objective. The percent area covered by Aβ plaques was determined using IPLab Spectrum 3.1 Image Analyzer software (Signal Analytics).
Statistical Analyses
The data were analyzed using a one-way analysis of variance and Tukey’s post-hoc comparison or a two-tailed Student’s t-test, where appropriate. Calculated comparisons of p < 0.05 were considered significant. All reported values represent the means ± S.E.
Results
ACE inhibitor treatment does not alter cerebral Aβ levels in APP transgenic mice without Aβ deposition
To determine the dose of ACE inhibitor required to block ACE enzymatic activity in the brain and periphery, we administered increasing concentrations of captopril, from zero to 2 g/l, in the drinking water of wild-type C57BL/6 mice. Captopril is reported to be brain-penetrating in rodents and humans and is among the most bioactive of the ACE inhibitors (Brown and Vaughan, 1998; Evered et al., 1980; Ohrui et al., 2004). In the brains of mice treated with varying levels of captopril, only the highest dose of captopril, at 2 g/l (approximately ten times the highest dose used in humans), showed a statistically significant decrease in ACE activity (Fig. 1A), resulting in a decrement of 28%. In contrast, kidney tissue ACE activity was largely inhibited (>87% of vehicle control) under any administered dose of captopril (Fig. 1B). These data suggest that, although captopril can potently inhibit ACE activity in the periphery, only a relatively small amount of the drug is able to access the central nervous system to inhibit brain ACE.
Figure 1. Reduction of tissue ACE activity by captopril.
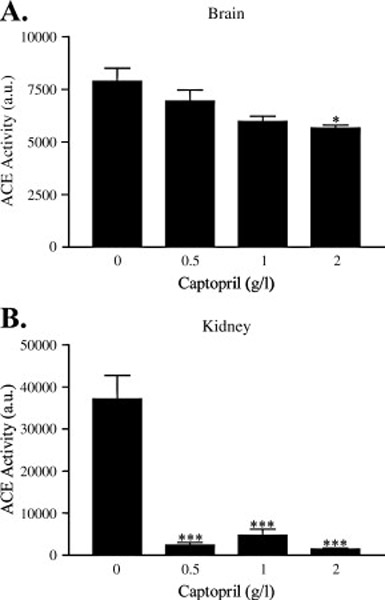
Wild-type C57BL/6 mice (n = 3 per condition) were orally administered the indicated doses of captopril and tissue harvested after 10 days of treatment. ACE activity was measured in brain (A) and kidney homogenates (B) after incubating 10 µg protein with 1 mM of the substrate Hip-His-Leu. Data represent the mean ± S.E. Compared to vehicle (0 g/l captopril) in each tissue, *, p < 0.05; ***, p < 0.001.
Hypertension is a common chronic medical condition, and many patients are treated with ACE inhibitors. Because we have previously found that ACE inhibition prevents Aβ catabolism in cell culture, we extended our studies to ask whether prolonged oral administration of captopril is able to modulate the ability of ACE to degrade Aβ in vivo. To this end, we administered captopril, at 2 g/l in drinking water, to 3×Tg-AD mice at an age at which Aβ is predominantly soluble. As an additional control group, to account for any unexpected effects of lowering blood pressure on Aβ levels, we administered losartan at 0.6 g/l in drinking water to 3×Tg-AD mice. Losartan is an angiotensin II receptor (type AT1) antagonist, and thus inactivates angiotensin II signaling one step downstream of ACE inhibition, producing clinical benefits similar to ACE inhibitors (Konstam et al., 2005).
Captopril, losartan, or vehicle (water) were administered to 3×Tg-AD mice for 28 days, then cerebral Aβ was measured to test for any alterations in Aβ metabolism. Because of its small active site and known activity against small peptides (Natesh et al., 2003), we hypothesized that ACE would primarily affect aqueous and/or detergent-soluble Aβ species rather than formed plaques. One cerebral hemisphere per mouse was biochemically fractionated into three distinct pools: the aqueous pool, representing soluble Aβ; the NP-40 detergent-soluble pool; and the guanidine-soluble pool, representing insoluble Aβ. ELISA analysis for Aβ showed no effect of ACE inhibitor treatment on Aβ metabolism in 3×Tg-AD mice, in any of the fractionated pools (Fig 2A–C). Further, no difference in Aβ species could be detected by three ELISAs that probed for Aβ1-Total (Fig 2 left panel), Aβ1–40 (Fig 2 middle panel) or Aβ1–42 (Fig 2 right panel). To test the prolonged effects of drug treatment on protease activity in these 3×Tg-AD mice, ACE activity was measured from protein homogenates prepared from the remaining cerebral hemispheres and kidneys in the absence of protease inhibitors. It was apparent that the prolonged captopril administration was effective at maintaining ACE inhibition in the kidney (Fig 3B), but not in the brain (Fig 3A) even at the high dose used, possibly due to the relatively low level of brain penetration and the high variability in ACE activity within this cohort of mice. Previous studies suggest that there may be a compensatory genetic co-regulation of ACE and NEP levels (Jalil et al., 2004; Jongun, 2004), such that low ACE activity may stimulate NEP expression. To exclude NEP upregulation as the cause of the observed preservation of Aβ levels between treatment groups, we tested NEP activity in kidney and brain tissue (Fig 3B) and found no difference between treatment groups. These data exclude the possibility of altered NEP activity and demonstrate that chronic treatment of young APP transgenic mice with an ACE inhibitor has no effect on the levels of non-deposited Aβ.
Figure 2. ACE inhibitor treatment does not affect cerebral levels of Aβ in the 3×Tg-AD mouse model.
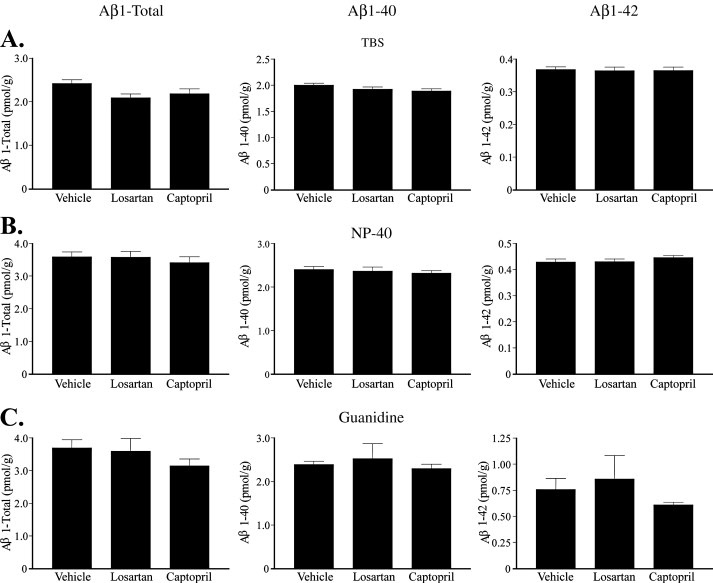
3×Tg-AD mice were treated with captopril (2 g/l, n = 11), losartan (0.6 g/l, n = 11) or vehicle (water, n = 13) for 28 days. Brains were fractionated into an aqueous TBS pool (A), an NP-40 detergent soluble pool (B), and a guanidine hydrochloride soluble pool (C). Aβ levels were measured by an ELISA specific for Aβ1-Total, Aβ1–40 and Aβ1–42 (left panel, middle panel and right panel, respectively). Data represent the mean ± S.E.
Figure 3. Effects of ACE inhibition on ACE and NEP activity.
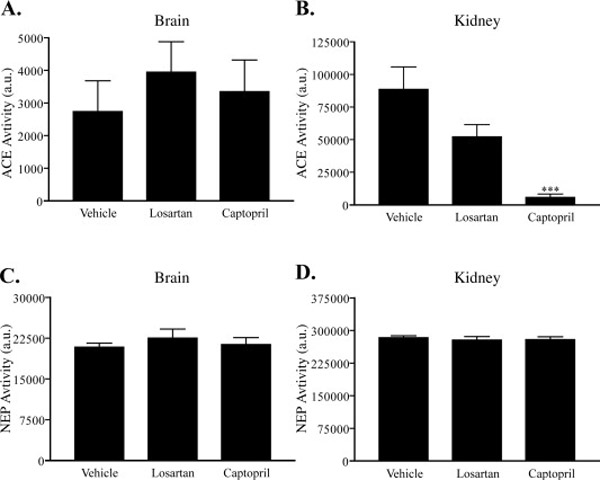
ACE activity was measured in the drug treated 3×Tg-AD brain (A) and kidney (B) tissue homogenates by incubating 10 µg protein with 1 mM of the substrate Hip-His-Leu. NEP enzymatic activity was measured in the same lysates by incubating 100 µg protein with 50 µM of the substrate DAGNPG in brain (C) and kidney (D). Data represent the mean ± S.E. Compared to vehicle, ***, p < 0.001.
ACE inhibitor treatment does not alter Aβ levels in the brain or periphery of APP transgenic mice with plaque deposition
Though ACE inhibition does not regulate cerebral levels of Aβ in young, plaque-free mice, it is possible that ACE may play a role in Aβ metabolism in aged mice with an advanced stage of plaque deposition, when other Aβ clearance mechanisms may be saturated. We examined this question by treating J20 APP transgenic mice with vehicle or captopril administered in drinking water at 2 g/l. Mice were treated for 28 days, and at the end of the treatment period tissue was harvested for analysis of Aβ levels by ELISA. Brains were fractionated into aqueous, NP-40 detergent-soluble and guanidine-soluble pools (Fig 4 A–C), identically to the 3×Tg-AD mice. Assaying these different biochemical pools for Aβ1-Total (Fig 4 left panel), Aβ1–40 (Fig 4 middle panel) and Aβ1–42 (Fig 4 right panel) showed no difference in any Aβ species. Thus, prolonged captopril administration did not affect Aβ levels in the cerebrum of aged APP transgenic mice with significant Aβ burden.
Figure 4. ACE inhibitor treatment does not affect central or peripheral levels of Aβ in the J20 mouse model.
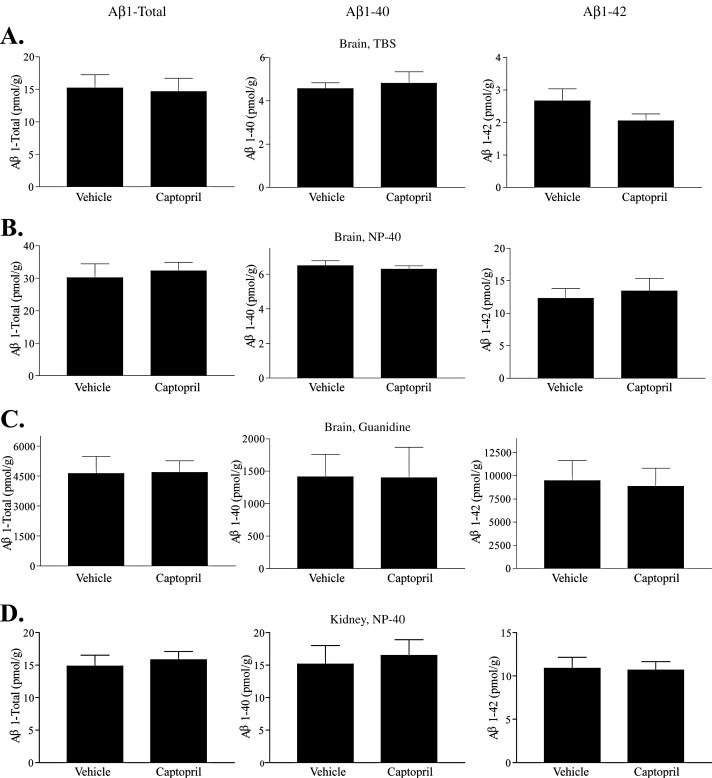
J20 mice were treated with captopril (2 g/l, n = 9) or vehicle (water, n = 10) for 28 days. Brains were fractionated into an aqueous TBS pool (A), an NP-40 detergent soluble pool (B), and a guanidine hydrochloride soluble pool (C). NP-40 soluble homogenate from the J20 kidney (D) were similarly analyzed for Aβ content. Aβ levels were measured by an ELISA specific for Aβ1-Total, Aβ1–-40 and Aβ1–42 (left panel, middle panel and right panel, respectively). Data represent the mean ± S.E.
Aβ is transported out of the central nervous system, where it is rapidly cleared by the peripheral organs (Shibata et al., 2000). To assess peripheral tissue levels of Aβ, the kidney was homogenized into an NP-40 detergent soluble pool. Kidney was chosen as the readout for peripheral tissue levels of Aβ because of its high endogenous expression of ACE protein and the nearly complete inhibition of kidney ACE activity by captopril administration. Despite prolonged and nearly complete ACE inhibition, we observed no difference in kidney Aβ levels between vehicle- and captopril-treated J20 APP transgenic mice (Fig 4D) in any species of Aβ tested. These data demonstrate that in aged APP transgenic mice, there is no alteration in Aβ levels in cerebral or peripheral tissue due to chronic ACE inhibitor administration.
Plaque deposition is not changed by ACE inhibitor treatment
Although we found no significant quantitative alteration in Aβ levels by ELISA in the J20 mice, chronic ACE inhibitor treatment could change the distribution or degree of plaque pathology through subtle changes in Aβ metabolism, vascular factors, or Aβ transport through the parenchyma or to the periphery. To examine this possibility, we analyzed the degree and localization of Aβ plaque burden in the J20 mice. Plaques immunoreactive for Aβ were found largely in the hippocampus and cortex (Fig 5), as expected (Mucke et al., 2000). Comparison of the degree of plaque deposition in the cortex (Fig 5A,C) and hippocampus (Fig 5B,D) using quantitative image analysis revealed no difference in Aβ immunoreactivity in either brain region (Fig 5E–F). We found no obvious differences in the type or distribution of plaques and no differences in the incidence of cerebral amyloid angiopathy. Therefore, we conclude that ACE inhibitor treatment does not change the distribution or degree of Aβ deposition in J20 APP transgenic mice.
Figure 5. ACE inhibitor treatment does not affect cerebral Aβ deposition.
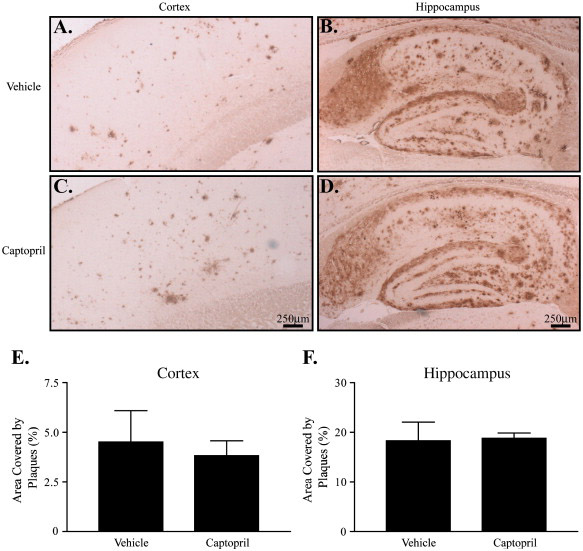
J20 mice treated for 28 days with vehicle (water, A–B, n = 4) or captopril (2 g/l, C–D, n = 4) were harvested and brains fixed for immunohistochemical analysis. Brain sections were stained for Aβ, and the plaques were visualized in cortex (A, C) and hippocampus (B, D). Sections shown are representative of the median degree of plaque burden for each group. The percent area covered by plaques in cortex (E) and hippocampus (F) was determined using computer-assisted image analysis. Scale bars, 250 µm. Data represent the mean ± S.E.
Discussion
A strong and growing body of genetic evidence links ACE polymorphisms to AD susceptibility, and alterations in ACE expression in post-mortem AD brain have been reported. We have previously demonstrated that overexpression of human ACE in a cell culture model results in the degradation and clearance of naturally secreted human Aβ and that this effect is attenuated by the ACE inhibitor captopril. These reports advanced the hypothesis that a genetic or pharmacologically-induced deficit in ACE activity could promote Aβ accumulation in the brain, which could thus elevate the risk of AD.
To study the effects of ACE inhibition on Aβ metabolism in vivo, we chronically treated two APP transgenic mouse lines with a 2 g/l dose of captopril. Patients undergoing treatment for hypertension take up to 600 mg/day of captopril (Lijnen et al., 1980). The dose of 2 g/l captopril administered to the mice in this study, resulting in a daily intake of 10–15 mg per mouse, is approximately 10 times the relative amount of drug used at the highest therapeutic doses in humans, accounting for a specific metabolic rate about 10 times higher in mice (de Cavanagh et al., 1997). The 2 g/l dosing regimen is consistent with previous studies examining chronic effects of captopril (Pfeffer et al., 1982), and a therapeutic lowering of blood pressure is achieved within 24 hrs of administration (Mattson and Krauski, 1998).
Here, we show that the prototypical ACE inhibitor, captopril, does not alter Aβ levels in cerebral or peripheral tissue of APP transgenic mice. The inability of significant ACE inhibition to modulate Aβ levels in the periphery may be due to the vast majority of peripheral Aβ being degraded by the liver (Ghiso et al., 2004), which notably does not express ACE or several other Aβ-degrading proteases, including neprilysin, that are important for Aβ clearance in the brain. We detect no differences in cerebral Aβ levels between control (either vehicle or the angiotensin II receptor antagonist losartan) and captopril administrations in mice harboring only non-deposited Aβ species. This finding cannot be due to upregulation of the Aβ-degrading protease neprilysin, as activity of this enzyme was not changed in the central nervous system or periphery of treated versus control mice. Similarly, in mice bearing significant plaque deposition and high levels of soluble Aβ, we find no differences in Aβ levels between control and captopril treated mice. Finally, we find no effect of chronic captopril administration on the deposition or distribution of Aβ plaques in aged APP transgenic mice.
Several studies have investigated the effects of ACE inhibitors on susceptibility to AD in human patients. These examinations vary in their conclusions from reporting no effect (Wolozin et al., 2000), to modest improvement (Ohrui et al., 2004), to a slightly elevated but non-significant hazard (Khachaturian et al., 2006) towards the risk and progression of AD. This lack of consensus demonstrates the need for further clinical investigation into this important issue, as ACE inhibitors are widely used for the treatment of hypertension and other disorders. Genetic studies of the ACE gene are similarly unsettled, though there is a consistently strong association of the I/D polymorphism with AD (Lehmann et al., 2005). Further scrutiny of the ACE locus may reveal the pathological polymorphism(s) that cause genetic association with AD, and such a discovery would inform future clinical and scientific inquiry into the mechanism by which ACE genetic variation modulates AD susceptibility.
It has been recently reported that a single acute administration of an ACE inhibitor, by both oral and intracerebroventricular routes, does not change Aβ levels in wild-type mice and young APP transgenic mice, and that there is no change in murine Aβ levels in ACE deficient mice (Eckman et al., 2006). The data we present are in agreement with these findings, although different experimental designs were employed which provide additional insight. In our experiments, we examined adult and aged mice that express human APP, and thus produce the human Aβ peptide, which has sequence and biochemical differences from murine Aβ. By examining aged APP transgenic mice with plaque deposition, we were also able to determine the effects of an ACE inhibitor on insoluble Aβ deposits. Finally, we orally administered captopril over a prolonged period of time to model the human situation, in which ACE inhibitors are taken chronically. Although we and others have previously shown that ACE inhibitors prevent ACE-mediated Aβ proteolysis in vitro, we did not find evidence of a role for ACE in Aβ metabolism in vivo, perhaps due to the lack of significant inhibition of brain ACE. It remains unclear whether, and in what time course or anatomical location, ACE activity regulates human Aβ levels in the central nervous system.
Our results show that prolonged captopril treatment does not cause accumulation of Aβ within the brain of APP transgenic mice. One explanation for this finding is the lack of inhibition of brain ACE by the drug. In a closely matched cohort of wild-type mice, we find a modest but significant reduction in brain ACE activity at the 2 g/l dose of captopril. However, when analyzing ACE activity in APP transgenic mice of slightly different ages and pooled from several litters, we find no significant inhibition of brain ACE activity. In contrast to the brain, kidney ACE activity was nearly completely inhibited in all experiments; nevertheless, kidney levels of Aβ in these human APP transgenic mice were not increased. Thus, even at the relatively high doses we employed, captopril showed limited ability to inhibit central nervous system ACE, perhaps secondary to poor brain penetration of the drug. It is possible that if ACE inhibitors better able to penetrate the brain were administered under experimental conditions similar to those used here, there could be some effect on brain Aβ catabolism. If such ACE inhibitors did decrease cerebral Aβ clearance, they may put individuals at risk for developing AD. We conclude from the present study that prolonged administration of the ACE inhibitor captopril, with its limited capacity to inhibit brain ACE, does not affect Aβ metabolism centrally or peripherally in APP transgenic mice.
Acknowledgments
We thank S. Schutz, C. Lemere and L. Jiang for assistance with immunohistochemistry, T. Ward and A. George for technical assistance, G. Shankar, I. Rappley and K. Dakin for critical scientific discussion, and N. Boucher for expert administrative support. This work was supported by the NIH Grant AG12749 (DJS), K08 NS046324 (WF) and a Pre-Doctoral Fellowship from the Harvard Center for Neurodegeneration and Repair (MLH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97:1411–1420. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- Caccamo A, et al. Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Coates D. The angiotensin converting enzyme (ACE) Int J Biochem Cell Biol. 2003;35:769–773. doi: 10.1016/s1357-2725(02)00309-6. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, et al. Enalapril and captopril enhance antioxidant defenses in mouse tissues. Am J Physiol. 1997;272:R514–R518. doi: 10.1152/ajpregu.1997.272.2.R514. [DOI] [PubMed] [Google Scholar]
- Eckman EA, et al. Regulation of Steady-state beta-Amyloid Levels in the Brain by Neprilysin and Endothelin-converting Enzyme but Not Angiotensin-converting Enzyme. J Biol Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- Eckman EA, et al. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- Evered MD, et al. Captopril given intracerebroventricularly, subcutaneously or by gavage inhibits angiotensin-converting enzyme activity in the rat brain. Eur J Pharmacol. 1980;68:443–449. doi: 10.1016/0014-2999(80)90419-7. [DOI] [PubMed] [Google Scholar]
- Falkevall A, et al. Degradation of the amyloid beta -protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006 doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- Farris W, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta -protein, and the beta -amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florentin D, et al. A highly sensitive fluorometric assay for "enkephalinase," a neutral metalloendopeptidase that releases tyrosine-glycine-glycine from enkephalins. Anal Biochem. 1984;141:62–69. doi: 10.1016/0003-2697(84)90425-1. [DOI] [PubMed] [Google Scholar]
- Ghiso J, et al. Systemic catabolism of Alzheimer's Abeta40 and Abeta42. J Biol Chem. 2004;279:45897–45908. doi: 10.1074/jbc.M407668200. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Selkoe DJ. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 2005;280:37644–37650. doi: 10.1074/jbc.M508460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, et al. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–47868. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- Iwata N, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Jalil JE, et al. Neutral endopeptidase and angiotensin I converting enzyme insertion/deletion gene polymorphism in humans. J Hum Hypertens. 2004;18:119–125. doi: 10.1038/sj.jhh.1001646. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongun L. Reciprocal regulation of angiotensin converting enzyme and neutral endopeptidase in rats with experimental hypertension. Physiol Res. 2004;53:365–368. [PubMed] [Google Scholar]
- Katzov H, et al. A cladistic model of ACE sequence variation with implications for myocardial infarction, Alzheimer disease and obesity. Hum Mol Genet. 2004;13:2647–2657. doi: 10.1093/hmg/ddh286. [DOI] [PubMed] [Google Scholar]
- Khachaturian AS, et al. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- Kolsch H, et al. ACE I/D polymorphism is a risk factor of Alzheimer's disease but not of vascular dementia. Neurosci Lett. 2005;377:37–39. doi: 10.1016/j.neulet.2004.11.062. [DOI] [PubMed] [Google Scholar]
- Konstam MA, et al. Comparison of losartan and captopril on heart failure-related outcomes and symptoms from the losartan heart failure survival study (ELITE II) Am Heart J. 2005;150:123–131. doi: 10.1016/j.ahj.2004.10.035. [DOI] [PubMed] [Google Scholar]
- Lehmann DJ, et al. Large Meta-Analysis Establishes the ACE Insertion-Deletion Polymorphism as a Marker of Alzheimer's Disease. Am J Epidemiol. 2005 doi: 10.1093/aje/kwi202. [DOI] [PubMed] [Google Scholar]
- Leissring MA, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Lijnen P, et al. Dose response in captopril therapy of hypertension. Clin Pharmacol Ther. 1980;28:310–315. doi: 10.1038/clpt.1980.167. [DOI] [PubMed] [Google Scholar]
- Lue LF, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson DL, Krauski KR. Chronic sodium balance and blood pressure response to captopril in conscious mice. Hypertension. 1998;32:923–928. doi: 10.1161/01.hyp.32.5.923. [DOI] [PubMed] [Google Scholar]
- Melchor JP, et al. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, et al. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesh R, et al. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;421:551. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- Oba R, et al. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci. 2005;21:733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- Oddo S, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ohrui T, et al. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology. 2004;63:1324–1325. doi: 10.1212/01.wnl.0000140705.23869.e9. [DOI] [PubMed] [Google Scholar]
- Patten RD, et al. Effects of angiotensin II receptor blockade versus angiotensin-converting-enzyme inhibition on ventricular remodelling following myocardial infarction in the mouse. Clin Sci (Lond) 2003;104:109–118. doi: 10.1042/CS20020219. [DOI] [PubMed] [Google Scholar]
- Pfeffer JM, et al. Regression of left ventricular hypertrophy and prevention of left ventricular dysfunction by captopril in the spontaneously hypertensive rat. Proc Natl Acad Sci U S A. 1982;79:3310–3314. doi: 10.1073/pnas.79.10.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigat B, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RA, et al. An improved fluorometric assay of rat serum and plasma converting enzyme. Hypertension. 1985;7:244–252. doi: 10.1161/01.hyp.7.2.244. [DOI] [PubMed] [Google Scholar]
- Savaskan E, et al. Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer's dementia. Neurobiol Aging. 2001;22:541–546. doi: 10.1016/s0197-4580(00)00259-1. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, et al. Boosting with intranasal dendrimeric Abeta1–15 but not Abeta1–15 peptide leads to an effective immune response following a single injection of Abeta1–40/42 in APP-tg mice. J Neuroinflammation. 2006;3:14. doi: 10.1186/1742-2094-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, et al. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker HM, et al. Plasmin deficiency does not alter endogenous murine amyloid beta levels in mice. Neurosci Lett. 2004;368:285–289. doi: 10.1016/j.neulet.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Wolozin B, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Yan P, et al. Matrix Metalloproteinase-9 Degrades Amyloid-beta Fibrils in Vitro and Compact Plaques in Situ. J Biol Chem. 2006;281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]