

Abstract
The Vaccine Research Center has developed a number of vaccine candidates for different diseases/infectious agents (HIV-1, Severe Acute Respiratory Syndrome virus, West Nile virus, and Ebola virus, plus a plasmid cytokine adjuvant—IL-2/Ig) based on a DNA plasmid vaccine platform. To support the clinical development of each of these vaccine candidates, preclinical studies have been performed in mice or rabbits to determine where in the body these plasmid vaccines would biodistribute and how rapidly they would clear. In the course of these studies, it has been observed that regardless of the gene insert (expressing the vaccine immunogen or cytokine adjuvant) and regardless of the promoter used to drive expression of the gene insert in the plasmid backbone, the plasmid vaccines do not biodistribute widely and remain essentially in the site of injection, in the muscle and overlying subcutis. Even though ∼ 1014 molecules are inoculated in the studies in rabbits, by day 8 or 9 (∼ 1 week postinoculation), already all but on the order of 104–106 molecules per microgram of DNA extracted from tissue have been cleared at the injection site. Over the course of 2 months, the plasmid clears from the site of injection with only a small percentage of animals (generally 10–20%) retaining a small number of copies (generally around 100 copies) in the muscle at the injection site. This pattern of biodistribution (confined to the injection site) and clearance (within 2 months) is consistent regardless of differences in the promoter in the plasmid backbone or differences in the gene insert being expressed by the plasmid vaccine. In addition, integration has not been observed with plasmid vaccine candidates inoculated i.m. by Biojector 2000 or by needle and syringe. These data build on the repeated-dose toxicology studies performed (see companion article, Sheets et al., 2006) to demonstrate the safety and suitability for investigational human use of DNA plasmid vaccine candidates for a variety of infectious disease prevention indications.
Keywords: DNA vaccines, HIV/AIDS, SARS, WNV, Ebola, DNA vaccine biodistribution, DNA vaccine integration, plasmid vaccines
DNA vaccines are a novel product class with limited (though ever increasing) clinical experience, beginning with clinical trials in the late 1990's (Ulmer et al., 1996). Thus, their development represents a unique preclinical challenge. The U.S. FDA (Food and Drug Administration) has provided manufacturers with guidance on toxicology studies they deem necessary to support the safety of this novel product class to enter clinical trials (CBER, 1996, 2005). Among these recommended studies are repeated-dose toxicology, biodistribution, and integration analyses. DNA vaccines consist of a closed circular bacterial plasmid(s) containing a bacterial origin of replication, (generally) a selectable marker such as an antibiotic resistance gene and a gene insert expressing a vaccine immunogen or cytokine adjuvant under the control of a (generally) strong mammalian promoter (often one from a human or animal virus) and polyadenylation signals for good transcriptional expression in the vaccinee. Frequently, the gene insert has been codon optimized for efficient translation in the species to be vaccinated.
The Vaccine Research Center (VRC) is developing several vaccine candidates for human diseases based on a DNA vaccine platform. These various candidate vaccines are intended for use in prevention of diseases from emergent viruses, such as the HIVAIDS, severe acute respiratory syndrome (SARS) virus, or West Nile virus (WNV), as well as for counterterrorism measures, such as vaccines against Ebola virus. An immune response is generated by the in vivo expression of the viral proteins after inoculation with the plasmid vaccine candidates encoding them. In addition, one plasmid described herein expresses a cytokine adjuvant. Therefore, the authors have performed numerous similar biodistribution and repeated-dose toxicology studies on similar DNA vaccines expressing a variety of vaccine immunogens and a cytokine adjuvant to support the preclinical development of each of these vaccine candidates. The experience thereby gained has permitted refinements of protocols over time, as well as eventual efficiencies in preparing regulatory submissions by providing more data on the safety of various different candidate vaccines. These similarities are observed even with vaccines for differing disease indications, built on similar, although not identical, plasmid vaccine platforms. This has expedited the clinical development of newer candidates with significant savings in time and resources.
A companion article in this issue provides the results of repeated-dose toxicology studies performed on the same vaccines for which the biodistribution and integration analyses are described in this article. Target organs for toxicity, other than the injection sites, were not identified in the toxicology studies, and this correlated well with the lack of distribution outside of the injection site by the vaccines, as described in this article.
MATERIALS AND METHODS
Plasmid Vaccines and Plasmid Cytokine Adjuvant
Product VRC-3900 (HIV-1).
This precursor plasmid vaccine construct (6438 bp) encodes the gag gene of the HXB2 strain of HIV-1 (Table 1). Briefly, it is built on the pVR1012x/s plasmid backbone containing a kanamycin resistance gene, the human cytomegalovirus (CMV) promoter/enhancer (containing the immediate early 5′ untranslated region and intron), and the bovine growth hormone (BGH) polyadenylation sequence. This backbone differs from a related backbone (Hartikka et al., 1996) used in products described below by removal of an XmnI site in the region following the BGH poly A sequence and replacement with an SfiI site. This was done to ensure that at least one SfiI site was present in each plasmid construct to permit use of SfiI in the integration studies. When the gene insert contained an SfiI site, the pVR1012 backbone was used. When it did not, the pVR1012x/s backbone, with the introduced SfiI site, was used. A synthetic version of the gag gene using codons optimized for expression in human cells was inserted into this plasmid backbone (pVR1012x/s). The nucleotide sequence of the synthetic gag gene shows little homology to the HXB2 gene, but the protein encoded is the same. This candidate vaccine did not enter clinical trials because development of multigenic constructs was pursued instead.
TABLE 1.
Materials and Methods—Products Tested
|
Product |
Study number |
Disease |
Genes |
Plasmid number |
Promoter/plasmid backbone |
|---|---|---|---|---|---|
| NA | A | HIV | Clade B gag | 3900 | CMV/pVR1012x/s |
| 1 | B, E, F, G, H | HIV | Clade B gag-pol | 4302 | CMV/pVR1012x/s |
| 2 | D | HIV | Clade B gag-pol-nef, env | 4306, 2805 | CMV/pVR1012, CMV/pVR1012x/s |
| 3 | See companion article | HIV | Clade B gag-pol, env | 4302, 2805 | CMV/pVR1012x/s (both) |
| 4 | D | Adjuvant | IL-2/Ig | 7000 | CMV/pVR1012x/s |
| 6 | D | HIV | Clade A gag-pol-nef, env Clade B gag-pol-nef, env Clade C gag-pol-nef, env | 4313, 5305, 4306, 2805 4311, 5309 | CMV/pVR1012x/s (env), CMV/pVR1012 (gpn) |
| 7 | C, I | Ebola | NP (Zaire) GP (Zaire, Sudan, and Ivory Coast) | 6401 6001, 6201, 6301 | CMV/pVR1012 (all) |
| 12 | E | Ebola | NP (Zaire) GP (Zaire, Sudan/Gulu) | 6402, 6605, 6204 | CMV/R (all) |
| 15 | F | SARS | Spike protein | 8318 | CMV/R |
| 17 | G | WNV | PreM and E | 8109 | CMV/pVR1012 |
| Comparator |
D |
Malaria |
Plasmodium falciparum Circumsporozoite protein |
VCL-2510 |
CMV |
Product 1 (HIV-1): VRC-4302.
This first-generation plasmid vaccine candidate (9170 bp) was generated by inserting into the pVR1012x/s plasmid backbone a synthetic human codon–optimized gag gene from the HXB2 strain of HIV-1 ligated in frame with sequences encoding a synthetic (human codon optimized) pol polyprotein from the NL4-3 strain of HIV-1. To allow for the natural translational frameshifting, the synthetic coding region of the last four amino acids of the nucleocapsid protein through the rest of gag plus an additional three amino acids from pol were replaced with the authentic viral sequences from NL4-3. This introduces the site required for frameshifting and restores the ability to express all the gag proteins. Mutations were introduced into the protease-, RT-, and integrase-encoding sequences to destroy the biological activities of these viral proteins for safety while retaining the maximal number of potential epitopes for immunogenicity when inoculated into humans as a vaccine candidate (Huang et al., 2001). A phase I trial of this product was completed, and this product also served as a comparator plasmid for later products tested in biodistribution studies.
Product 2 (HIV-1).
The product is a combination of two plasmids, VRC-2805 and VRC-4306. VRC-2805 (6869 bp) consists of a HIV-1 clade B env gene inserted into the pVR1012x/s backbone, described above. The env gene contains the following modifications: a human codon–optimized sequence from HXB2 into which sequences from BaL were inserted to change the tropism from the CCR4 receptor to the CXCR5 receptor. The chimeric env gene was truncated to include the entire SU protein–coding region and a portion of TM, including the fusion domain, the transmembrane domain, and regions important for oligomerization. A portion of the fusion/cleavage domain has been deleted, as has been the interspace between heptads 1 and 2. This version of env is referred to as gp145Δcfi (Chakrabarti et al., 2002). VRC-4306 (9790 bp) encodes human codon–optimized gag, pol, and nef genes expressed as a fusion protein. Deletions have been introduced into the protease-, RT-, and integrase-coding regions to destroy the biological activities of these viral proteins for safety reasons. In addition, the authentic viral sequences were included in the region to allow for frameshifting (as described above for Product 1 [HIV-1]). The gag portion of the fusion protein is from the HXB2 strain, and the pol and nef portions are from the NL4-3 strain (the entire nef including the initial ATG is included in the construct). This was inserted into the pVR1012 backbone. This HIV-1 clade B combination product was not tested in clinical trials on its own because a multiclade approach was chosen for clinical development. However, the two components of this vaccine candidate are included among the components of a product that proceeded into clinical trials (see description of Product 6 [HIV-1] below for more details).
Product 3 (HIV-1).
This product is a combination of two plasmids. VRC-2805 is a component of Product 2 (HIV-1) described above. VRC-4302 is Product 1 (HIV-1) described above. This HIV-1 clade B Product 3 also did not proceed to clinical trials because a multiclade approach was chosen for clinical development.
Product 4 (cytokine adjuvant plasmid).
VRC-7000 (6086 bp) encodes a fusion protein human IL-2/Ig inserted into the pVR1012x/s backbone. The entire coding region of the human IL-2 cDNA is included (native sequence without mutations), as is the Fc portion of a human IgG2 (Barouch et al., 2004).
Product 6 (HIV-1).
This product is a combination of six plasmids. VRC-2805 (clade B env gene) and VRC-4306 (clade B gag-pol-nef) are described above in Product 2 (HIV-1). VRC-4311 (9786 bp) consists of a human codon–optimized gene encoding a gag-pol-nef fusion protein (modified as described for VRC-4306) from a clade C sequence identified as GenBank accession number U52953. The stop codon TAG was removed from the gag-pol gene, and the nef gene from the same virus was ligated in. The fusion sequence was inserted into the pVR1012 backbone. VRC-5309 (6829 bp) consists of a human codon–optimized gene encoding the env protein (modified as described for VRC-2805 to gp145Δcfi) from HIV-1 clade C 97ZA012 strain inserted into the pVR1012x/s backbone. VRC-4313 (9783 bp) consists of a human codon–optimized gene encoding a gag-pol-nef fusion protein (modified as described for VRC-4306) from a HIV-1 clade A strain identified as GenBank accession number AF004885 (gag-pol) and AF069670 (nef) inserted in the pVR1012 backbone. VRC-5305 (6836 bp) consists of a human codon–optimized gene encoding the env protein (modified as described for VRC-2805 to gp145Δcfi) from HIV-1 clade A 92rw020 strain inserted into the pVR1012x/s backbone. This product did not proceed into clinical trials although a product consisting of four of the six plasmids (VRC-2805, VRC-4306, VRC-5309, and VRC-5305) contained in this product was tested in a completed phase I trial and is being tested in other ongoing phase I trials. The basis for deciding to clinically develop this four-plasmid product instead of Product 6 (HIV-1) was as a result of murine (Kong et al., 2003) and nonhuman primate (NHP, unpublished data) immunogenicity studies. Further NHP studies led to the development of another six-plasmid product containing the CMV/R promoter (see below in Product 12 [Ebola] for description) and separating the gag, pol, and nef onto separate plasmids (Barouch et al., 2005). The preclinical studies described herein and in the companion article for Product 6 (HIV-1), as well as those for Product 12 (Ebola), along with phase I clinical data, supported the development of this newer generation six-plasmid product which has also entered phase I testing (Martin et al., 2005) and is proceeding into advanced clinical development as a prime for an adenovirus type 5-vectored boost HIV vaccine regimen.
Product 7 (Ebola).
This first-generation Ebola product (Sullivan et al., 2000; Xu et al., 1998) consists of a combination of four plasmids. VRC-6401 (7329 bp) consists of a synthetic human codon–optimized gene expressing the nucleoprotein (NP) of the Zaire strain of Ebola virus inserted into the pVR1012x/s backbone. VRC-6201 (7087 bp) consists of a synthetic human codon–optimized gene expressing the full-length glycoprotein (GP) of the Sudan strain of Ebola virus inserted into the pVR1012x/s backbone. VRC-6301 (7036 bp) consists of a synthetic human codon–optimized gene expressing the full-length GP protein of the Ivory Coast strain of Ebola virus inserted into the pVR1012x/s backbone. VRC-6001 (7188 bp) consists of a synthetic human codon–optimized gene expressing the full-length GP protein of the Zaire strain of Ebola virus inserted into the pVR1012x/s backbone. However, this product did not proceed to clinical trial because of observed in vitro cytotoxicity with the full-length GP constructs (Sullivan et al., 2005) and the theoretical safety concerns this phenomenon raised; modified GP vectors were developed.
Product 12 (Ebola).
This second-generation Ebola product consists of a combination of three plasmids. The backbone, referred to as CMV/R, used for each of these plasmids differs from the pVR1012x/s and pVR1012 plasmid backbones in that it substitutes the Human T-Lymphotrophic Virus (HTLV-1) Long Terminal Repeat (LTR) R-U5 region for the CMV immediate early region 1 enhancer to improve gene expression (Barouch et al., 2005). VRC-6402 (6625 bp) consists of a synthetic human codon–optimized gene expressing the NP protein of the Zaire strain of Ebola virus. VRC-6605 (6337 bp) consists of a synthetic human codon–optimized gene expressing a transmembrane-deleted GP protein of the Zaire strain of Ebola virus. VRC-6204 (6327 bp) consists of a synthetic human codon–optimized gene expressing the transmembrane-deleted GP protein of the Sudan/Gulu strain of Ebola virus. A phase I trial of this product has been completed and represents the first clinical trial of an Ebola virus vaccine candidate in the world.
Product 15 (SARS).
VRC-8318 (8164 bp) encodes a human codon–optimized gene expressing the S glycoprotein from the Urbani strain of SARS in the CMV/R backbone described above. The transmembrane region and a portion of the cytoplasmic domain were included (Yang et al., 2004). This first-generation SARS product is being tested in an ongoing clinical trial and represents the first SARS vaccine candidate to be tested in clinical trial in the United States.
Product 17 (WNV).
VRC-8109 (6982 bp) encodes human codon–optimized WNV preM and E protein genes from the NY99 strain in the pVR1012 backbone. In addition, a leader sequence, which has also been human codon optimized, from the SA-14 isolate of Japanese encephalitis virus has been included. This first-generation WNV is being tested in an ongoing human clinical trial.
Comparator Plasmid
The earlier studies involved comparison to a DNA plasmid vaccine from the U.S. Navy's malaria program, which had been previously tested preclinically and clinically. This product is briefly described in Epstein et al. (2002, 2004). Later studies involved comparison to Product 1 (HIV-1, VRC-4302) described above.
Delivery
Many of the studies were performed with the Biojector 2000 (referred hereafter as Biojector) device, a needleless injection system cleared by FDA for i.m. and s.c. injections of liquid medications, including vaccines. The liquid vaccine is forced through a tiny orifice held against the skin creating a very fine, high-pressure stream that penetrates the skin, depositing the vaccine in the tissue beneath. The system has three components: the reusable device, a sterile single-use disposable needle-free syringe, and a CO2 cartridge. Earlier studies in mice were performed with needle and syringe because the precise device to be used in our clinical trials was not suitable for the size of the species without modification.
Biodistribution Study Designs
Different groups of animals were inoculated for biodistribution studies than for repeated-dose toxicology studies for the following reasons. In the biodistribution analyses, the animals were only inoculated once to determine where the inoculum subsequently biodistributed, whereas in the repeated-dose toxicology studies, the animals were repeatedly dosed. The timing of terminations in the biodistribution studies (generally 1 week and 1 and 2 months post-inoculation) was markedly different from the repeated-dose inoculations and timing of terminations (generally 2 days and 2 weeks after last inoculation) in the repeated-dose toxicology studies.
Studies A, B, and C were performed by BioReliance (Rockville, MD) and involved inoculation of 6- to 7-week-old male and nonpregnant female Hsd:ICR (CD-1) mice with 100 mcg (on the order of 1011 molecules) of DNA plasmid vaccine by either the i.m. or i.v. (data not shown) routes. These were compared to inoculation of phosphate-buffered saline (PBS) by each route. Animals were inoculated at one time point, and five animals per gender per inoculation route (test article; one animal per gender per inoculation route control) were terminated at study days 8 and 50. The following organs were collected for biodistribution analyses: gonads, brain, kidneys, mesenteric lymph nodes, lung, liver, right thigh muscle (injection site in case of i.m. inoculation), bone marrow (left femur), heart, and blood. Tissues were snap frozen in liquid nitrogen after being placed in sterile vials. In addition to tissues being collected for biodistribution, animals were monitored for morbidity and mortality twice daily and for clinical signs of toxicity (body position; locomotor activity; secretions from eyes, nose, or mouth; coat condition; respiration; excretions; muscle tone; body tremors) daily (data not shown). Body weights were measured at days 1, 8, and 50 (prior to termination; data not shown). Gross pathology was observed at termination (data not shown). These studies were performed in compliance with Good Laboratory Practices (GLP) regulations (21 CFR 58).
Studies D, E, F, and G were performed by GeneLogic (formerly Therimmune) and involved inoculation of 12- to 13-week-old New Zealand white rabbits with 2 mg (moderate human dose, on the order of 1014 molecules) of DNA plasmid delivered i.m. by the Biojector needleless injection device, which is the method also used for injection of the candidate vaccines in clinical studies. In each study, comparison was made to PBS and to a comparator plasmid (VCL-2510 [malaria] for Study D, VRC-4302 [HIV-1] for Studies E, F, and G) for which there was prior biodistribution, toxicology, and integration data, as well as clinical safety data. Animals were inoculated at one time point and terminated by sodium thiopental injection (Study D), Nembutal sodium injection (Studies F and G), or sodium pentobarbital injection (Study E) and exsanguination at days 8 or 9, 30 or 31, and 60 or 61. The variance in study days for termination between studies was based on pragmatic scheduling considerations at the animal facility, and the time points were approximatly 1 week and 1 and 2 months post-inoculation on study day 1. The designs of these studies were consistent with FDA recommendations current at the time of study initiation, as well as the refinement of protocols as experience was gained by the VRC. The following organs were collected for biodistribution analyses: blood, gonads, liver, thymus (in Studies F and G only), heart, lung, adrenal glands (in Studies F and G only), kidney, spleen, mesenteric lymph nodes (Study D), right and left popliteal lymph nodes (collected separately in Studies F and G) or contralateral popliteal lymph nodes (in Studies D and E), subcutis at injection site, thigh muscle at injection site, bone marrow (from left femur in Studies F and G), and brain. Paired organs were processed together. Tissues and bone marrow cells were snap frozen in liquid nitrogen after being placed in sterile vials and stored at − 70°C. In addition, animals were monitored for morbidity (tremors, convulsions, salivation, diarrhea, lethargy, coma, and atypical behavior) and mortality twice daily and for clinical signs of toxicity (evaluation of skin and fur characteristics, eye and mucous membranes, respiratory, circulatory, automonic and central nervous systems, and somatomotor and behavior patterns) prior to dosing, weekly, and at termination (data not shown). Body weights were taken prior to dosing, weekly, and at termination (fasted), and food consumption was measured daily (data not shown). These studies were performed in compliance with GLP regulations.
Integration Study Design
Studies H and I were performed at the University of Michigan (in-life portion) and by Althea Technologies, Inc. (San Diego, CA) (DNA extraction and PCR analysis). They involved inoculation of 6- to 7-week-old female CD-1 mice (n = 9 in Study H, n = 10 in Study I) with 100 mcg (split into two sites per animal) i.m. into the right and left quadriceps muscles. At 28 days postinoculation, injected muscle tissue was harvested. Specimens were pooled (all right muscles together, all left muscles together), minced, placed into extraction buffer (0.5 mg/ml proteinase K, 50mM Tris [pH 8.0], 100mM EDTA, 100mM NaCl, 0.5% Tween 20) and incubated overnight at 56°C. DNase-free RNAse A (0.3 mg/ml) was added and incubated at 37°C for 30 min. Each specimen was extracted three times with equal volumes of buffered phenol, then chloroform. DNA was precipitated with two volumes of ethanol at room temperature. DNA was pelleted, rinsed in 70% ethanol, and air-dried at room temperature. Pellets were resuspended in 2.4 ml 10mM Tris (pH 8.0), and concentrations were determined by UV spectrophotometer. Thirty mcg of right or left quadriceps muscle DNA were loaded into each of 24 agarose tube gels, for a total of 720 mcg. The gels were run at 100 V using field-inversion gel electrophoresis for 3 h and using bacteriophage lambda cut with HindIII and KpnI as size markers. These pooled, purified specimens were used for PCR analysis. Additionally, the purified DNA was digested with 10 units SfiI/mcg DNA, overnight at 50°C. After digestion, the DNA was precipitated and subjected to a second round of purification (∼ 50 mcg total DNA for each pooled specimen). This was again extracted from the gel and used for PCR analysis. These studies were not conducted in compliance with GLP, but as research studies.
Tissue Processing and DNA Extraction for Studies A, B, and C
Genomic DNA (gDNA) was isolated from tissues using the Promega Wizard Genomic DNA Purification Kit. DNA was diluted with sterile nuclease-free water to a final concentration of 0.1 mcg/mcL for use. Ten mcL of sample was tested per PCR reaction.
Tissue Processing and DNA Extraction for Studies D, E, F, G, H, and I
Generally, 200 mg of tissue were processed for DNA extraction. Tissues were physically homogenized and subjected to digestion with proteinase K. DNA was extracted with the BioRobot M48 Workstation (Qiagen, Valencia, CA) using reagents and protocols recommended by the manufacturer. A naïve tissue sample was included with each run of the BioRobot to serve as a sentinel control for contamination. The concentration of the eluted DNA was determined by UV spectrophotometry and adjusted to a final concentration suitable for quantitative PCR (qPCR).
qPCR Analysis for Studies A, B, and C
A real-time quantitative modification of the TaqMan PCR technique in which the amplicon is detected by a sequence-specific fluorogenic probe was used. Primers and probes were selected using the Primer Express Primer Design Software (PE Applied Biosystems, Foster City, CA). The forward primer used was 5′-TGGGATCTCCACGCGAAT-3′, the reverse primer was 5′-GGAAGCTCCGCCGCTACC-3′, and the probe was 5′-CCATGTCCGGAACACGTACCCGA-3′ labeled with the 6-Carboxyfluorescein (6-FAM) reporter dye (PE Applied Biosystems, Foster City, CA). Amplification of the target sequence was performed in the ABI PRISM 7700 Sequence Detector System using 1× Universal TaqMan buffer containing AmpErase UNG and AmpliTaq Gold DNA Polymerase with primers at a final concentration of 0.4μM and the probe at 80nM. Tissue was separately spiked with 100 copies of the plasmids of interest to monitor qPCR inhibition. Controls in which there was no DNA, a negative control (mouse gDNA), the spiked controls, and a standard curve (two series of 10-fold template dilutions of the plasmids of interest from 100 to 105 copies) were also run. Assay performance characteristics were established as follows. Values of less than one copy were considered not biologically relevant and were scored as negative. The limit of detection (LOD) of the assay was considered to be 1 copy, and the limit of quantitation (LOQ, bound of linear range) of the assay was considered to be > 10 copies. Such specimens were scored as positive. Therefore, values between one and nine copies were regarded as nonquantifiable. The qPCR in Studies A, B, and C was performed by BioReliance.
qPCR Analysis for Studies D, E, F, G, H, and I
A TaqMan qPCR assay was designed to target the bovine growth hormone polyadenylation signal sequence, an element common to the plasmid backbone of all constructs. Primers and probe were designed using Primer Express software (Applied Biosystems, Inc., Foster City, CA). The forward primer used was 5′-TGAAGAATTGACCCGGTTCCT-3′, the reverse primer was 5′-GTACTTTAGCGGGTGGGATTGA-3′, and the probe was 5′-FAM-TTCTCTGTGACACACCCTGTCCACGC-TAMRA-3′. TaqMan reactions were performed in a 96-well plate using the ABI PRISM 7700 instrument. Amplification of the target sequence was performed in duplicate reactions each containing up to 1 μg gDNA from tissue, and 2× TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) with primers and probe at final concentrations of 600nM and 200nM, respectively. A third replicate reaction was performed spiked with 100 copies of the target sequence to monitor qPCR inhibition. Cycling conditions were 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 30 s and 63°C for 1 min. Quantification of the target sequence in each specimen was determined using a standard curve of plasmid DNA diluted in a background of gDNA isolated from a naïve animal. Standards, controls containing no template, sentinel extraction controls, and background gDNA controls were all run in duplicate reactions. Assay performance characteristics were established, and the assay was qualified for use with each transgene insert and animal model. The LOD of this assay was 10 copies of target sequence of gDNA extracted from tissues. The LOQ of this assay was 50 copies of target sequence/mcg gDNA extracted from tissues. qPCR analyses in Studies D, E, F, G, H, I, and J were performed by Althea Technologies, Inc.
Statistical Analyses on the PCR Results
Wilcoxon rank-sum tests were used to compare the distributions at the first and last time points. Two-sided p values from this procedure are reported for each test article, using both the thigh muscle and the subcutis data, where available. For readings that were reported as below the LOD or LOQ, a set value just below the limit was inserted; the use of nonparametric, rank-based tests allowed these discrete observations to be included in the comparison of the distributions. To adjust for the two comparison sites, a threshold of 0.025 was used as a cutoff for statistical significance. Larger p values are simply reported as “Not Significant.” For graphical displays, data were transformed using logarithms due to the strongly skewed distributions and non-constant variance.
RESULTS
As a general pattern, plasmid remains at the site of injection in the muscle and overlaying subcutis through which the vaccine is injected and does not biodistribute widely throughout the animals. Sporadically, a few animals (rabbits inoculated with 2 mg DNA plasmid) had low-level signals (low hundreds or at most thousands of copies) in other tissues but at day 8 or 9, the injection site in virtually all animals had tens of thousands to millions of copies. By day 30/31, few, if any, tissues other than the injection site muscle and subcutis had any positive signals. However, many, but generally not all, animals still had quantifiable signals (hundreds, thousands of copies) in the injection sites. By day 60/61, almost all remaining signals were observed in the subcutis at levels of a few hundred copies or less. This pattern was observed for all plasmid vaccines tested regardless of backbone, promoter, or gene insert.
The data and results of statistical analyses for the muscle and subcutis specimens are shown in Figures 1, 2, and 3. Table 2 shows the number of animals inoculated i.m. which had a quantitatively positive signal (regardless of the actual quantity) at the final time points for both genders of animals in all tissues tested. Both genders were grouped because no gender differences were noted. Only sporadically quantifiably positive signals were observed in tissues other than at the injection site.
FIG. 1.
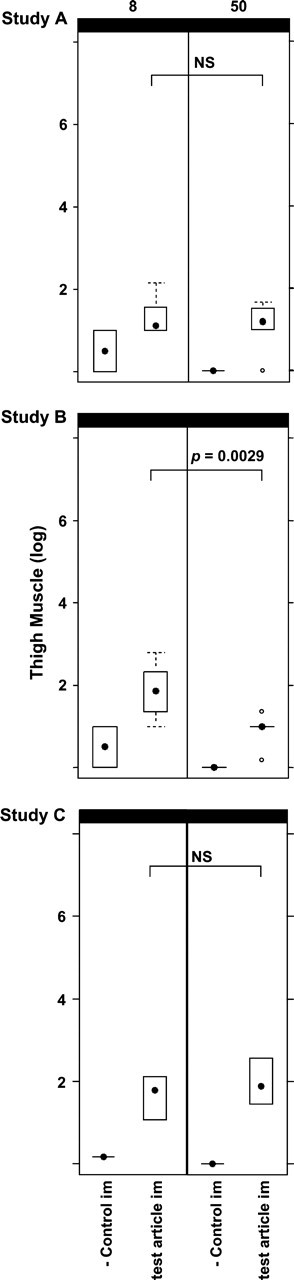
Box-and-whisker plots for Studies A, B, and C. Plasmid copy levels, as detemined by PCR, in the thigh muscles (site of i.m. injections) of placebo control (n = 2) and test article (n = 10) inoculated animals are shown. Day 8 results are shown on the left side of the figure and day 50 results on the right. The results of statistical analyses comparing the test article copy numbers from day 50 to day 8 are also shown; NS = not significant.
FIG. 2.
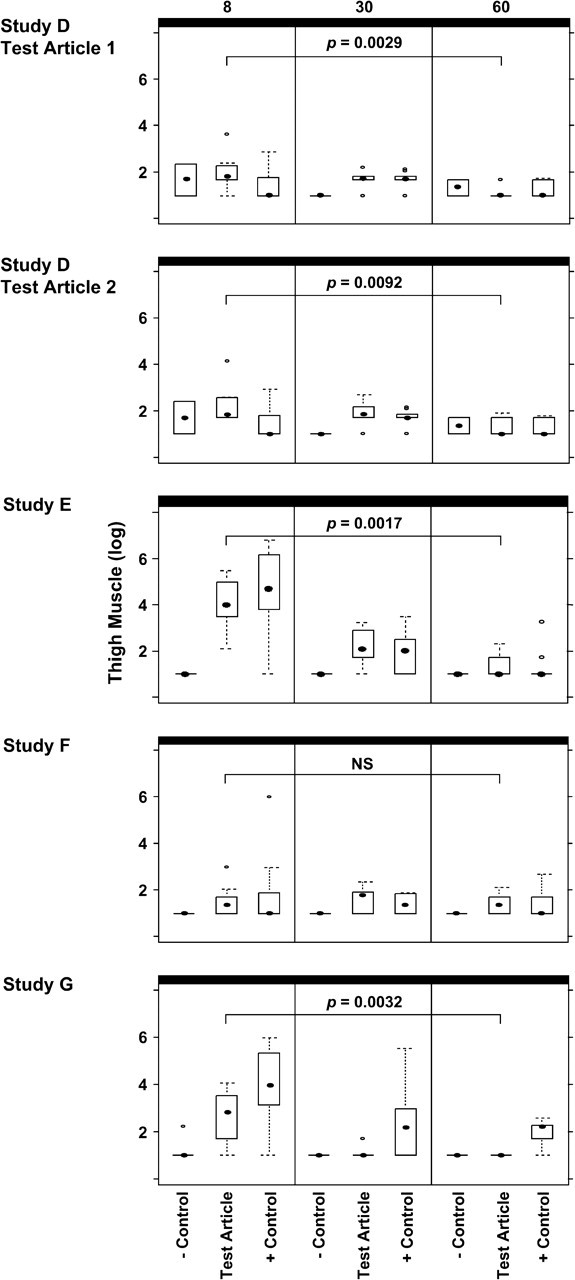
Box-and-whisker plots for Studies D, E, F, and G. Plasmid copy levels as determined by PCR, in the thigh muscles of placebo control, test article, and comparator plasmid-inoculated (+ control) animals are shown. Day 8/9 results are shown on the left side of the figure, day 30/31 results in the middle, and day 60/61 on the right. The results of statistical analyses comparing the test article copy numbers from day 60/61 to day 8/9 are also shown; NS = not significant.
FIG. 3.
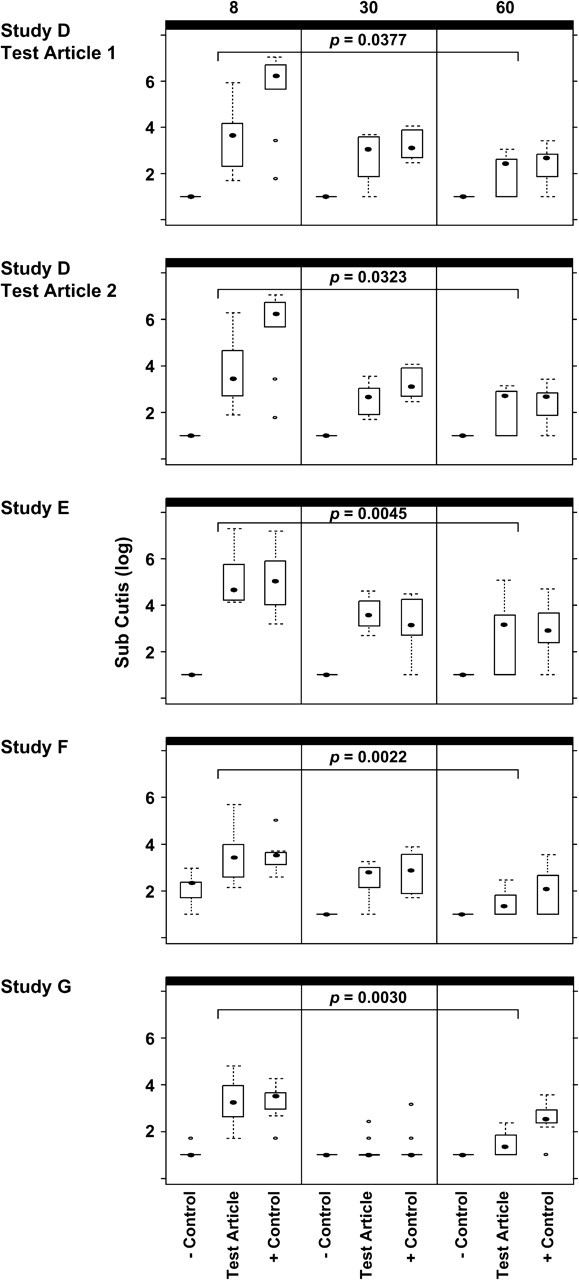
Box-and-whisker plots for Studies D, E, F, and G. Plasmid copy levels as determined by PCR, in the subcutis specimens of placebo control, test article, and comparator plasmid-inoculated (+ control) animals are shown. Day 8/9 results are shown on the left side of the figure, day 30/31 results in the middle, and day 60/61 on the right. The results of statistical analyses comparing the test article copy numbers from day 60/61 to day 8/9 are also shown.
TABLE 2.
Quantifiably positive PCR results in all tissues in all studies at the final study timepoint
|
|
|
Injection site muscle |
Injection site sub-cutis |
Adrenal glands |
Blood |
Bone marrow |
Brain |
Gonads |
Heart |
Kidneys |
Liver |
Lungs |
Lymph node— mesenteric |
Lymph node—right popliteal |
Lymph node—left popliteal |
Spleen |
Thymus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study A | Placebo (n = 2) | 0 | * | * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | * | * |
| Test article (n = 10) | 6 | * | * | 8 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | * | * | * | * | |
| Study B | Placebo (n = 2) | 0 | * | * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | * | * |
| Test article (n = 10) | 1 | * | * | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | * | * | * | * | |
| Study C | Placebo (n = 2) | 0 | * | * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | * | * |
| Test article (n = 10) | 8 | * | * | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | * | * | |
| Study D | Placebo (n = 2) | 0 | 0 | * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | 0 | 0 | * |
| Test article 1 (n = 10) | 0 | 7 | * | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | * | * | 0 | 0 | * | |
| Test article 2 (n = 10) | 2 | 7 | * | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | 0 | 0 | * | |
| Comparator (n = 10) | 2 | 8 | * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | 0 | 0 | * | |
| Study E | Placebo (n = 2) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | 0 | 0 |
| Test article (n = 10) | 2 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | * | * | 0 | 0 | |
| Comparator (n = 10) | 1 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | * | * | 0 | 0 | |
| Study F | Placebo (n = 2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Test article (n = 10) | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Comparator (n = 10) | 2 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Study G | Placebo (n = 2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Test article (n = 10) | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | |
| Comparator (n = 10) | 7 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| VRC-4302 |
n = 40** |
9 (23) |
25 (83) |
0 |
0 |
1 (3) |
1 (3) |
0 |
0 |
1 (3) |
0 |
0 |
1 (3) |
0 |
0 |
0 |
0 |
#—Absolute # quantifiably positive.
#(#)—Absolute # (%).
N.D.=Not done.
Cumulative across studies, n = 30 for subcutis, adrenals, spleen, and thymus; n = 20 for pop. LN; n = 40 in all other tissues.
The range of values among the animals in a particular study for each tissue are as follows: Injection site muscle—10's to 1000's; Injection site sub-cutis—100's-100,000's; all other tissues—10's to 100's.
The PCR assays used for all studies were qualified and thus the performance parameters well described. When PCR values were obtained that were between the LOD and LOQ, it is impossible to tell if they represent true positives or false positives. In Figures 1, 2, and 3, such specimens were given a set value just below the limits in the statistical analyses of positive tissues and means of quantities detected. In Table 2, only quantifiably positive specimens were included, and these indeterminate specimens were not reported.
Study A: Biodistribution of HIV Vaccine Candidate—Product VRC-3900
Four groups were inoculated in this study. Two groups received PBS, either i.v. (data not shown) or i.m., and the other two groups received 100 mcg VRC-3900, either i.v. (data not shown) or i.m. PCR and statistical analysis results are shown in Figure 1 and/or Table 2.
Study B: Biodistribution of HIV Vaccine Candidates—Product 1
Four groups were inoculated in this study. Two groups received PBS, either i.v. (data not shown) or i.m., and the other two groups received 100 mcg of Product 1 (HIV-1), either i.v. (data not shown) or i.m. PCR and statistical analysis results are shown in Figure 1 and/or Table 2.
Study C: Biodistribution of Ebola Vaccine Candidate—Product 7
Four groups were inoculated in this study. Two groups received PBS, either i.v. (data not shown) or i.m., and the other two groups received 100 mcg of Product 7 (Ebola), either i.v. (data not shown) or i.m. PCR and statistical analysis results are shown in Figure 1 and/or Table 2.
Study D: Biodistribution of HIV Vaccine Candidates—Product 2 (HIV-1) Adjuvanted with Product 4 (Cytokine Adjuvant, Test Article 1); Product 6 (HIV-1, Test Article 2)
Four groups were inoculated in this study. One group received PBS, one group received 2 mg of Product 2 (HIV-1) adjuvanted with Product 4 (cytokine adjuvant, test article 1), one group received 2 mg of Product 6 (HIV-1, test article 2), and one group received 2 mg of control plasmid VCL-2510 (malaria). PCR and statistical analyses results are shown in Figures 2 and 3 and/or Table 2.
Study E: Biodistribution of Ebola Vaccine Candidate—Product 12
Three groups were inoculated in this study. One group received PBS, one group received 2 mg of Product 12 (Ebola), and the final group received 2 mg of Product 1 (HIV-1) as a comparator plasmid. PCR and statistical analyses results are shown in Figures 2 and 3 and/or Table 2.
Study F: Biodistribution of SARS Vaccine Candidate—Product 15
Three groups were inoculated in this study. One group received PBS, one group received 2 mg of Product 15 (SARS), and the final group received 2 mg of Product 1 (HIV-1) as a comparator plasmid. PCR and statistical analyses results are shown in Figures 2 and 3 and/or Table 2.
Study G: Biodistribution of WNV Vaccine Candidate—Product 17
Three groups were inoculated in this study. One group received PBS, one group received 2 mg of Product 17 (WNV), and the final group received 2 mg of Product 1 (HIV-1) as a comparator plasmid. PCR and statistical analyses results are shown in Figures 2 and 3 and/or Table 2.
Study H: Integration Analysis for HIV Vaccine Candidate—Product 1 and Study I—Integration Analysis for Ebola Vaccine Candidate—Product 7
While target DNA was detected in DNA extracted from the pooled injection site specimens prior to separation and purification of high-molecular weight DNA, no target was detected after either primary or secondary high-molecular weight DNA separation and purification. Thus, no more than five copies of plasmid DNA could be integrated into 1 mcg of high-molecular weight mouse gDNA. From these negative results, it was concluded that there was a lack of integration.
DISCUSSION
Over the course of these studies, a consistent pattern of biodistribution (confined to the site of injection) and clearance has been observed when DNA plasmid vaccines at doses up to 2 mg are inoculated i.m. by needle and syringe or Biojector, regardless of the expressed gene insert or the promoter in the plasmid backbone used to drive that expression. These studies have demonstrated that plasmid vaccines do not widely biodistribute and remain at the site of injection (the muscle and overlying subcutis). No differences were noted in these biodistribution and clearance patterns between genders. Furthermore, plasmid DNA is virtually never found in gonad specimens and in the sporadic cases where a signal was seen, it was very low (often nonquantifiable) and could be the result of inadvertent contamination in the course of collection of the tissues during necropsy despite scrupulous efforts to avoid it. These findings should allay the theoretical concerns that have been voiced about the potential for plasmid vaccine products to biodistribute to reproductive organs.
Although rigorous measures are undertaken to prevent cross-contamination (changing of gloves, equipment, etc.), the PCR readout is so exquisitely sensitive that the smallest contamination during necropsy and tissue collection would be detected. In fact, it seems likely that all the sporadically positive tissues (i.e., tissues other than the muscle and subcutis at the site of injection) are not indicative of any biodistribution of DNA plasmid into those tissues but are rather the result of unavoidable low-level contamination or, alternatively, are false positives of the assay. This is a limitation when using such an exquisitely sensitive readout.
In addition, these studies demonstrate that plasmid vaccine candidates clear relatively rapidly. Although ∼ 1014 molecules are inoculated in the studies in rabbits, even by day 8 or 9 (∼ 1 week post-inoculation), all but on the order of 104–106 copies per microgram of DNA extracted from tissue have already been cleared from the site of injection. Over the course of 2 months, all the inoculum clears from the muscle of most animals, and the few animals in which a signal remains, generally in the subcutis, the copy numbers are on the order of 102–103 per microgram of DNA extracted from tissue. We do not at this point have data to explain why more plasmid remains in the subcutis/skin than in the muscle which receives the bolus of the inoculum. However, this phenomenon has been seen consistently in our data and that of others (personal communications, Chris Butler) whenever the subcutis/skin specimen has been tested. One possibility is that the subcutis/skin consists mainly of adipose tissues (cells which are large in comparison to the nuclei and which have large storage vacuoles) and cornified dermal layers (cells with no nuclei). It may be that the plasmid that remains has deposited into a portion of the cell (i.e., not the nucleus) in which, or outside of cells where, it does not get processed (i.e., expressed or degraded). We are considering further experiments to address this phenomenon.
One concern that has been expressed about DNA plasmid vaccines is the possibility that they might integrate into the host genome. When given i.m., the integration studies provided no evidence of any integration in the tissue where the largest amount of plasmid is inoculated. This profile may differ by delivery method (Wang et al., 2004).
One benefit of conducting these studies on an array of products is that it has provided eventual efficiencies in preparing regulatory submissions by providing more data on the safety and similarity of patterns of biodistribution of various candidate vaccines. These similarities are observed even with vaccines for differing disease indications, built on similar, although not identical, plasmid vaccine platforms. One example of this increased efficiency achieved is the continuous bridging of the biodistribution of newer generation products to an initial plasmid vaccine (Product 1 [HIV-1], a first-generation prototype), by including an arm in each new study for comparison to this product. Doing this has permitted bridging to an integration study and phase I clinical safety data, which had been obtained by the time of regulatory submission of the newer candidates. When biodistribution patterns and clearance patterns of a newer generation product were seen to be similar to Product 1 (HIV-1), even for another disease indication, integration studies were not required to be repeated for each candidate. As another example of this increased efficiency, although the product tested in Study D did not go forward to clinical trials, this study along with a toxicology study on the same product described in the companion article in this issue supported the conduct of clinical trials with two other products. One of these products is a subset of Product 6 (HIV-1, consisting of four of the six plasmids). The other product is another six-plasmid product, which consists of three plasmids (the clades A, B, and C env genes) expressing the same proteins and three plasmids expressing the clade B gag, pol, and nef genes on separate plasmids (rather than as a fusion protein). This newer generation six-plasmid product contains a different promoter (the CMV/R promoter) than the original Product 6 (HIV-1), but preclinical and clinical data from Product 12 (Ebola, also using the CMV/R promoter), the preclinical data for Product 6 (HIV-1), and the clinical data for the four-plasmid product supported the regulatory approval of the current six-plasmid construct without having to repeat preclinical biodistribution and repeated-dose toxicology studies. These regulatory efficiencies save months in the clinical development pathway of new candidate vaccines. Given the urgency of public health crises such as the HIV/AIDS epidemic with 14,000 new infections daily and the threat of global spread of newly emerging viral infections like SARS, this shortened development time is crucial.
The designs of Studies F and G represent the most refined biodistribution studies of those performed and were built on the experience of the prior studies, as well as with guidance from FDA to meet their regulatory requirements. If future biodistribution studies of new vaccine candidates are required, the study design of these studies will likely be followed.
The integration studies were designed early in development of the multitude of vaccine candidates in the VRC pipeline. While they might not represent the design of an integration study that would be performed today, if additional studies were required, these integration studies were accepted by the FDA at the time they were submitted to support conduct of clinical trials with those early vaccine candidates as well as the subsequent candidates that have been bridged back to those data. The primary difference that would be introduced in future integration studies would be to use a larger species (rabbits), so that doses more comparable to the human dose can be delivered and tissue specimens will be of sufficient size that they would not need to be pooled in order to get sufficient amounts of high-molecular weight DNA to perform the analyses.
Overall, we conclude from these studies that the pattern of biodistribution (confined to the site of injection) and clearance (within 2 months) of DNA plasmid vaccines at doses up to 2 mg delivered by Biojector or needle and syringe is consistent regardless of differences in the promoter in the plasmid backbone or in the gene being expressed by the plasmid vaccine or cytokine adjuvant. Thus, it should not be necessary to repeat biodistribution studies of newer vaccine candidates, if delivered i.m. by either of these devices. These studies have demonstrated that theoretical safety concerns expressed about DNA plasmid vaccines, e.g., that they might biodistribute to the gonads or that they might integrate, have not been borne out, when the vaccines were delivered i.m. with these devices. The data from these studies have been successfully used in submissions to a regulatory agency (U.S. FDA) to support the conduct of clinical trials with a number of vaccine candidates for differing disease indications (HIV-1, WNV, SARS, and Ebola). In addition, the biodistribution and integration studies build on the data acquired from companion repeated-dose toxicology studies (see companion article in this issue) to demonstrate the safety of these vaccine candidates and their suitability for investigational human use.
Acknowledgments
We would like to thank the following individuals: Mario Roederer for insightful comments on data presentation, John Jessop for insightful comments on study reports, Toni Miller and Brenda Hartman for their excellent assistance with figures, and Darlene Fitch for preparing data tables for statistical analysis. We would also like to acknowledge the staff at BioReliance, particularly Audrey Chang, responsible for the PCR analyses, for their conduct of the early studies (A, B, and C) and the staff at GeneLogic and Althea Technologies, Inc., for their conduct of the rest of the studies. Further, we would like to acknowledge Rick Stout (Bioject, Tualatin, Oregon) for providing the Biojector devices and the staff of Vical, Inc. (San Diego, CA) for manufacturing all clinical trial materials under contract to the National Institute of Allergy and Infectious Diseases. This research was supported by the Intramural Research Program of the National Institutes of Health, Vaccine Research Center, NIAID.
References
- Barouch, D., Truitt, D. M., and Letvin, N. L. (2004). Expression kinetics of the interleukin-2/immunoglobulin (IL-2/Ig) plasmid cytokine adjuvant. Vaccine 22,3092–3097. [DOI] [PubMed] [Google Scholar]
- Barouch, D., Yang, Z.-Y., Kong, W.-P., Korioth-Schmitz, B., Sumida, S. M., Truitt, D. M., Kishko, M. G., Arthur, J. C., Miura, A., Mascola, J. R., et al. (2005). An HTLV-1 regulatory element enhances the immunogenicity of HIV-1 DNA vaccines in mice and nonhuman primates. J. Virol. 79,9694–9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CBER. (1996). Points to Consider on Plasmid DNA Vaccines for Preventive Infectious Disease Indications. CBER.
- CBER. (2005). Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. CBER.
- Chakrabarti, B. K., Kong, W.-P., Wu, B.-Y., Yang, Z.-Y., Friborg, J., Ling, X., King, S. R., Montifiori, D. C., and Nabel, G. J. (2002). Modifications of the human immunodeficiency virus envelope glycoprotein enhances immunogenicity for genetic immunization. J. Virol. 76,5357–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, J. E., Gorak, E. J., Charoenvit, Y., Wang, R., Freydberg, N., Osinowo, O., Richie, T. L., Stoltz, E. L., Trespalacios, F., and Nerges, J. (2002). Safety, tolerability, and lack of antibody responses after administration of a PfCSP DNA malaria vaccine via needle or needle-free jet injection, and comparison of intramuscular and combination intramuscular/intradermal routes. Hum. Gene Ther. 13,1551–1560. [DOI] [PubMed] [Google Scholar]
- Epstein, J. E., Charoenvit, Y., Kester, K. E., Wang, R., Newcomer, R., Fitzpatrick, S., Richie, T. L., Tornieporth, N., Heppner, D. G., and Ockenhouse, C., (2004). Safety, tolerability, and antibody responses in humans after sequential immunization with a PfCSP DNA vaccine followed by the recombinant protein vaccine RTS,S/AS02A. Vaccine 22,1592–1603. [DOI] [PubMed] [Google Scholar]
- Martin J. E., Enama M. E., Koup R. A., Bailer R. T., Moodie Z., Roederer M., Nabel G. J., and Graham B. S. (2005) VRC 004: Safety and Immunogenicity of a Multiclade HIV-1 DNA Vaccine in Healthy Uninfected Adults (VRC-HIVDNA009-00-VP). J. Allergy. Clin. Immunol. 115, 892. [Google Scholar]
- Hartikka, J., Sawdey, M., Cornefert-Jensen, F., Margalith, M., Barnhart, K., Nolasco, M., Vahlsing, H. L., Meek, J., Marquet, M., and Hobart, P. (1996). An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum. Gene Ther. 7,1205–1217. [DOI] [PubMed] [Google Scholar]
- Huang, Y., Kong, W.-P., and Nabel, G. J. (2001). Human immunodeficiency virus type 1-specific immunity after genetic immunization is enhanced by modification of Gag and Pol expression. J. Virol. 75,4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, W.-P., Huang, Y., Yang, Z.-Y., Chakrabarti, B. K., Moodie, Z., and Nabel, G. J. (2003). Immunogenicity of multiple gene and clade human immunodeficiency virus type 1 DNA vaccines. J. Virol. 77,12764–12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, R. L., Stein, J., Manetz, T. S., Andrews, C., Bailer, R., Rathmann, J., and Gomez, P. L. (2006). Toxicological Safety Evaluation of DNA Plasmid Vaccines Against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile Virus is Similar Despite Differing Plasmid Backbones or Gene Inserts. [DOI] [PMC free article] [PubMed]
- Sullivan, N. J., Peterson, M., Yang, Z. Y., Kong, W. P., Duckers, H., Nabel, E., and Nabel, G. J. (2005). Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 79,547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, N. J., Sanchez, A., Rollin, P. E., Yang, Z. Y., and Nabel, G. J. (2000). Development of a preventive vaccine for Ebola virus infection in primates. Nature 408,605–609. [DOI] [PubMed] [Google Scholar]
- Title 21 Code of Federal Regulations (CFR). Part 58.
- Ulmer, J. B., Sadoff, J. C., and Liu, M. A. (1996). DNA vaccines. Curr. Opin. Immunol. 8,531–536. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Troilo, P. J., Wang, X., Griffiths, T. G., Pacchione, S. J., Barnum, A. B., Harper, L. B., Pauley, C. J., Niu, Z., Denisova, L., et al. (2004). Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 11,711–721. [DOI] [PubMed] [Google Scholar]
- Xu, L., Sanchez, A., Yang, Z., Zaki, S. R., Nabel, E. G., Nichol, S. T., and Nabel, G.J. (1998). Immunization for Ebola virus infection. Nat. Med. 4,37–42. [DOI] [PubMed] [Google Scholar]
- Yang, Z.-Y., Kong, W.-P., Huang, Y., Roberts, A., Murphy, B. R., Subbarao, K., and Nabel, G. J. (2004). A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 428,561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]