

Abstract
SHP-1 and SHP-2 are two SH2 domain-containing tyrosine phosphatases with major pathological implications in cell growth regulating signaling. They share significant overall sequence identity, but their biological functions are often opposite. SHP-1 is generally considered as a negative signal transducer while SHP-2 as a positive one. However, the precise role of each enzyme in shared signaling pathways is not well defined. In this study, we investigated the interaction of these two enzymes in a single cell system by knocking down their expressions with siRNAs and analyzing the effects on epidermal growth factor signaling. Interestingly, knockdown of either SHP-1 or SHP-2 caused significant reduction in the activation of ERK1/2 but not Akt. Furthermore, SHP-1, SHP-2, and Gab1 formed a signaling complex, and SHP-1 and SHP-2 interact with each other. The interaction of SHP-1 with Gab1 is mediated by SHP-2 since it was abrogated by knockdown of SHP-2 and SHP-2, but not SHP-1, binds directly to tyrosine phosphorylated Gab1. Together, the data revealed that both SHP-1 and SHP-2 have a positive role in epidermal growth factor-induced ERK1/2 activation and that they act cooperatively rather than antagonistically. The interaction of SHP-1 and SHP-2 may be responsible for hitherto unexpected novel regulatory mechanism of cell signaling by tyrosine phosphatases.
Introduction
SHP-1 and SHP-2 are Src homology 2(SH2) domain-containing tyrosine phosphatases. SHP-1 is highly expressed in hematopoietic cells and, at a lower level, in various nonhematopoietic cells, while SHP-2 is widely expressed (1). Both SHP-1 and SHP-2 have important pathological implications. In mice, mutation of the SHP-1 gene is responsible for the motheaten and viable motheaten phenotype (2, 3). In humans, reduction of SHP-1 gene expression is observed in natural killer cell lymphomas as well as other types of lymphomas/leukemias (4, 5). Methylation of the SHP-1 promoter causes loss of SHP-1 expression in malignant T- lymphoma cells (6). On the other hand, activation mutation of SHP-2 causes Noonan syndrome (7), an autosomal dominant disorder characterized by dysmorphic facial features, proportionate short stature and heart disease (most commonly pulmonic stenosis and hypertrophic cardiomyopathy). This gain-of-function mutation of SHP-2 is also associated with sporadic juvenile myelomonocytic leukemia, myelodysplasic syndrome, acute lymphoblastic leukemia, and acute myelogenous leukemia (8–11). Furthermore, SHP-2 is an intracellular target of Helicobacter pylori CagA protein (12) that is associated with severe gastritis and gastric cancer. SHP-1 and SHP-2 share over 55% sequence identity and are regulated by similar mechanisms. However, their biological functions are quite different (1). Motheaten and viable motheaten mice, which have mutations of the SHP-1 gene, develop a severe autoimmune and immunodeficiency syndrome characterized by increased numbers of many hematopoietic cells (2, 3). SHP-1 is thus generally considered as a negative regulator of cell proliferation. SHP-1 also dephosphorylates receptors of growth factors, cytokines, and antigens and tyrosine-phosphorylated proteins associated with these receptors, and therefore, it is often defined as a negative signal transducer. SHP-2, on the other hand, is generally considered as a positive signal transducer. Functional knockout of the Shp-2 gene in the mouse causes death of embryos at mid-gestation (13). Cells expressing a catalytically inactive cysteine-to-serine mutant of SHP-2 and those derived from SHP-2 knockout mice exhibited reduced activation of signal transduction pathways induced by growth factors and cytoskine (1, 14). SHP-2 also has a role in angiotensin II signaling which may be responsible for the defects in heart development associated with its mutation (15). The general concept is that SHP-1 and SHP-2 has opposite roles in cell signaling. However, there are data suggesting a positive role of SHP-1 in growth signaling as revealed by over-expression of a catalytically inactive C-to-S mutant of SHP-1 (16–18). In this study, we employed small interfering RNAs (siRNAs) to knock down the expression of SHP-1 and SHP-2 in a single cell system and analyzed the effects on EGF-induced cell signaling. Our results demonstrate that both SHP-1 and SHP-2 play a positive role in EGF signaling and interact with each other.
Experimental Procedures
Materials
Rabbit polyclonal antibodies against SHP-1, SHP-2, GAPDH, and ERK1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Gab1 and anti-phospho-ERK1/2 were from Upstate Biotechnology, Inc. (Lake Placid, NY) and Cell Signaling, Inc. (Beverly, MA), respectively. Recombinant SHP-1, SHP-2, maltose-binding protein (MBP) fusion protein of SHP-1 and glutathione-S-transferase (GST) fusion protein of SHP-2 were expressed and purified as previously described (19–22). Pre-designed SHP-1 and SHP-2 siRNA oligos were purchased from Ambion Inc. (Austin, TX). Vector-based siRNAs targeted at TGCGGCTGACATTGAGAAC of SHP-1 (ref 23) and CAGGAACTGAAATACGACG of SHP-2 were made by using the pSUPER-neo vector (OligoEngine, Seattle, WA) according to the manufacturer’s protocol.
Cell culture, transient transfection, stimulation, and lysis
We used colorectal adenocarcinoma Caco-2 cells to study the role of SHP-1 and SHP-2 in growth factor signaling. These cells express both of the phosphatases, and respond well to EGF stimulation. The cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Burlington, Ontario) containing 10% fetal bovine serum. The cells were transiently transfected with SHP-1 and SHP-2 siRNAs (Ambion) or pSuper RNAi constructs in the presence of lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. After transfection, the cells were cultured for 48 hr in a complete medium and then in a serum-free medium for 16 hr before stimulation with 50 ng/ml EGF for various periods of time. After washing with cold PBS three times, the cells were lysed in a cell lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 5 mM 2-mercaptoethanol, 50 mM NaF, 10 ug/ml leupeptin, 10 ug/ml aprotinin, 1 mM p-aminophenylmethanesulfonyl fluoride, and 1% (w/v) NP-40 buffer. The lysates were cleared by centrifugation at 15,000 ×g for 10 min at 4°C.
Immunoprecipitation and Immunoblotting
For immunoprecipitation, cell lysates (1.5 mg protein/ml) were pre-cleaned by protein A/G Sepharose (Santa Cruz Biotechnology) and then incubated with primary antibodies bound to protein A/G Sepharose at 4°C. After extensive washing with the aforementioned cell extraction buffer, proteins in the immunoprecipitates were separated on SDS gels and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% bovine serum albumin and then probed with specific primary antibodies followed by horse radish peroxidase-conjugated goat secondary antibodies. Detection was made by using the ECL system (Amersham Biosciences).
Expression of a GST fusion protein carrying the SHP-2-binding motifs of Gab1
Tyr-657 and Tyr-689 of Gab1 are known to be the binding sites for SHP2 (24). A C-terminal fragment (aa 635–724) of Gab1 that carries these two sites was expressed in E. coli cells as a GST fusion by using the pGex-2T vector. Tyrosine phosphorylation of the fusion protein was achieved by co-expressing an active form of c-Src in E. coli cells as we described for PZR in our earlier studies (25). The protein was purified by using glutathione-Sepharose. The non-phosphorylated and phosphorylated fusion proteins were designated GST-Gab1CT and GST-pGab1CT, respectively.
GST fusion protein pull-down assays
To study the interaction of SHP-1 with SHP-2, GST and GST-SHP-2 immobilized on glutathionesepharose were incubated with purified MBP and MBP-SHP-1 at 4 °C for 1 hr in a binding buffer containing 20 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, and 100 mM NaCl. To investigate the interaction of Gab1 with SHP-1 or SHP-2, GST-Gab1CT and GST-pGab1CT were immobilized on glutathionesepharose and incubated with purified SHP-1 or SHP-2 in the above buffer system. After extensive wash with the binding buffer, bound and unbound materials were separated on SDS gels and visualized by Coomassie blue staining or Western blotting analyses.
Results
Suppression of SHP-1 and SHP-2 expressions by siRNAs
Many previous studies on the role of SHP-1 and SHP-2 in cell signaling have employed over-expressing dominant negative mutants and fibroblast cells derived from knockout mice. While over-expression of dominant negative mutants may have non-specific effects, the knockout cells express truncated forms of the enzymes (1). The RNAi technique has been widely used to study the loss-of-function phenotype of genes. In this study, we first employed commercially available double strand RNAs to knock down the expression of SHP-1 and SHP-2 in Caco2 cells. Results are shown in Fig. 1. SHP-1 siRNA specifically suppressed the expression of SHP-1 by 80% but had no effect on that of SHP-2. Conversely, the siRNA of SHP-2 specifically knocked down the expression of SHP-2 by 70% but left SHP-1 intact. We also examined the protein levels and GAPDH and ERK1/2 which were not affected by the siRNAs either. The data suggest that these siRNAs worked very efficiently to suppress the expression of SHP-1 and SHP-2. This provides an excellent system to study the function of these enzymes in parallel.
Fig. 1.
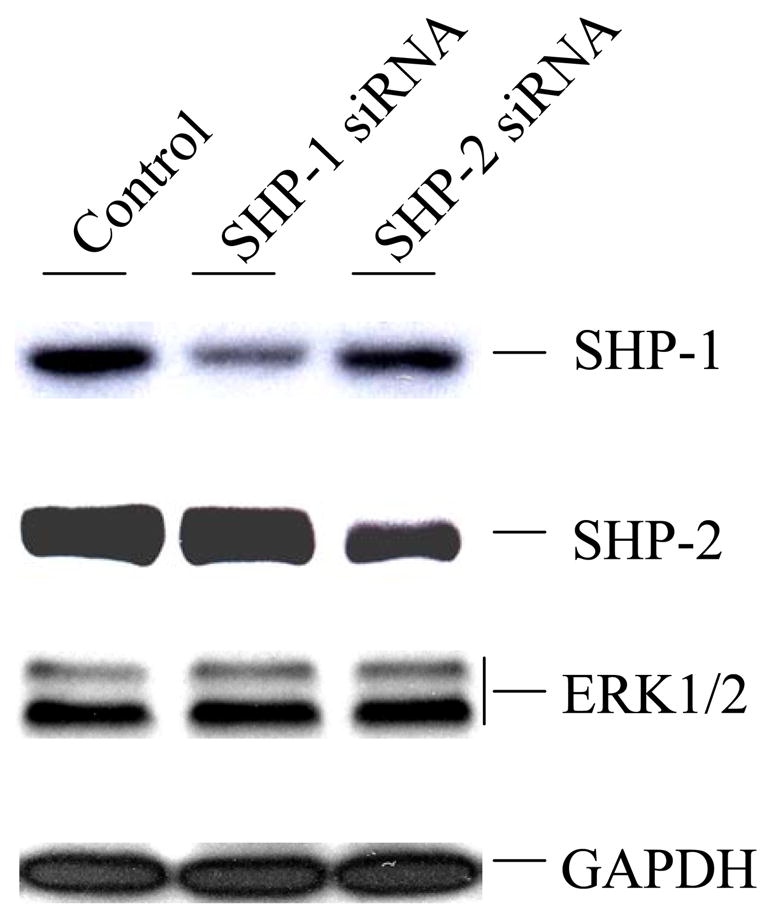
Knockdown of SHP-1 and SHP-2 expressions in Caco2 cells by siRNA. Caco2 cells (30–40% confluency) were transfected with the indicated siRNAs in the presence of lipofectamine 2000 as described in Experimental Procedures. Cell extracts containing equal amounts of total proteins were separated on 10% polyacrylamide gels, transferred to PVDF membranes, and subjected to Western blot analysis with antibodies against SHP-1, SHP-2, ERK1/2, and GAPDH as indicated.
Knockdown of SHP-1 or SHP-2 reduces EGF-induced activation of ERK1/2 but not of Akt
We treated the cells with EGF and analyzed the activation of down-stream signaling pathways. We employed phospho-specific antibodies to determine the activation of ERK1/2 and Akt. As shown in Fig. 2, knockdown of either SHP-1 or SHP-2 significantly reduced the activation of ERK1/2. The magnitude of ERK1/2 activation was reduced by about 60%. These data are very similar to those observed with cells over-expressing dominant negative mutants of SHP-1 or SHP-2 (16, 26). Thus, it furthers the positive role of both enzymes in EGF-induced ERK activation. We also analyzed the activation of Akt by using phosphospecific Akt antibodies. Interestingly, knockdown of either enzyme showed no significant effects on Akt. This suggests that SHP-1 and SHP-2 specifically regulate EGF activation of the ERK1/2 pathway but not that of Akt. To rule out possible non-specific effects of the siRNA oligos, we also employed vector-based siRNAs to knock down the expression of SHP-1 and SHP-2. Data shown in Fig. 3 demonstrated that the siRNA constructs also caused specific suppression of SHP-1 or SHP-2 expression and decreases in the EGF-induced activation of ERK1/2.
Fig. 2.

Effects of SHP-1 and SHP-2 knockdowns on EGF-induced activation of ERK1/2. Caco2 cells were treated as described in Fig. 1 except that immediately before extraction cells were stimulated with 50 ng/ml of EGF for the indicated periods of time. Cell extracts were subjected to Western blot analysis with specific antibodies that detect the indicated proteins. The line graphs in the right panel are quantitative representations of phospho-ERK2 and phospho-Akt levels (normalized with total ERK2 and Akt, respectively) based on gel scanning data from three independent experiments. Data represent relative band intensity with error bars denoting standard deviation.
Fig. 3.
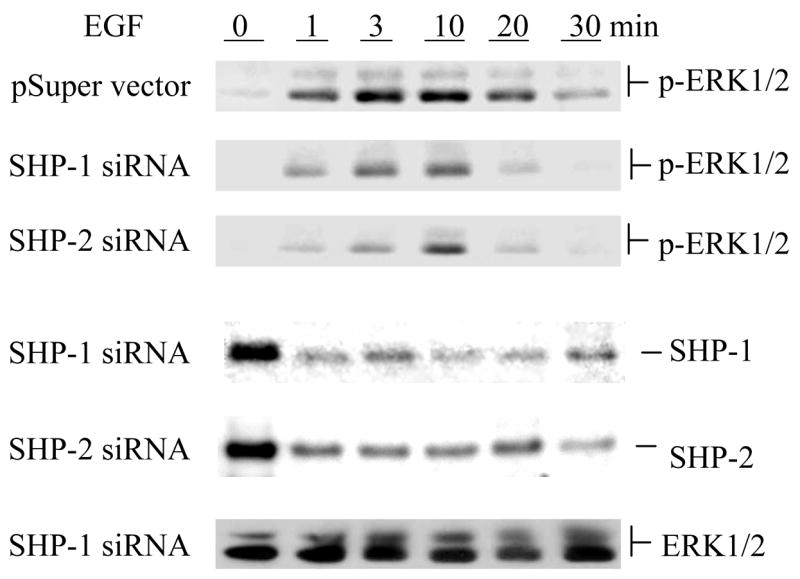
Verification of the specific effects of SHP-1 and SHP-2 knockdowns on EGF-induced activation of ERK1/2 by using vector-based siRNAs. Caco2 cells were transfected with empty pSure-neo vector or constructs carrying SHP-1 or SHP-2 siRNAs as described in Experimental Procedures. Cells were serum-starved and then stimulated with 50 ng/ml of EGF for the indicated periods of time. Cell extracts were subjected to Western blot analysis with specific antibodies that detect the indicated proteins.
SHP-1 and SHP-2 form a signaling complex with Gab1
It has been well accepted that signal transduction is carried through formation of multiple signaling complexes. This is particularly true for SHP-1 and SHP-2 since both enzymes stay largely inactive in cytosol due to their internal suppressed structures (27, 28). It is believed that they are activated by binding to tyrosine phosphorylated proteins (1). A major binding protein of SHP-2 involved in growth signaling is Gab1, an adaptor protein that undergoes tyrosine phosphorylation upon EGF stimulation (29). Binding of SHP-2 to Gab1 has been well documented. However, whether SHP-1 has a similar property is not clear. We thus performed immunoprecpitation to examine the interaction of SHP-1 and SHP-2 with Gab1. As shown in Fig. 4, SHP-2 was indeed co-immunoprecipitated with Gab1. Interestingly, SHP-1 was also found in the immuno-complex. The association of both SHP-1 and SHP-2 with Gab1 was markedly enhanced upon EGF stimulation. As with the activation of other signaling events, this association is transient. It peaked at 3 min and declined thereafter. Taken together, these biochemical analyses suggest that both SHP-1 and SHP-2 are linked to the EGF signaling pathway through Gab1.
Fig. 4.
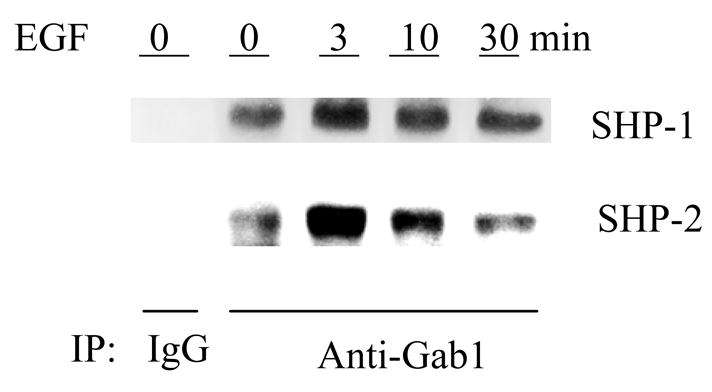
Association of SHP-1 and SHP-2 with Gab1. Subconfluenet Caco2 cells were serum-starved for 16 hr and then left untreated or treated with 50 ng/ml of EGF for the indicated periods of time. Cell extracts were immunoprecipitated with anti-Gab1 antibody. The immunoprecipitates were analyzed for the presence of SHP-1 and SHP-2 by Western blotting with antibodies against SHP-1 and SHP-2 as indicated. An irrelevant rabbit IgG was used as control.
SHP-2 directly interacts with SHP-1 and mediates the interaction of SHP-1 with Gab1
As an SH2 domain-containing protein, it is not surprising that SHP-1 also binds Gab1. The question is whether SHP-1 and SHP-2 affect each other in binding to Gab1. To address this issue, we repeated the above immunoprecipitation experiments with cells which had either SHP-1 or SHP-2 knocked down. Intriguing results emerged which showed distinct functions of SHP-1 and SHP-2. Whereas knockdown of SHP-1 had no effects on the association of SHP-2 with Gab1, knockdown of SHP-2 totally blocked the interaction of SHP-1 with Gab1 (Fig. 5). This suggests that SHP-2 likely mediates the interaction of SHP-1 with Gab1. The C-terminal portion of Gab1 contains two SHP-2 binding sites that resemble the so-called immunoreceptor tyrosine-based inhibition motifs found in many inhibitory receptors (29). It has been known that both SHP-1 and SHP-2 can bind to these motifs through their SH2 domains (1). To verify that the association of SHP-1 with Gab1 is not through their direct interaction, we performed binding assays with purified proteins. For this purpose, a C-terminal fragment of Gab1 (aa 635–724) that carries the two SHP-2 binding sites was expressed in E. coli cells as a GST fusion protein. When co-expressed in E. coli cells with c-Src as described for PZR in our earlier studies (25), the fusion protein was heavily phosphorylated on tyrosine. The phosphorylation presumably occurs on the SHP-2 binding sites because the Gab1 part of the fusion protein contains only two tyrosyl residues and GST per se could not be phosphorylated (25). After immobilization on glutathione Sepharose beads, the phosphorylated fusion protein GST-pGab1CT, but not the non-phosphorylated GST-Gab1CT, readily pulled down purified SHP-2. In contrast, neither could pull down SHP-1 (Fig. 6). These data indicate that SHP-1 does not directly bind to Gab1 through the mechanism used by SHP-2. One natural question then is whether SHP-2 carries SHP-1 to Gab1. We thus analyzed the co-immunoprecipitation of SHP-1 and SHP-2. Indeed, SHP-1 and SHP-2 formed a constitutive complex which was not affected by EGF stimulation (Fig. 7A). To prove a direct interaction between these two enzymes, we also performed in vitro pull-down assays with purified enzymes. As shown in Fig. 7B, a GST fusion protein of SHP-2 specifically pulled down a MBP fusion protein of SHP-1. In control experiments with GST or MBP alone, no binding was found. Additional experiments showed that GST-SHP-2 also effectively pulled down SHP-1 expressed as a non-fusion protein (data not shown). This provides evidence that SHP-1 and SHP-2 directly interact with each other. Since the SHP-1 and SHP-2 fusion proteins purified from E. coli cells were not phosphorylated, this interaction of SHP-1 and SHP-2 apparently does not involve tyrosine phosphorylation.
Fig. 5.
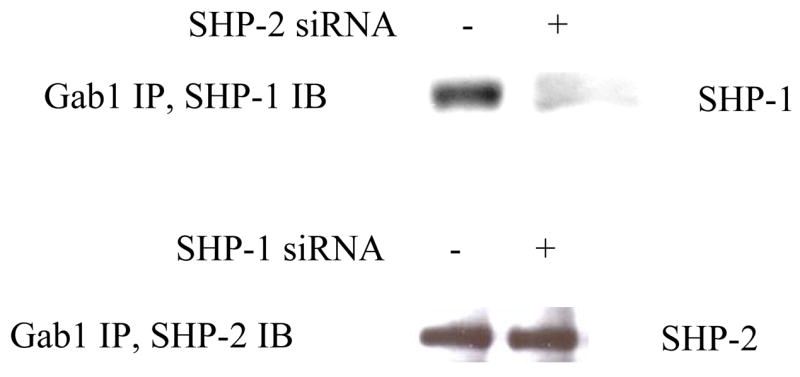
Disruption of Gab1/SHP-1 association by knockdown of SHP-2 expression. Caco2 cells were treated with SHP-1 or SHP-2 siRNA as described in Fig. 1. Cell extracted were immunoprecipitated with anti-Gab1 antibody, and the immunoprecipitates were subjected to Western blotting analysis with anti-SHP-1 or anti-SHP-2 antibodies as indicated.
Fig. 6.
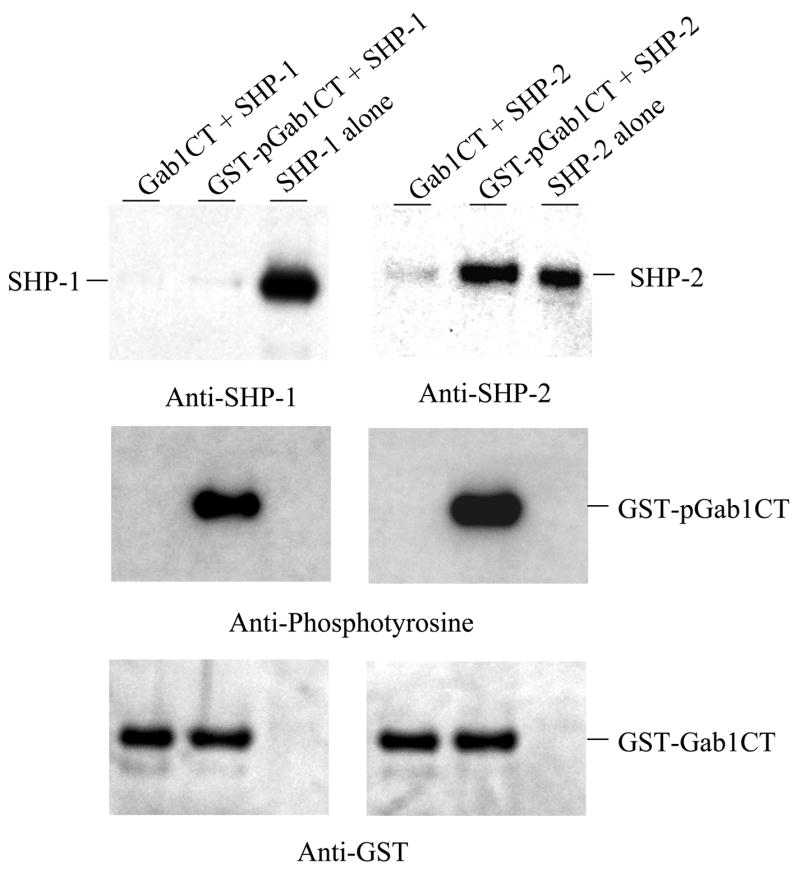
Direct binding of SHP-2 but not SHP-1 to tyrosine-phosphorylated Gab1. Non-phosphorylated GST-Gab1CT and phosphorylated GST-pGab1CT were immobilized onto glutathione-Sepharose beads and then incubated with purified SHP-1 and SHP-2. Proteins bound to the beads were analyzed by Western blotting with antibodies against SHP-1, SHP-2, phosphotyrosine, and GST. Purified SHP-1 and SHP-2 were loaded as controls.
Fig. 7.
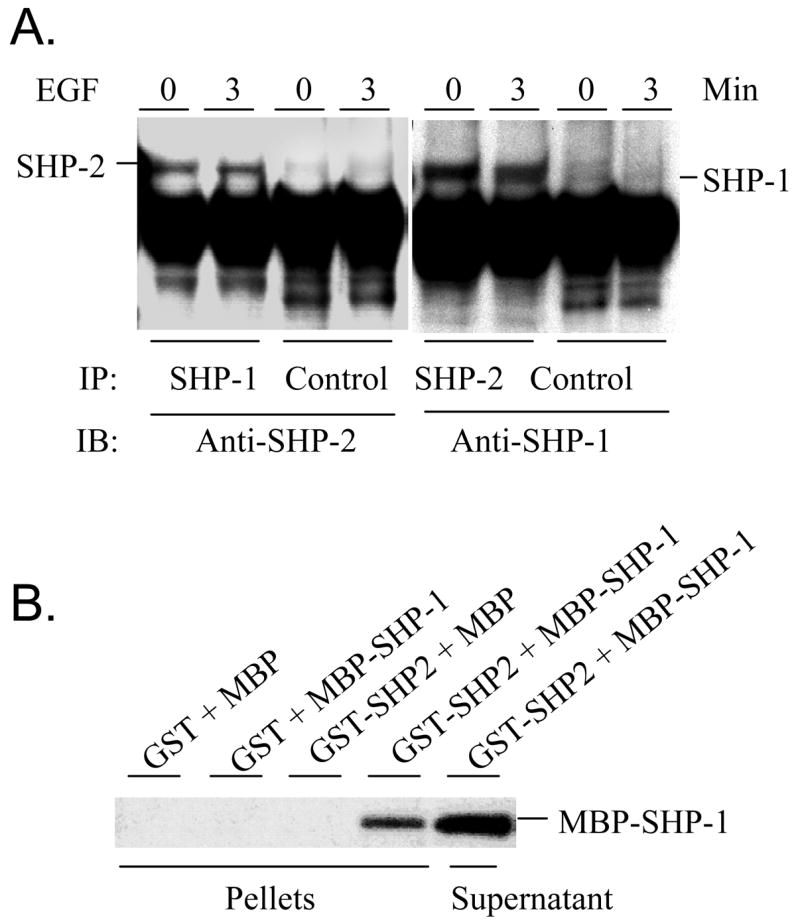
Direct interaction of SHP-1 with SHP-2. A. Subconfluent Caco2 cells were serum-starved for 16 hr and then left untreated or treated with 50 ng/ml of EGF for 3 min. Cell extracts were immunoprecipitated with anti-SHP-1 or ant-SHP-2 antibodies, and the immunoprecipitates were subjected to Western blotting analyses with antibodies against SHP-1 and SHP-2 as indicated. An irrelevant rabbit IgG was used as controls. B. GST and GST-SHP-2 immobilized onto glutathione-Sepharose beads were incubated with purified MBP or MBP-SHP-1. Proteins bound to the beads and remained in supernatants were analyzed by Western blotting with an anti-SHP-1 antibody.
Discussion
SHP-1 has been generally considered as a negative signal transducer, essentially as an antagonist of SHP-2. The present study suggests that this is a misconception. One of the reasons for such a misconception may be because studies on SHP-1 and SHP-2 to date were performed separately without due attention to their interaction. Our present results obtained with cells (Caco 2) expressing both SHP-1 and -2 demonstrate that like SHP-2, SHP-1 is also required for growth factor-induced activation of ERK1/2. Importantly, we have demonstrated a direct interaction of SHP-1 and SHP-2, implying that SHP-1 plays a complicated role in regulating the function of SHP-2. In C. elegans and Drosophila, there is only one SH2 domain-containing tyrosine phosphatase which has an essential role in embryonic development (30, 31). Mammals have two such enzymes. It should not be surprising that they have complementary roles in cell signaling. Although SHP-1 is highly expressed in hematopoietic cells, it is also widely expressed in other cell types. In fact, the expression of SHP-1 is controlled by two mutually exclusive cis promoters in the Shp-1 gene which give rise to either hematopoietic or non-hematopoietic form of SHP-1 (32). These two forms differ only in the first three amino acid residues at the N-terminus, and they presumably have similar activity and functions. In many non-hematopoietic cells (e.g., Caco 2 cells), the level of SHP-1 and SHP-2 appears to be comparable. The role of SHP-1 in immuno-response has been clearly demonstrated by the phenotype of motheaten and viable motheaten mice which bears natural mutation of SHP-1. At the biological function level, the concept of SHP-1 as a negative regulator mainly comes from the fact that the SHP-1 deficient mice have enhanced proliferation of hematopoietic cells (except for the red cell lineage) and that their lymphocytic cells exhibit hyper-response to antigens (1–3). At the cell signaling level, the negative role of SHP-1 is attributable to its ability to cause dephosphorylation of growth factor and cytokine receptors and associated tyrosine kinases. However, it should be noted that SHP-2 also dephosphorylates growth factor receptors but it enhances rather than inhibits the downstream signal transduction (33). We believe that the intensity of signal transduction activation may not be proportional to the level of tyrosine phosphorylation of upstream receptors at a steady state but to the dynamics of the phosphorylation. Of course, the site of phosphorylation also matters. Therefore, SHP-1 should not be viewed as a negative signal transducer simply because it dephosphoryaltes growth factor receptors. Our present study suggests SHP-1 and SHP-2 have a similar function in EGF signaling and that their roles are not redundant. They rely on each other to regulate cell signaling. In this regard the present results may also have therapeutic implications since diseases associated with loss-of-function of one enzyme can be treated by inhibiting the other enzyme.
By knocking down the expression of SHP-1 and SHP-2, our study also suggests that SHP-1 and SHP-2 specifically regulate the ERK1/2 pathway but not the Akt pathway. The detailed mechanism by which SHP-1 and SHP-2 regulate EGF-induced ERK activation remains unknown. It may not be a simple dephosphorylation of phosphorylated EGF receptor by SHP-1 or SHP-2. The ERK activation signaling pathway consists of upstream components Grb2, SOS, Ras, Raf, and MEK. Grb2 functions by directly binding to growth factor receptors or other associated adaptor proteins (e.g., Gab1 and SHC) thereby recruiting SOS. SOS, in turn, activates Ras, which leads to activation of the downstream cascade Raf/MEK/ERK1/2. SHP-2 is known to be required for maintaining normal levels of Ras activation. Ras is activated by guanine nucleotide exchange factor SOS but inactivated by GTPase-activating protein GAP. One scenario that SHP-2 promotes the ERK activation pathway is to prevent activation of GAP by dephopshorylating the phosphorylation site responsible for recruitment and activation of GAP. As a phosphatase, SHP-1 may play a similar role. In all, SHP-1 and SHP-2 presumably function by dephosphorylating some key components of the signaling pathways thereby facilitating signal transduction. However, their physiological substrates have not been clearly defined. This may be due to the transient nature of phosphorylation of these substrates which makes its detection and quantification difficult.
Signal transduction is carried through formation of multiple signaling complexes involving adaptors and scaffold proteins.The Gab family adapter proteins are scaffolding adapter molecules that display sequence similarity with Drosophila DOS (daughter of sevenless), which is initially identified as a substrate for Corkscrew, the SHP-2 homolog in Drosophila, and they contain a pleckstrin homology domain and binding sites for SH2 and SH3 domains thereby recruiting various signaling proteins (29). Studies in multiple systems have implicated that Gab1 has a critical role in signaling via many growth factor and cytokine receptors. Gab1-deficient mouse embryos die in utero with multiple defects in placental, heart, skin, and muscle development similar to phenotypes observed in mice lacking signals of EGF, platelet-derived growth factor, and hepatocyte growth factor pathways (34). Binding of SHP-2 with Gab1 has been well defined and is believed to be critical for Gab1-mediated signal transduction, and Gab1 mutants lacking SHP-2 binding sites are unable to activate ERK1/2 and downstream transcription factors (24, 29). In fact, a major function of SHP-2 is to dephosphorylate the GAP binding sites on Gab1 thereby disengaging GAP and sustaining Ras activation (35). Our study suggests that SHP-1 also binds to Gab1 but through SHP-2. Participation of both SHP-1 and SHP-2 in the Gab1 signaling complex presumably provides more versatilities and flexibility for cells to control signal transduction. First, interactions of SHP-1 and SHP-2 may affect each other’s catalytic activity. Second, SHP-1 and SHP-2 may have different substrate specificity and thus preferentially act on different proteins. Even with shared substrates, they may have different catalytic efficiencies (19, 20). Taken together, a fine tune of cell signaling can be achieved by increasing or decreasing the activity of either SHP-1 or SHP-2.
Acknowledgments
This work was supported by grants HL058205 (to T. Inagami) and HL076309 (to ZJ Zhao) from the National Institutes of Health.
Abbreviations
- EGF
epidermal growth factor
- MBP
maltose-binding protein
- GST
glutathione-S-transferase
- ERK
extracellular signal-regulated kinase
- Gab
Grb2-associated binder
- siRNA
small interfering RNA
References
- 1.Neel BG, Gu H, Pao L. Trends Biochem Sci. 2003;28:284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 2.Tsui HW, Siminovitch KA, Souza L, Tsui FWL. Nature Genetics. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 3.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 4.Wu C, Sun M, Liu L, Zhou GW. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 5.Oka T, Yoshino T, Hayashi K, Ohara N, Nakanishi T, Yamaai Y, Hiraki A, Sogawa CA, Kondo E, Teramoto N, Takahashi K, Tsuchiyama J, Akagi T. Am J Pathol. 2001;159:1495–505. doi: 10.1016/S0002-9440(10)62535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Raghunath PN, Vonderheid E, Odum N, Wasik MA. Am J Pathol. 2000;157:1137–46. doi: 10.1016/S0002-9440(10)64629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, Van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 8.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 9.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Blood. 2004;103:2325–31. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Arico M, Masera G, Basso G, Sorcini M, Gelb BD, Biondi A. Blood. 2004;104:307–13. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 11.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du J, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG. Cancer Res. 2004;64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 12.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. Science. 2002;295:683–6. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 13.Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T. EMBO J. 1997;16:2352–64. doi: 10.1093/emboj/16.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng GS. Exp Cell Res. 1999;253:47–54. doi: 10.1006/excr.1999.4668. [DOI] [PubMed] [Google Scholar]
- 15.Tang H, Zhao ZJ, Landon EJ, Inagami T. J Biol Chem. 2000;275:8389–96. doi: 10.1074/jbc.275.12.8389. [DOI] [PubMed] [Google Scholar]
- 16.Su L, Zhao Z, Bouchard P, Banville D, Fischer EH, Krebs EG, Shen SH. J Biol Chem. 1996;271:10385–90. doi: 10.1074/jbc.271.17.10385. [DOI] [PubMed] [Google Scholar]
- 17.Krautwald S, Buscher D, Kummer V, Buder S, Baccarini M. Mol Cell Biol. 1996;16:5955–5963. doi: 10.1128/mcb.16.11.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You M, Zhao Z. J Biol Chem. 1997;272:23376–23381. doi: 10.1074/jbc.272.37.23376. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z, Bouchard P, Diltz CD, Shen SH, Fischer EH. J Biol Chem. 1993;268:2816–2820. [PubMed] [Google Scholar]
- 20.Zhao Z, Larocque R, Ho WT, Fischer EH, Shen SH. J Biol Chem. 1994;269:8780–8785. [PubMed] [Google Scholar]
- 21.Law CL, Sidorenko SP, Chandran KA, Zhao Z, Shen SH, Fischer EH, Clark EA. J Exp Med. 1996;183:547–60. doi: 10.1084/jem.183.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu F, Zhao R, Peng Y, Guerrah A, Zhao ZJ. J Biol Chem. 2001;276:29479–84. doi: 10.1074/jbc.M104428200. [DOI] [PubMed] [Google Scholar]
- 23.Chen D, Iijima H, Nagaishi T, Nakajima A, Russell S, Raychowdhury R, Morales V, Rudd CE, Utku N, Blumberg RS. J Immunol. 2004;172:3535–3543. doi: 10.4049/jimmunol.172.6.3535. [DOI] [PubMed] [Google Scholar]
- 24.Cunnick JM, Mei L, Doupnik CA, Wu J. J Biol Chem. 2001;276:24380–7. doi: 10.1074/jbc.M010275200. [DOI] [PubMed] [Google Scholar]
- 25.Zhao R, Fu X, Teng L, Li Q, Zhao ZJ. J Biol Chem. 2003;278:42893–42898. doi: 10.1074/jbc.M306136200. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Z, Tan T, Wright JH, Diltz CD, Shen SH, Krebs EG, Fischer EH. J Biol Chem. 1995;270:11765–69. doi: 10.1074/jbc.270.20.11765. [DOI] [PubMed] [Google Scholar]
- 27.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, Zhou GW. J Biol Chem. 2003;278:6516–20. doi: 10.1074/jbc.M210430200. [DOI] [PubMed] [Google Scholar]
- 29.Nishida K, Hirano T. Cancer Sci. 2003;94:1029–33. doi: 10.1111/j.1349-7006.2003.tb01396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutch MJ, Flint AJ, Keller J, Tonks NK, Hengartner MO. Genes Dev. 1998;12:571–85. doi: 10.1101/gad.12.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perkins LA, Larsen I, Perrimon N. Cell. 1992;70:225–36. doi: 10.1016/0092-8674(92)90098-w. [DOI] [PubMed] [Google Scholar]
- 32.Banville D, Stocco R, Shen SH. Genomics. 1995;27:165–73. doi: 10.1006/geno.1995.1020. [DOI] [PubMed] [Google Scholar]
- 33.Zhao RJ, Zhao ZJ. Biochem J. 1999;338:35–39. [PMC free article] [PubMed] [Google Scholar]
- 34.Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. Nature. 1996;379:560–4. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 35.Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. J Biol Chem. 2005;280:5350–5360. doi: 10.1074/jbc.M410012200. [DOI] [PubMed] [Google Scholar]