

Abstract
Central cholinergic signaling has long been associated with aspects of memory, motivation, and mood, each affected functions in neuropsychiatric disorders such as schizophrenia. In this chapter, we review evidence related to the core hypothesis that dysregulation of central cholinergic signaling contributes to the pathophysiology of schizophrenia. Although central cholinergic circuits are resistant to simplification—particularly when one tries to parse the contributions of various classes of cholinergic receptors to disease related phenomena—the potential role of ACh signaling in Schizophrenia pathophysiology deserves careful consideration for prospective therapeutics. The established role of cholinergic circuits in attentional tuning is considered along with recent work on how the patterning of cholinergic activity may modulate corticostriatal circuits affected in schizophrenia.
I. Introduction
Cholinergic innervation of cortical and striatal brain areas is extensive and diffuse, as are both pre- and postsynaptic targets for acetylcholine (ACh) interaction. Receptors for ACh (AChRs) come in two broad classes—ionotropic (nicotinic) and metabotropic (muscarinic)—each class having multiple subtypes with both opposing and synergistic actions. Activation of these receptors regulates neuronal excitability by interaction with pre- and postsynaptically localized ACh-binding sites. ACh can act as a tonic, diffuse signal, modulating the release of ACh and other transmitters, including dopamine, glutamate, and GABA. Alternatively, ACh can exert its effects via highly localized and directed interactions with neuronal AChRs to increase or decrease neuronal firing.
The complexity of CNS cholinergic circuits and signaling mechanisms produces a system in which origins and end results may be easier to appreciate than intervening steps. It is clear that ACh, released from the cholinergic inputs of the basal forebrain, striatal, and the pontomesencephalic (PM) areas, plays an important role in supporting neurocognitive and motivational functions of the prefrontal cortical, hippocampal, and ventral tegmental projections to the striatum (for reviews see Cragg, 2006; Gotti and Clementi, 2004; chapter by Martin and Freedman, this volume; Mesulam, 2004; Sarter et al., 2005; Smythies, 2005; Wonnacott et al., 2005). In addition, there is considerable evidence that events which reduce the amount of ACh at cholinergic targets may contribute to functional deficits—including deficits related to schizophrenia (Hyde and Crook, 2001; Sarter et al., 2005; chapter by Martin and Freedman, this volume). But considerable confusion sets in when one tries to extract exactly how the intervening steps, with activation of muscarinic and/or nicotinic receptors and consequent changes in downstream circuits, are integrated to elicit the broad spectrum of effects modulated by cholinergic signaling.
Some of the confusion arises from attempts to reconcile the varying “anticholinergic” properties of antipsychotic medications with data on the effects of muscarinic agonists per se. Further confusion arises from the fact that commonly used cholinergic ligands may be less specific in their binding properties than previously thought: indeed, some compounds traditionally considered selective muscarinic antagonists may function as partial agonists or antagonists of other ACh (nicotinic and muscarinic) receptor subtypes. Finally, schemes that overemphasize the role of a particular ACh-signaling pathway to the exclusion of others, rather than viewing the function of cholinergic circuits as the result of the summation of actions of ACh at all of its receptors, may do more to confuse than enlighten. Anatomical and functional data underscore the interaction of cholinergic circuits with other neurotransmitter systems (Smiley et al., 1999). Indeed, interaction of ACh with its full panoply of receptor sites elicits substantive changes in the synaptic transmission of dopamine, glutamate, serotonin, and GABA in a variety of brain regions.
We will first provide a thumbnail sketch of cholinergic circuits and then examine how they are involved in functions relevant to schizophrenia with focus on interactions with dopamine- and glutamate-mediated signaling. We will then review developmental/genetic, pathological, and pharmacological evidence for potential cholinergic contributions to schizophrenia. Although cholinergic signaling may not be the major site of circuit dysregulation underlying the etiology of schizophrenia, more knowledgeable manipulation of cholinergic systems may provide an untapped reservoir of considerable therapeutic potential in the treatment of the positive and negative symptoms of this complex disease.
II. ACh in Brain Regions Implicated in Schizophrenia
Central cholinergic circuits participate in aspects of memory formation, motivational and volitional behaviors, and affect. Each of these functions is altered in neuropsychiatric disorders, including schizophrenia. Cholinergic neurons in the CNS make up for any apparent deficit in numbers by projecting to a broad swath of cerebral cortical mantle, select portions of the temporal lobe, and by their profuse axonal arborizations throughout the corpus striatum. The schematic diagram presented in Fig. 1 attempts to bring some order to the cholinergic chaos by corralling the diverse targets of ACh innervation into a manageable subset of brain regions strongly implicated in schizophrenia. Our focus is represented in primary colors: red for cholinergic neuronal groups and a subset of their projections, and yellow for the chosen cholinoceptive targets—brain regions that have been examined in detail in recent circuit analyses and that will be the focus of this chapter. Obviously this degree of simplification endangers the generality of our considerations—for example, there is little discussion of cholinergic signaling in amygdala, or in cingulate or somatosensory cortex—areas of study that have contributed important progress to our understanding of central cholinergic coding. Such omissions are not intended to infer relative impact on the field, but rather reflect the limits of time, space, and comprehension of the authors.
Fig. 1.
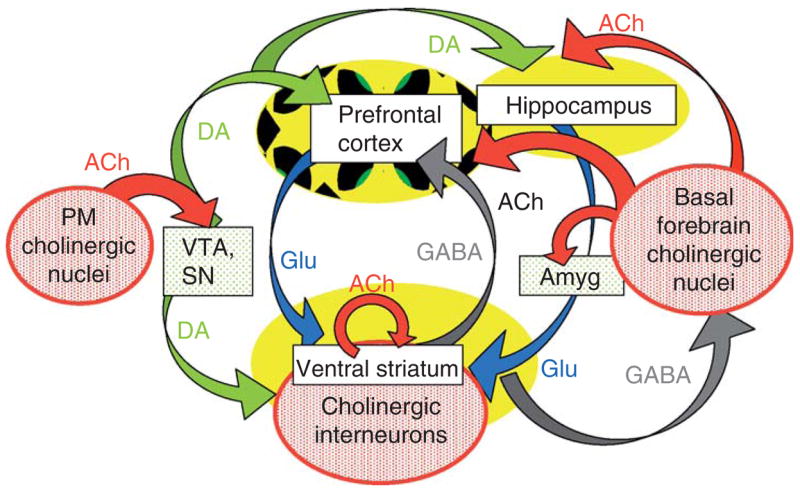
Schematic diagram of cholinergic circuits (in red) and their projections within a subset of key brain regions affected in SZ. Cholinergic inputs to prefrontal cortex and hippocampus arise primarily from the basal forebrain group, including the septal cholinergic neurons, the nbM, the preoptic and diagonal band nuclei. Other contributors to the forebrain ACh group are neurons within the substantia innonimata and ventral pallidum. The second major subgroup of ACh-containing neurons, the pontomesencephalic (PM) cholinergic neurons, provides input to brainstem aminergic nuclei (e.g., VTA, SN, and raphe). Cholinergic interneurons intrinsic to the basal ganglia are thought to modulate the relative impact of glutamatergic, dopaminergic, and GABAergic circuits within the ventral striatum. Potential mechanisms of cholinergic regulation of neuronal excitability in prefrontal cortex and hippocampus are also discussed in the text.
Overall, there are three major groups of cholinergic neurons and interneurons within the primate brain. Cholinergic inputs to prefrontal cortex and hippocampus arise primarily from the basal forebrain group, which includes the medial septal cholinergic neurons, the nucleus basalis of Meynert (nbM) and the preoptic and diagonal band nuclei (Fig. 1, right). Other contributors to the forebrain ACh groups are neurons within the substantia innominata and ventral pallidum. The relative contribution of cholinergic versus noncholinergic neurons to each of the basal forebrain nuclei ranges from <5% to 90% in human brain, with the nbM as the highest density forebrain cholinergic nucleus (Mesulam et al., 1984; reviewed in Hyde and Crook, 2001). Other major targets of the basal forebrain groups include the amygdala, olfactory bulb, and hypothalamus (Woolf, 1991).
The second major subgroup of ACh-containing neurons (Fig. 1, left), the PM cholinergic neurons, provides input to brainstem aminergic nuclei (e.g., VTA, SN, raphe) as well as to the cerebellum, thalamus, and hypothalamus (Woolf, 1991). In addition, the PM group of neurons project to the basal forebrain cholinergic neurons, thereby coordinating central cholinergic modulation of brainstem, midbrain, and forebrain circuits.
The final major group of cholinergic neurons consists of the ACh interneurons that are intrinsic to the basal ganglia. These intrastriatal neurons modulate the relative impact of multiple glutamatergic and dopaminergic circuits on the medium spiny GABAergic projection neurons of the striatum: our focus will be on the role of cholinergic circuits in the regulation of ventral striatal signaling.
A. Cholinergic Pathways Within the Ventral Striatum
Cholinergic neurons within the striatum are typically large, aspiny neurons that comprise 1–5% of the striatal interneurons, varying somewhat with species and consideration of dorsal versus ventral regions. The extensive arborizations of the striatal cholinergic interneurons throughout the corpus striatum provide a tonic level of ACh release, with the ultimate concentration of extracellular ACh being set (and reset) by the interplay of local ACh release and the activity of the omnipresent acetylcholinesterase (AChE).
Striatal cholinergic neurons are characterized by their tonic activation profile, and periods of relatively low activity (referred to as “pauses”) can be associated with salience and prediction of reward (Cragg, 2006; Graybiel et al., 1994). The phasic lulls and peaks of the striatal cholinergic activity, which alters local ACh and choline concentrations, are influenced by corticostriatal projections from hippocampal subicular and prefrontal glutamatergic neurons (Fig. 1, in blue), as well as from amygdala, cingulate, and other cerebral and temporal cortices (not shown). In the rodent, the bulk of the hippocampal projections to the nucleus accumbens arise from ventral rather than dorsal hippocampal areas analogous to the more anterior portions of the primate hippocampus.
Dopaminergic inputs to the ventral striatum arise from the ventral tegmentum (VTA) and substantia nigra (SN), which themselves are recipients of cholinergic projections from PM neurons. Activation of a variety of pre- and postsynaptic dopamine receptors strongly regulates the release of ACh and the excitability of striatal cholinergic interneurons (Maurice et al., 2004; Wang et al., 2006). In addition, local circuits of opiate peptide and GABAergic neurons influence the net levels of striatal cholinergic tone.
Even this, admittedly limited, summary of key regulators of the spatial and temporal profile of ACh-mediated signaling in striatum reveals considerable complexity. Nevertheless, recent progress in dissecting the interaction of cholinergic circuits with dopaminergic and glutamatergic inputs to the striatum inspires considerable hope that we may be approaching a bona fide understanding of how ACh works at least in one region that is known to effect in schizophrenia and that is particularly high in ACh tone (see below and Cragg, 2006; Calabresi et al., 2000; Wilson, 2006; Wonnacott, 2005 for reviews; Wang et al., 2006).
B. Cholinergic Projections in Prefrontal Cortex and Hippocampus
The principal source of cholinergic input to the PFC and hippocampus is from the basal forebrain nuclei, with particularly strong contributions from the medial septum in rodent and from the nbM in human brain. The primate prefrontal cortex receives a fairly homogeneous cholinergic input with the highest density of cholinergic marker-positive fibers in layers I, II, and V (Lewis, 1990; Smiley et al., 1997). Cholinergic axons within the cerebral cortex of human brain are studded with numerous en passant swellings that serial EM reveals as primarily asymmetric type synapses. Close appositions of cholinergic synaptic profiles in cortex (Mesulam, 1999, 2004; Smiley et al., 1997) as well as the prevalence of cholinergic marker-positive swellings in the vicinity of pyramidal and nonpyramidal neurons in PFC and hippocampus is consistent with proposed modulatory effects of ACh on both excitatory and inhibitory cortical circuits (Mansvelder et al., 2006). Likewise, evidence has accrued that the release of ACh per se is likely subject to cholinergic, as well as dopaminergic, synaptic tuning in both PFC and hippocampus (DeBoer et al., 1996; Moore et al., 1999).
III. Physiology of ACh Circuits and Signaling in Brain Regions Implicated in Schizophrenia Pathology
A. ACh Receptors in the CNS
When clinicians and patients contemplate the “anticholinergic” side effects of various drugs, they often focus on the diverse and distressing array of peripheral autonomic cholinergic actions, including alterations in gastrointestinal function, nausea, and changes in appetite. In fact, the binding sites with which cholinergic drugs interact in the CNS are just as diverse as those in the periphery and often more accessible than expected, despite the blood–brain barrier. Any careful deliberation on ACh-binding sites in the CNS must include ACh-degradative, synthetic, and transporter proteins, as well as the multimembered muscarinic and nicotinic receptor subtypes (for reviews see Calabresi et al., 2000; Cobb and Davies, 2005; Gotti and Clementi, 2004; Mansvelder et al., 2006; Newhouse et al., 2004; Sarter et al., 2005). Pharmacological agents originally identified for their activity as AChE inhibitors (such as physostigmine and galantamine) are now known to act as partial agonists or antagonists of specific subtypes of CNS nicotinic AChRs (nAChRs). The oldest and most established “antimuscarinic” agent, atropine, blocks multiple classes of nAChRs at submicromolar concentrations—well within the clinically relevant and experimentally typical range of doses (Zwart et al., 1999). Carbamylcholine is more than a muscarinic agonist—it gates deliciously long openings of nAChRs. Finally, the activation of different types of pre-synaptic nicotinic and muscarinic receptors can facilitate or depress the release of ACh itself (see below).
Awareness of the complexities in the number and pharmacodiversity of ACh-binding partners in the CNS is essential to evaluating the past and present literature on ACh circuits and signaling. Humbling though this may be, we are actually well positioned to do so: the last 20 years have yielded impressive advances in understanding the differential regulation, expression, targeting, and function of the many muscarinic (at least 5 genes, so far) and nicotinic (11 subunit genes) receptors (see below). With this knowledge in hand, we need to reassess the effects of the pharmaceuticals we have and work toward the development of agents that more selectively manipulate the synthesis, release, and binding(s) of ACh.
A quick primer then, on the most important of ACh-binding sites, from information largely extracted from the following reviews: Calabresi et al. (2000); Gotti and Clementi (2004); Laviolette and van der Kooy (2004); MacDermott et al. (1999); Mansvelder et al. (2006); Sarter and Parikh (2005); Smythies (2005); Wonnacot et al. (2005).
Choline acetyltransferase (ChAT ): This enzyme is responsible for ACh synthesis. The regulation of ChAT gene expression in the CNS is thought to be coordinated with that of vAChT, by virtue of a common “cholinergic locus” promoter. However, the distribution of these two proteins between somatodendritic and axonal domains may be regulated independently.
ACh esterase (AChE): This binds ACh with micromolar affinity and is considered the principal degradative activity for ACh. AChE is one of the fastest turnover rate enzymes identified and is located primarily at intraneuronal and extracellular sites. Despite its preeminence as “the AChE,” recent work deleting AChE-encoding genes revealed that butyrylcholinesterase (BuChE) activity, which is associated with glial cells rather than neurons, can maintain grossly normal ACh balance. So BuChE is another ACh-binding partner to bear in mind.
The vesicular ACh transporter (vAChT ): This binds ACh with submicromolar affinity and translocates it into vesicular compartments within cholinergic neurons.
Muscarinic (metabotropic) AChRs: At least five genes are identified to date (M1–M5); M1, M2, and M4 subtypes predominate in the CNS. These ACh-binding proteins are coupled to a variety of G-proteins resulting in the activation or inhibition of an even wider variety of enzymatic and ion channel targets. Note that in the CNS, only a subset of the muscarinic-binding sites are postsynaptic; other subtypes of muscarinic receptors are targeted to axonal/presynaptic sites where they modulate the release of glutamate, dopamine, and ACh, among other key players.
Nicotinic (ionotropic) AChRs: Twelve subunit genes (α2–α10; β2–β4) encode a group of proteins that are faintly related to—and pharmacologically very distinct from—the renowned muscle-type nicotinic receptors. Drugs that interact with subtypes of neuronal nicotinic receptors (e.g., nicotine, hexamethonium) barely touch the muscle receptor and vice versa. Also important to note is that in the CNS, nAChRs, just like muscarinic receptors, are targeted to pre- as well as postsynaptic locations. In fact, the role of presynaptic nicotinic receptors as modulators of dopamine, glutamate, GABA, serotonin, and ACh release is so prevalent in the CNS that their contribution as postsynaptic receptors is often overlooked (but see Frazier et al., 1998; Jones and Yakel, 1997)!
In sum, a circumspect evaluation of how the dysregulation of cholinergic circuits may be involved in the pathophysiology requires recognition that ACh targets are many, perhaps not as pharmacologically distinct as previously considered and at pre-, post-, and perisynaptic locations. Viewing the function of cholinergic circuits as the result of the summation of actions of ACh at all of its receptors, although initially daunting, may resolve some apparent conflicts in the literature and guide the way to new therapeutic approaches (for reviews see Calabresi et al., 2000; Gotti and Clementi, 2004; Laviolette and van der Kooy, 2004; MacDermott et al., 1999; Mansvelder et al., 2006; Sarter and Parikh, 2005; Sarter et al., 2005; Smythies, 2005; Wonnacot et al., 2005).
B. Physiology of Ach Circuits in Striatum
The striatum is established stomping grounds for fans of central cholinergic circuits and ACh signaling. Although the numbers of cholinergic neurons in the striatum are small, they are the foremost, if not the exclusive, source of the high-pack cholinergic inputs in mammalian striatum. As discussed above, striatal cholinergic neurons are characterized by their large size, aspiny appearance, and tonic activation profile (hence the names ASpN and TANS neurons; Fig. 2). Changes in the activity profile of striatal TANS, referred to as “pauses,” are thought to arise in part from the slowing of autonomous pacemaker activity and in part to local changes in dopamine, glutamate, and GABA signaling (Cragg, 2006; Maurice et al., 2004; Wang et al., 2006). The association between changes in striatal cholinergic “tone” and salience/reward prediction has continued to stoke the fire of physiologists’ interests in the workings of striatal ACh circuits (see Cragg, 2006 for review; Apicella, 2002; Maurice et al., 2004; Wang et al., 2006 for recent highlights).
Fig. 2.
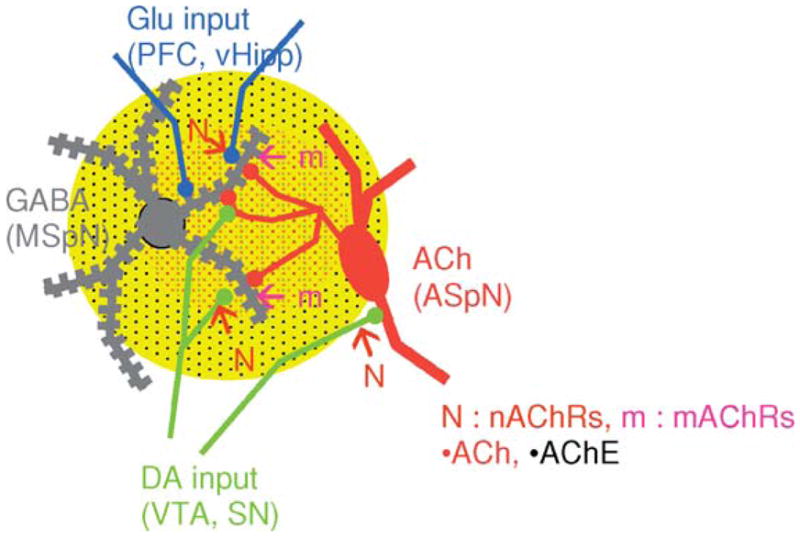
Schematic diagram of an aspiny cholinergic neuron (ASpN) and its projections to convergent sites of glutamatergic and dopaminergic input on striatal GABAergic medium spiny projection neurons (MSpN). Changes in local [ACh], from pauses in the firing of these TANS, modulate the net output of the striatum by interactions with ACh receptors and binding sites in pre-, post-, and perisynaptic compartments.
Perhaps the best studied, although still mechanistically mysterious, role of cholinergic circuits in striatum is in their reciprocal interactions with dopaminergic inputs from the VTA and SN. Recent studies add new dimensions to prior evidence that ACh acts as a key regulator of striatal output by influencing the activity of GABAergic medium spiny neurons (MSpNs, Fig. 2). The newest twist is that ACh is likely to exert its modulatory control on striatal activity through interaction with both pre- and postsynaptic, nicotinic and muscarinic receptors (Fig. 2). Presynaptic nicotinic receptors have long been implicated in the regulation of striatal dopamine release, with reports identifying some of the nAChR subtypes involved in aspects of nicotine addiction in staggering detail (for review see Wonnacott et al., 2005; Tapper et al., 2004 for recent highlights). The vague term “regulation” was intentionally employed because one of the points of controversy has been whether dopamine release in striatum is enhanced or depressed by nicotine. It turns out that the answer may be both, depending on the frequency of firing of the dopamine neurons (see Cragg, 2006 for review; Partridge et al., 2002; Rice and Cragg, 2004; Zhang and Sulzer, 2004; Zhou et al., 2001). The effects of dopamine receptor agonists on modulating the release of ACh in striatum (as well as in PFC and in hippocampus, see below) are also well established (DeBoer et al., 1996). But new results reveal that depending on the type and location of the dopaminergic and muscarinic receptors activated, the net effect may be to stably enhance or depress the activity of the GABAergic MSpNs (Cragg, 2006; Wang et al., 2006; Wilson, 2006;). Indeed, the potential for mutual tuning of TANS and DANS seems more than flexible enough to account for the differences in valence and timing of all of the synaptic changes observed in striatum (Calabresi et al., 2000; Cragg, 2006; Maurice et al., 2004; Wang et al., 2006).
C. Physiology of Ach Circuits in PFC and Hippocampus
The role of cholinergic signaling in aspects of memory and cognition are typically attributed to the broad spectrum of effects that ACh elicits in altering the excitability of prefrontal cortical and hippocampal circuits (for reviews see Albuquerque et al., 1996; Buzsaki, 2002; Levin et al., 2006; Mansvelder et al., 2006; Newhouse et al., 2004; Picciotto, 2003; Role and Berg, 1996; Sacco et al., 2004; Sarter et al., 2005; Smythies, 2005; Wonnacott et al., 2005). Analysis of the pros and cons of the many theories on how ACh actually does what it does to regulate synaptic efficacy in these regions, as with striatum, is best served by considering the potential interaction of ACh with each of its five major sets of binding partners: ChAT, AChE, VAChT, muscarinic, and nAChRs (see discussion above).
ACh transmission in cortex and hippocampus likely involves both localized release and tonic or “volume” transmission (Cobb and Davies, 2005; Vizi and Kiss, 1998). Activation of presynaptic ACh receptors modulates the release of glutamate, ACh, and dopamine in PFC and hippocampus (Colgin et al., 2003; Laplante et al., 2004; Lucas-Meunier et al., 2003), enhancing or depressing transmission depending on the flavor(s) of AChRs expressed (Mansvelder et al., 2006; Sarter and Parikh, 2005; Wonnacott et al., 2005). Postsynaptic mAChRs and nAChRs have also been implicated in the modulation of PFC and hippocampal circuits (Cobb and Davies, 2005; Frazier et al., 1998; Ji et al., 2001; Jones and Yakel, 1997).
Perhaps the most important (albeit still controversial in detail) role of ACh circuits in cortex and in hippocampus is in the regulation of theta rhythm oscillatory activity (Buzsaki, 2002; Calabresi et al., 2000; Cobb and Davies, 2005; Hasselmo, 2005; Lee et al., 2005). Theta-frequency band oscillations constitute a prominent network pattern in all mammals, including humans. Theta activity has been proposed to underlie everything from temporal cooperativity of cortical and subcortical networks to coordinate modifications of synaptic connections within cortex and hippocampus per se (Buzsaki, 2002; Calabresi et al., 2000; Cobb and Davies, 2005; Hasselmo, 2005). In any case, there is no doubt that cholinergic circuits, specifically the septal cholinergic projections, play an essential role in theta oscillations, as selective lesion of the ACh synthesizing neurons in the medial septal/diagonal band nuclei abolishes hippocampal theta. Discrepancies arise in interpretation of studies that manipulate ACh by different pharmacological means—that is, M1-AChR versus AChE antagonists—which imply that other types of muscarinic and/or nicotinic AChRs may be involved (Buzsaki, 2002; Ji et al., 2001).
IV. Developmental and Genetic Deficits in Schizophrenia That May Influence Function and Assembly of Cholinergic Systems
Schizophrenia is widely viewed as a neurodevelopmental disease, resulting from a combination of environmental challenges acting on susceptible genotypes. Significant progress has been made at identifying both relevant environmental and genetic risk factors (Bresnahan et al., 2005; Harrison and Weinberger, 2005), although we have yet to make progress at understanding how these factors interact to dysregulate relevant circuits in the developing brain. The vertebrate forebrain contains relatively few cholinergic neurons, yet this population exerts widespread modulatory control over essentially all striatal–cortical networks.
Experimental studies have shown that during development, the cholinergic system is especially sensitive to environmental insults (e.g., ethanol, lead, organophosphates, tobacco smoke; Eriksson et al., 2001; Reddy et al., 2003; Robinson, 2002; Thomas et al., 2000). These and other insults would certainly have the potential to interact with genetic vulnerabilities affecting brain development to produce deficits which could contribute to disease states.
A. Development of Cholinergic Systems
Forebrain cholinergic neurons arise early in telencephalic development (~E10 in mouse which approximately corresponds to gestational day 40 in humans; Clancy et al., 2001) in the medial ganglionic eminence, a ventricular/subventricular neurogenic zone that appears as a thickening along the ventral/medial wall of the ventricle (Brady et al., 1989; Furusho, 2006; Marin et al., 2000; Olsson et al., 1998; Semba et al., 1988). Presumptive forebrain projection cholinergic neurons migrate from the MGE (and possibly from the anterior entopeduncular/preoptic area) radially to take up locations in basal forebrain nuclei (medial septum, magnocellular nucleus, diagonal band of Broca), whereas the striatal cholinergic interneurons migrate tangentially from the MGE, occupying dispersed sites throughout the striatal plate. Subsequent to this early birth and migration from the MGE, 1–2 weeks pass before these neurons undergo maturation into cholinergic neurons (Aznavour et al., 2005; Berger-Sweeney, 2003; Mechawar and Descarries, 2001). This delay allows time for other populations of predominantly GABAergic neurons to emerge from the MGE and LGE and occupy their appropriate positions throughout the striatum and cortex for radial population of the neocortex with pyramidal cells and for the proper targeting of axons from the dorsal thalamus to project through the striatal region to innervate cortical structures (Flames et al., 2004; Lopez-Bendito et al., 2006; van Vulpen and van der Kooy, 1998). Some investigators propose that during this period the presumptive cholinergic neurons provide instructive signals that guide the targeting and differentiation of later born striatal populations (Berger-Sweeney, 2003; Hohmann, 2003; Hohmann and Berger-Sweeney, 1998).
During the first two postnatal weeks, the cholinergic interneurons elaborate robust networks of axons locally within the striatum, whereas the forebrain cholinergic neurons elaborate a wide array of axonal projections that target all neocortical regions (prefrontal, sensory, and motor) and the hippocampal formation. As a result these two relatively small populations of cholinergic neurons maximize their ability to interact with striatal–cortical networks. It is likely that the delay between the migration of newly formed cholinergic neurons from neurogenic zones and the elaboration of axonal projections plays a critical role in properly controlling the final wiring of the forebrain cholinergic system.
A number of recent studies using molecular genetic approaches in mice are beginning to clarify the developmental processes that determine the specification of forebrain cholinergic neurons. In particular, the basics of a cholinergic transcription factor code are emerging. A variety of experimental approaches has demonstrated that expression of several transcription factors is important (Mash 1, Olig2, Lmx7, Lmx8) or essential (Nkx2.1) for generating forebrain cholinergic neurons (Bachy and Retaux, 2006; Furusho et al., 2006; Marin et al., 2000; Mori et al., 2004; Zhao et al., 2003). Thus, the combined expression of these factors, and their target genes, probably accounts for much of the intrinsic identity of the cholinergic phenotype. However, these studies have not yet distinguished between factors that determine how a newborn cholinergic neuron migrates from the MGE (radially into basal septal regions or tangentially into the striatum), or whether an individual cholinergic neuron will elaborate a spatially restricted axonal network, as is the case of the striatal interneurons, or a broadly targeted set of cortical projections.
B. Potential Role of Neuregulin 1
Neuregulin 1–ErbB signaling plays multiple critical roles in proper development of the neocortex, guiding both the tangential migration of MGE-derived GABAergic interneurons (Flames et al., 2004) and proper navigation of axonal projections from the dorsal thalamus into the cortex (Lopez-Bendito et al., 2006). Whether neuregulin also guides the migration and/or axon projections of forebrain cholinergic neurons is not known. We have seen apparent decreases in the numbers of specific populations of forebrain interneurons in adult mice that are heterozygous for an isoform specific, targeted mutation in the neuregulin 1 gene (Wolpowitz et al., 2000; Johnson, Talmage, and Role, Unpublished data).
Maturation and maintenance of cholinergic neurons depends on a number of extracellular signaling molecules. Of particular relevance to our discussion of cholinergic signaling and schizophrenia are nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and cortical steroids. NGF, BDNF, and glucocorticoids regulate the expression of ChAT, enhance the connectivity of, and promote the survival of cholinergic neurons (Fagan et al., 1997; Grosse et al., 2005; Guijarro et al., 2006; Johnston et al., 1987; Mobley et al., 1986; Phillips et al., 2004; Sofroniew et al., 2001; Takahashi, 1998; Takahashi and Goh, 1998; Ward and Hagg, 2000). Reports have demonstrated altered regulation of NGF (Parikh et al., 2003), BDNF (Weickert et al., 2003, 2005), and the HPA axis (Corcoran et al., 2001, 2003) in schizophrenics. Whether these changes alter corticostriatal cholinergic tone remains to be seen.
Beyond general effects on synaptic structures, there is no clear evidence linking the products of identified schizophrenia susceptibility genes with the cholinergic system, with the notable exception of the neuregulin 1 gene. Neuregulin 1 has been linked to schizophrenia in multiple populations, and disease-associated changes in the relative expression of different neuregulin 1 isoforms is seen in the DFPLC and hippocampus. Neuregulin 1 isoforms play important roles in neurodevelopment, in particular in the patterning of the neocortex (Flames et al., 2004; Lopez-Bendito et al., 2006). At present, these latter roles for neuregulin 1 have focused on tangential migration of cortical interneurons and axonal projections from the dorsal thalamus to the neocortex. Given the relative spatial and temporal parallels between these events (GABAergic interneurons originate in the MGE during an overlapping time frame with the striatal cholinergic interneurons) and the reported decreases in the numbers of ventral striatal cholinergic inter-neurons in postmortem tissue from schizophrenics, it is important that studies of the role of neuregulin 1 in forebrain development be extended to the cholinergic system as well.
A more direct association between neuregulin 1 and cholinergic signaling exists at the level of the expression of the nAChRs. Two families of neuregulin 1 isoforms were identified originally by virtue of their ability to regulate the expression of nAChRs at peripheral synapses (Falls et al., 1993; Yang et al., 1998). Subsequently, a number of investigators have demonstrated that neuregulin also can increase the synaptic expression of α7-containing nAChRs at central synapses (Kawai et al., 2002; Liu et al., 2001), and our laboratories have extended this story by demonstrating that neuregulin 1 signaling also regulates presynaptic expression and targeting of the α7 nAChRs (Role and Talmage, unpublished). These latter studies are particularly intriguing in light of the well-documented deficits in α7 nAChRs in schizophrenics, the association of this deficit with defects in P50 measures of auditory gating in schizophrenics and their first degree relatives, and the association of α7 subunit gene promoter polymorphisms with these deficits (see chapter by Martin and Freedman, this volume; Leonard et al., 1996, 2002).
V. Clinical and Preclinical Evidence for Deficits in Components of Brain Cholinergic Systems in Schizophrenia
There are numerous examples of deficits in components of brain cholinergic systems that have been linked with schizophrenia. They range from abnormal expression of receptors of various subtypes through decreases in cholinergic neurons in key areas.
A. Deficits in Components of Muscarinic Cholinergic Transmission
Several studies give evidence of alteration of muscarinic ACh receptors in the brains of schizophrenics. The majority of evidence in this realm points to decrements in binding suggestive of a decrease in available m1 muscarinic-binding sites in prefrontal cortex and hippocampus.
Using [(123)I ]IQNB SPECT, one group found decreased muscarinic receptor availability in unmedicated patients with schizophrenia as compared to controls in a variety of cortical and subcortical brain regions (Raedler et al., 2003). Another group used GTP-γS binding to distinguish M2 and M3 muscarinic receptors and found no change in postmortem cortex from patients with schizophrenia as compared to controls, while finding a reduction in M1 (Scarr et al., 2006). Other groups have found decreased pirenzapine binding in the hippocampus in brains of patients with schizophrenia (Crook et al., 2000) and decreased M1 receptor mRNA in dorsolateral prefrontal cortex (Dean et al., 2002) but not caudate nucleus (Dean et al., 2000).
At the level of genetic findings, there is evidence for linkage of an M1 polymorphism to decreased performance on the Wisonsin Card Sort Test (Liao et al., 2003). In addition, there is evidence that an M5 polymorphism confers susceptibility to schizophrenia. Interestingly, the M5 variant seems to confer risk only in combination with a nicotinic α7 polymorphism (De Luca et al., 2004).
Finally, it has been reported that circulating antibodies to the M1 receptor can be identified in the serum of some schizophrenic patients. These antibodies displace tritiated pirenzapine and have agonist-like properties at the M1 receptor in in vitro assays (Borda et al., 2002).
B. Deficits in Components of Nicotinic Cholinergic Transmission
There is also ample evidence of alterations of nAChRs in the brains of patients with schizophrenia. The most notable set of findings pertains to decrements in α7-containing nAChRs and their function in sensory gating. These studies are dealt with in detail in the subsequent chapter by Martin and Freedman, this volume.
Apart from the well-defined case of α7-containing nicotinic receptors, more global changes in nicotinic cholinergic receptors have been observed as well. One group observed decreased high-affinity nicotine and epibatidine binding in postmortem brains from patients with schizophrenia as compared to controls (Breese et al., 2000). The differences were seen in hippocampus, cortex, and caudate in the subgroup of patients versus controls who smoked. The same group reported increases in receptor sites in nicotine and haldol treated rats (Breese et al., 2000). However, another group found elevation of nicotine binding in the striatum of patients with schizophrenia (Court et al., 2000) and minimal changes in α-bungarotoxin binding in the thalamus (Court et al., 1999).
C. Deficits in Cholinergic Innervation
Besides decrements in receptors for ACh, one group has observed a reduction in numbers of cholinergic interneurons in the ventral striatum (Holt et al., 1999, 2005), but not in other striatal regions. However, cortical cholinesterase and ChAT activity is not reduced in the brains of patients with schizophrenia (Haroutunian et al., 1994).
D. Summary
There are numerous findings of abnormalities in the expression or distribution of many components of cholinergic systems in the brains of patients with schizophrenia, the bulk of which would be expected to lead to decrements in cholinergic neurotransmission. It is unclear whether all of these are primary deficits or in some cases downstream effects of other lesions. At least some cholinergic deficits may have to interact with deficits in other systems in order to confer disease vulnerability. However, despite these uncertainties, the preponderance of evidence points toward possible roles for abnormalities in cholinergic systems participating in the pathophysiology of schizophrenia.
VI. Evidence for Cholinergic Contributions to Schizophrenia Pathophysiology from Clinical and Preclinical Psychopharmacology
Correlation of the efficacy of medications used to treat target symptoms of schizophrenia with effects of those medications on cholinergic systems has been another way in which investigators have attempted to infer potential roles for cholinergic systems in the pathophysiology of schizophrenia. The complex receptor-binding properties of antipsychotic medications and complex relationships of target symptoms and medication side effects have made it difficult to make such inferences in a clear and convincing way. That said, there seems to be ample evidence that well-chosen cholinergic targets have potential to be therapeutic targets.
A. ACh Release, Muscarinic Blockade, Partial Agonists, and “Atypicality”
Preclinical studies have demonstrated that many antipsychotic medications cause the release of ACh in cortical (Ichikawa et al., 2002; Li et al., 2005) and hippocampal regions ( Johnson et al., 2005; Shirazi-Southall et al., 2002). While a broad range of antipsychotic medications have been shown to release cortical and hippocampal ACh, olanzapine and clozapine have been shown to be especially potent in this regard. Interestingly, these are the two antipsychotic medications which seem to have greater efficacy against a broader range of symptoms than other antipsychotic medications (Kane et al., 1988, 2001; Lieberman et al., 2003, 2005).
To some extent, antipsychotic-induced release of cortical ACh correlates with M2 binding affinity; antipsychotic medications with less M2 binding tend to be less potent inducers of cortical ACh release ( Johnson et al., 2005). Observations such as these combined with the observation that both clozapine and olanzapine bind muscarinic receptors with high affinity have led to the idea that anticholinergic properties of antipsychotic medications might be responsible for ACh release, and further might be correlated with “atypicality.” This scenario is plausible in that muscarinic receptors can serve as inhibitory presynaptic auto-receptors, providing a potential mechanism by which their blockade could augment ACh release.
However, olanzapine and clozapine may be only weakly antimuscarinic at the level of clinical symptoms, with fewer anticholinergic side effects than expected based on their potent in vitro displacement of muscarinic ligands (Bymaster et al., 1996). This discrepancy is likely due both to subtype selectivity at muscarinic sites, and to partial agonist activity at M4 and M2 receptors (Bymaster et al., 2003; Michal et al., 1999).
Interestingly, clozapine has cognition impairing properties in mice, but the effect is complex, in that clozapine reduces scopolamine-induced impairment while its direct effects on cognition are reversed by cholinesterase inhibitors. This pattern of reduction of antagonist effects and reversal by treatments that increase the endogenous ligand is highly suggestive of partial agonist effects (Ninan and Kulkarni, 1996) or of interaction of ACh with other types of muscarinic and/or nicotinic receptors (see above). Of note, at least two other compounds, xanomeline (reviewed in Mirza et al., 2003) and PTAC (Bymaster et al., 1999), show antipsychotic-like activity in animal models and are also partial agonists at M4 and M2 receptors.
A further wrinkle in this data is introduced by the fact that both clozapine and olanzapine still release cortical and hippocampal ACh in mice lacking M2 and M4 receptors (Bymaster et al., 2003). As such, neither blockade nor partial agonism at these sites appears to be required for ACh release. In view of recent studies implicating both presynaptic nicotinic and dopamine receptors in the modulation of ACh release, the potential contribution of these pathways to the effects of clozapine and olanzapine should, perhaps, be considered.
B. Both Procholinergic and Anticholinergic Compounds May Ameliorate or Worsen Different Symptom Domains
Adding further to confusion about the role of cholinergic transmission in schizophrenia, muscarinic agonists and antagonists exert opposing effects on various schizophrenia symptom clusters. There has been extensive interest in this area because of the use of medications with “anticholinergic” properties as antipsychotics (e.g., chlorpromazine, perphenazine) and because of the use of medications such as benztropine and biperiden to counter the parkinsonian side effects of traditional neuroleptic antipsychotics. For some time, the prevailing opinion seems to have been that anticholinergic effects were fairly neutral in relation to symptoms of schizophrenia, but closer examination has revealed a more complicated picture.
Several studies showed some increase in positive symptoms (hallucinations and delusions) when anticholinergic medications were added to neuroleptics. In a placebo-controlled trial, procyclidine or placebo was added to flupenthixol in a group of 36 patients, with the subsequent finding that those receiving procyclidine had more positive symptoms than those receiving placebo ( Johnstone et al., 1983). Another study compared symptoms in 47 patients receiving neuroleptics during periods with and without treatment with benztropine or trihexyphenidyl. Again, the anticholinergic medication was associated with an increase in positive symptoms (Singh et al., 1987). A study showed that biperiden increased positive symptoms in a small group of schizophrenic patients when added during a medication free period, strengthening the case that the symptomatic worsening is a direct anticholinergic effect rather than one that depends on interaction with the effects of other medications (Tandon et al., 1991).
Anticholinergic compounds such as benztropine have been shown to cause memory impairment (Brebion et al., 2004; Tune et al., 1982). In one small study, higher serum anticholinergic levels correlated with worsened recall but improved reaction time (Strauss et al., 1990). It is possible that improvements in reaction time represent an anti-parkinsonian effect as all patients in the study were taking antipsychotic medications. In another study, single injections of benztropine or glycopyrrolate impaired free recall (McEvoy and Freter, 1989). Overall, it appears likely that anticholinergic medications could worsen some cognitive deficits in schizophrenia.
At the same time, there is a small body of evidence showing that anticholinergic may decrease some negative symptoms in schizophrenia. This provides an instance in which the domain of cognitive symptoms and the domain of negative symptoms can be separated from one another in part based on how they are affected by pharmacological treatment.
In one study, treatment of a small number of patients with trihexyphenidyl resulted in decreases in affective flattening, avolition, and anhedonia/asociality (Tandon et al., 1992). In another study by the same group, biperiden also reduced negative symptoms (while increasing positive symptoms; Tandon et al., 1991). Abuse of trihexyphenidyl and benztropine has at times been cited as “self-medication” of negative symptoms by patients with schizophrenia, and indeed in one study those who abused these medications tended to have higher Brief Psychiatric Rating Scale (BPRS) scores and more negative symptoms than those who did not abuse anticholinergic medications (Zemishlany et al., 1996).
One might speculate that actions of anticholinergic medications against negative symptoms reflect activity in psychomotor circuits that are parallel in some way to the motor circuits in which anticholinergic medications oppose the parkinsonian actions of neuroleptics.
Conversely, muscarinic agonists have been proposed to have antipsychotic activity and may have potential effects against positive symptoms with particular attention paid to the compound xanomeline (reviewed in Bymaster et al., 2002, 2003).
Recent findings on the function of muscarinic receptors in the striatum may shed some light on these apparent paradoxes. The recent paper by Wang and coworkers (also discussed in Section III.B of this chapter) shows that D2 dopamine receptors serve to inactivate striatal cholinergic interneurons which signal to M1 muscarinic receptors on MSpNs. In essence, muscarinic stimulation and blockade of dopamine are functional equivalents in this circuit. The muscarinic receptors reduce intracellular calcium in the MSpNs, which in turn reduces the production of endocannabinoids, reducing depolarization induced suppression and long-term depression of neurotransmission. In summary, cholinergic activation in this circuit results in more activity of MSpNs in the indirect pathway—and subsequently less release of inhibition of the corticostriatal pathways that may convert drives and feelings into actions and perceptions. One might predict that this action would reduce positive symptoms in schizophrenia in a manner analogous to that of D2 blockade by neuroleptics, but one might also predict that in some components of these pathways, the same set of actions could result in an increase in negative symptoms (Wang et al., 2006).
While far from clear, the body of evidence on effects of muscarinic agonists and antagonists in schizophrenia, and on the cholinergic binding properties of antipsychotic medications seems to point to a complex role for cholinergic transmission in different domains of psychopathology with loci of action in the brain likely to depend on the specific symptom domain examined. A challenge to neuropyschopharmacologists will be to find ways to balance and dissociate beneficial and harmful effects of blocking and enhancing cholinergic transmission at muscarinic receptors.
C. Nicotine Ameliorates a Wide Range of Deficits Seen in Schizophrenia
A large part of the pharmacological evidence pointing to potential cholinergic roles in schizophrenia pathophysiology concerns salutary effects of nicotine in patients with schizophrenia. Nicotine affects a wide range of symptom domains and neuropsychological findings. We will give a broad sampling of the data here, and refer the reader to the subsequent chapter by Martin and Freedman, this volume, for an in-depth review of data on nicotinic receptors and the processing of sensory information in schizophrenia.
Nicotine has been shown to improve abnormalities in smooth pursuit eye movement and saccades during visual tracking (Avila et al., 2003; Depatie et al., 2002; Larrison-Faucher et al., 2004; Sherr et al., 2002). The improvement in saccades was independent of the smoking status of the patients, thus addressing the possibility that nicotine’s effect resulted directly from the elevated incidence of smoking by people with schizophrenia. Nicotine also improved sustained attention in these visual tasks (Avila et al., 2003; Depatie et al., 2002). The effects of nicotine on performance of visual tracking and tasks of visual attention may involve hippocampus and cingulate gyrus (Levin et al., 2006; Newhouse et al., 2004; Tanabe et al., 2006).
In another domain, nicotine improved performance of tasks involving working memory in schizophrenic subjects, enhancing task-related activation of thalamus and anterior cingulated cortex as seen on fMRI (Jacobsen et al., 2004).
Nicotine has also been shown to reverse haloperidol-induced impairments in reaction time and working memory (Levin et al., 1996). Nicotine nasal spray improved delayed recognition and spatial working memory in schizophrenic patients (Myers et al., 2004; Smith et al., 2006). Interestingly, nicotine may actually impair working memory in otherwise healthy smokers (Park et al., 2000) suggesting an inherent difference in nicotine responses in the brains of persons with schizophrenia (discussed in Mansvelder et al., 2006; Newhouse et al., 2004).
While numerous studies show beneficial effects of administered nicotine in schizophrenia, the reverse is not true, that is to say, nicotine withdrawal did not increase positive symptoms in a group of patients with schizophrenia who quit smoking. Increases in negative symptoms were modest and transient (Dalack et al., 1999). In considering these results, it may be important to take into account that the frequent high peaks of nicotine delivered by smoking may not be of sustained therapeutic benefit as compared to other systems of delivery.
Finally, the fairly extensive data on sensory processing and nicotine is perhaps the best pharmacological evidence for a role of cholinergic systems in schizophrenia. It is made stronger by the linkage of a polymorphism in the α7 nicotinic receptor subunit gene to sensory gating deficits in patients with schizophrenia and their relatives (Freedman et al., 2003; see chapter by Martin and Freedman, this volume). This topic is reviewed in detail in the subsequent chapter by Martin and Freedman, this volume. Overall, and in contrast to the data on muscarinic AChRs, evidence to date strongly supports the notion that treatments that interact with nAChRs have almost uniformly ameliorative effects on symptoms of schizophrenia.
D. Despite Clear Effects of Other Cholinergic Compounds, Cholinesterase Inhibitors Are Not Proven Adjuncts in the Treatment of Schizophrenia
Further pharmacological evidence concerning cholinergic participation in the pathophysiology of schizophrenia comes from studies in which patients were treated with medications from the family of AChE inhibitors which were initially developed to treat Alzheimer’s disease (Coyle and Kershaw, 2001; Crismon, 1994; Dooley and Lamb, 2000; Jann, 2000). The effects of these medications in patients with schizophrenia are equivocal at best.
There are several case reports describing improvement in negative and cognitive symptoms of individual patients with cholinesterase inhibitors (Rosse and Deutsch, 2002, using galantamine). There are some positive open label trials of rivastigmine (Lenzi et al., 2003; Mendelsohn et al., 2004) in which patients show improvements on standard-rating scales such as Positive and Negative Symptom Scale (PAANS) or BPRS. The study by Lenzi et al. (2003) was focused on quality of life measures. The study by Mendelsohn et al. (2004) was limited to patients with comorbid dementia, so it may be difficult to generalize the result to the schizophrenic population as a whole.
Some positive findings involving cholinesterase inhibitors were in studies that focused on a specific neurocognitive endophenotype rather than on clinical outcome as a whole. One group found that donepezil normalized fMRI findings on a verbal fluency task (Nahas et al., 2003) and another group found that rivastigmine improved performance on a sustained attention task (Aasen et al., 2005).
On the negative side, several reports of placebo-controlled, double-blind crossover trials of donepezil show no efficacy against symptoms of schizophrenia. These studies were all done using donepezil as a neuroleptic augmentation treatment. Some (Mazeh et al., 2006; Stryjer et al., 2003) were in elderly patients or patients with known comorbid dementia. Others were in a general population of stable schizophrenic patients (Friedman et al., 2002; Stryjer et al., 2004; Tugal et al., 2004). No effects were seen on positive, negative, or cognitive symptoms. All of the studies were fairly small, and in some cases there is concern about potential confounding effects of concurrent nicotine use by patients.
Overall, at this time there is little evidence to suggest significant benefits of cholinesterase inhibitors in schizophrenia, especially to patients who do not suffer from comorbid dementia. In some respects, given what we have outlined about the complexity of cholinergic systems, it is not surprising that a class of medications which brings about global increases in ACh levels would have modest effects; in many brain locations, presynaptic inhibition of release may compensate for decreased degradation when ACh levels rise. Thus, given the evidence of effects of treatments targeting nicotinic and muscarinic receptors, it would seem unwarranted to view the modest effects of cholinesterase inhibitors as evidence against participation of cholinergic systems in schizophrenia pathophysiology.
VII. Conclusions
Proper function of cholinergic systems in the brain is essential for a variety of neurocognitive tasks that are impaired in schizophrenia including attention, volition, working memory, assignment of salience, and the processing of sensory information.
While it is unlikely that cholinergic deficits alone account for any particular symptom domain in schizophrenia, there is ample evidence that schizophrenia is associated with genetic changes and brain abnormalities that can influence both the development and function of cholinergic systems, and that interaction of cholinergic deficits with deficits in other systems has the potential to produce disease symptoms.
Perhaps the clearest case of a cholinergic deficit is that of abnormality in the control of α7 nicotinic receptor expression conferring deficits in sensory gating and vulnerability to schizophrenia (cf chapter by Martin and Freedman, this volume). However, it is likely that nicotine has other important sites of action relating to schizophrenia, and that muscarinic effects on corticostriatal and other circuits are of independent import.
Inferences made from clinical and preclinical psychopharmacological data about the performance of cholinergic systems in schizophrenia are fraught with difficulty and do not point to a simple dysregulation of cholinergic transmission at a single brain location. Rather, there are numerous points where dysfunction of particular components of cholinergic signaling can contribute to symptoms or where medications can ameliorate (or worsen) symptoms regardless of an intrinsic cholinergic deficit. In some instances, these points are uncomfortably close to one another and the effects of cholinergic signaling may be arrayed in opposite directions. In other instances, manipulation of cholinergic systems at one site may be undone by effects of the same manipulation at a distant site. The challenge for neurobiologists and pyschopharmacologists is to find ways to refine our interventions in this complex system and to develop compounds or combinations of compounds which can target sites of interest, without substituting one set of impairments for another.
References
- Aasen I, Kumari V, Sharma T. Effects of rivastigmine on sustained attention in schizophrenia: An FMRI study. J Clin Psychopharmacol. 2005;25:311–317. doi: 10.1097/01.jcp.0000169267.36797.76. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Bonfante-Cabarcas R, Marchioro M, Matsubayashi H, Alkondon M, Maelicke A. Nicotinic acetylcholine receptors on hippocampal neurons: Cell compartment-specific expression and modulatory control of channel activity. Prog Brain Res. 1996;109:111–124. doi: 10.1016/s0079-6123(08)62093-2. [DOI] [PubMed] [Google Scholar]
- Apicella P. Tonically active neurons in the primate striatum and their role in the processing of information about motivationally relevant events. Eur J Neurosci. 2002;16:2017–2026. doi: 10.1046/j.1460-9568.2002.02262.x. [DOI] [PubMed] [Google Scholar]
- Avila MT, Sherr JD, Hong E, Myers CS, Thaker GK. Effects of nicotine on leading saccades during smooth pursuit eye movements in smokers and nonsmokers with schizophrenia. Neuropsychopharmacology. 2003;28:2184–2191. doi: 10.1038/sj.npp.1300265. [DOI] [PubMed] [Google Scholar]
- Aznavour N, Watkins KC, Descarries L. Postnatal development of the cholinergic innervation in the dorsal hippocampus of rat: Quantitative light and electron microscopic immunocytochemical study. J Comp Neurol. 2005;486:61–75. doi: 10.1002/cne.20501. [DOI] [PubMed] [Google Scholar]
- Bachy I, Retaux S. GABAergic specification in the basal forebrain is controlled by the LIM-hd factor Lhx7. Dev Biol. 2006;291:218–226. doi: 10.1016/j.ydbio.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J. The cholinergic basal forebrain system during development and its influence on cognitive processes: Important questions and potential answers. Neurosci Biobehav Rev. 2003;27:401–411. doi: 10.1016/s0149-7634(03)00070-8. [DOI] [PubMed] [Google Scholar]
- Borda T, Perez Rivera R, Joensen L, Gomez RM, Sterin-Borda L. Antibodies against cerebral M1 cholinergic muscarinic receptor from schizophrenic patients: Molecular interaction. J Immunol. 2002;168:3667–3674. doi: 10.4049/jimmunol.168.7.3667. [DOI] [PubMed] [Google Scholar]
- Brady DR, Phelps PE, Vaughn JE. Neurogenesis of basal forebrain cholinergic neurons in rat. Brain Res Dev Brain Res. 1989;47:81–92. doi: 10.1016/0165-3806(89)90110-7. [DOI] [PubMed] [Google Scholar]
- Brebion G, Bressan RA, Amador X, Malaspina D, Gorman JM. Medications and verbal memory impairment in schizophrenia: The role of anticholinergic drugs. Psychol Med. 2004;34:369–374. doi: 10.1017/s0033291703008900. [DOI] [PubMed] [Google Scholar]
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, Marks MJ, Collins AC, Leonard S. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–364. doi: 10.1016/S0893-133X(00)00121-4. [DOI] [PubMed] [Google Scholar]
- Bresnahan M, Schaefer CA, Brown AS, Susser ES. Prenatal determinants of schizophrenia: What we have learned thus far? Epidemiol Psichiatr Soc. 2005;14:194–197. doi: 10.1017/s1121189x00007946. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Calligaro DO, Falcone JF, Marsh RD, Moore NA, Tye NC, Seeman P, Wong DT. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology. 1996;14:87–96. doi: 10.1016/0893-133X(94)00129-N. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Shannon HE, Rasmussen K, DeLapp NW, Ward JS, Calligaro DO, Mitch CH, Whitesitt C, Ludvigsen TS, Sheardown M, Swedberg M, Rasmussen T, et al. Potential role of muscarinic receptors in schizophrenia. Life Sci. 1999;64:527–534. doi: 10.1016/s0024-3205(98)00597-9. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder C, Ahmed S, McKinzie D. Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1:163–181. doi: 10.2174/1568007024606249. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder CC, Tzavara E, Nomikos GG, Calligaro DO, McKinzie DL. Muscarinic mechanisms of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1125–1143. doi: 10.1016/j.pnpbp.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Acetylcholine-mediated modulation of striatal function. Trends Neurosci. 2000;23:120–126. doi: 10.1016/s0166-2236(99)01501-5. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Davies CH. Cholinergic modulation of hippocampal cells and circuits. J Physiol. 2005;562:81–88. doi: 10.1113/jphysiol.2004.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Kramar EA, Gall CM, Lynch G. Septal modulation of excitatory transmission in hippocampus. J Neurophysiol. 2003;90:2358–2366. doi: 10.1152/jn.00262.2003. [DOI] [PubMed] [Google Scholar]
- Corcoran C, Gallitano A, Leitman D, Malaspina D. The neurobiology of the stress cascade and its potential relevance for schizophrenia. J Psychiatr Pract. 2001;7:3–14. doi: 10.1097/00131746-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Corcoran C, Walker E, Huot R, Mittal V, Tessner K, Kestler L, Malaspina D. The stress cascade and schizophrenia: Etiology and onset. Schizophr Bull. 2003;29:671–692. doi: 10.1093/oxfordjournals.schbul.a007038. [DOI] [PubMed] [Google Scholar]
- Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, Kerwin R, Perry R, Perry E. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: Alpha-bungarotoxin and nicotine binding in the thalamus. J Neurochem. 1999;73:1590–1597. doi: 10.1046/j.1471-4159.1999.0731590.x. [DOI] [PubMed] [Google Scholar]
- Court JA, Piggott MA, Lloyd S, Cookson N, Ballard CG, McKeith IG, Perry RH, Perry EK. Nicotine binding in human striatum: Elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease and in relation to neuroleptic medication. Neuroscience. 2000;98:79–87. doi: 10.1016/s0306-4522(00)00071-3. [DOI] [PubMed] [Google Scholar]
- Coyle J, Kershaw P. Galantamine, a cholinesterase inhibitor that allosterically modulates nicotinic receptors: Effects on the course of Alzheimer’s disease. Biol Psychiatry. 2001;49:289–299. doi: 10.1016/s0006-3223(00)01101-x. [DOI] [PubMed] [Google Scholar]
- Cragg SJ. Meaningful silences: How dopamine listens to the ACh pause. Trends Neurosci. 2006;29:125–131. doi: 10.1016/j.tins.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Crismon ML. Tacrine: First drug approved for Alzheimer’s disease. Ann Pharmacother. 1994;28:744–751. doi: 10.1177/106002809402800612. [DOI] [PubMed] [Google Scholar]
- Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B. Decreased muscarinic receptor binding in subjects with schizophrenia: A study of the human hippocampal formation. Biol Psychiatry. 2000;48:381–388. doi: 10.1016/s0006-3223(00)00918-5. [DOI] [PubMed] [Google Scholar]
- Dalack GW, Becks L, Hill E, Pomerleau OF, Meador-Woodruff JH. Nicotine withdrawal and psychiatric symptoms in cigarette smokers with schizophrenia. Neuropsychopharmacology. 1999;21:195–202. doi: 10.1016/S0893-133X(98)00121-3. [DOI] [PubMed] [Google Scholar]
- De Luca V, Wong AH, Muller DJ, Wong GW, Tyndale RF, Kennedy JL. Evidence of association between smoking and alpha7 nicotinic receptor subunit gene in schizophrenia patients. Neuropsychopharmacology. 2004;29:1522–1526. doi: 10.1038/sj.npp.1300466. [DOI] [PubMed] [Google Scholar]
- Dean B, Crook JM, Pavey G, Opeskin K, Copolov DL. Muscarinic1 and 2 receptor mRNA in the human caudate-putamen: No change in m1 mRNA in schizophrenia. Mol Psychiatry. 2000;5:203–207. doi: 10.1038/sj.mp.4000684. [DOI] [PubMed] [Google Scholar]
- Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2002;7:1083–1091. doi: 10.1038/sj.mp.4001199. [DOI] [PubMed] [Google Scholar]
- DeBoer P, Heeringa MJ, Abercrombie ED. Spontaneous release of acetylcholine in striatum is preferentially regulated by inhibitory dopamine D2 receptors. Eur J Pharmacol. 1996;317:257–262. doi: 10.1016/s0014-2999(96)00761-3. [DOI] [PubMed] [Google Scholar]
- Depatie L, O’Driscoll GA, Holahan AL, Atkinson V, Thavundayil JX, Kin NN, Lal S. Nicotine and behavioral markers of risk for schizophrenia: A double-blind, placebo-controlled, cross-over study. Neuropsychopharmacology. 2002;27:1056–1070. doi: 10.1016/S0893-133X(02)00372-X. [DOI] [PubMed] [Google Scholar]
- Dooley M, Lamb HM. Donepezil: A review of its use in Alzheimer’s disease. Drugs Aging. 2000;16:199–226. doi: 10.2165/00002512-200016030-00005. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Ankarberg E, Viberg H, Fredriksson A. The developing cholinergic system as target for environmental toxicants, nicotine and polychlorinated biphenyls (PCBs): Implications for neurotoxicological processes in mice. Neurotox Res. 2001;3:37–51. doi: 10.1007/BF03033229. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- Flames N, Long JE, Garratt AN, Fischer TM, Gassmann M, Birchmeier C, Lai C, Rubenstein JL, Marin O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004;44:251–261. doi: 10.1016/j.neuron.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Ross RG, Waldo MC, Stevens KE, Adler LE, Leonard S. The genetics of sensory gating deficits in schizophrenia. Curr Psychiatry Rep. 2003;5:155–161. doi: 10.1007/s11920-003-0032-2. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Adler DN, Howanitz E, Harvey PD, Brenner G, Temporini H, White L, Parrella M, Davis KL. A double blind placebo controlled trial of donepezil adjunctive treatment to risperidone for the cognitive impairment of schizophrenia. Biol Psychiatry. 2002;51:349–357. doi: 10.1016/s0006-3223(01)01342-7. [DOI] [PubMed] [Google Scholar]
- Furusho M, Ono K, Takebayashi H, Masahira N, Kagawa T, Ikeda K, Ikenaka K. Involvement of the Olig2 transcription factor in cholinergic neuron development of the basal forebrain. Dev Biol. 2006;293:348–357. doi: 10.1016/j.ydbio.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: From structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Grosse G, Djalali S, Deng DR, Holtje M, Hinz B, Schwartzkopff K, Cygon M, Rothe T, Stroh T, Hellweg R, Ahnert-Hilger G, Hortnag H. Area-specific effects of brain-derived neurotrophic factor (BDNF) genetic ablation on various neuronal subtypes of the mouse brain. Brain Res Dev Brain Res. 2005;156:111–126. doi: 10.1016/j.devbrainres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Guijarro C, Rutz S, Rothmaier K, Turiault M, Zhi Q, Naumann T, Frotscher M, Tronche F, Jackisch R, Kretz O. Maturation and maintenance of cholinergic medial septum neurons require glucocorticoid receptor signaling. J Neurochem. 2006;97:747–758. doi: 10.1111/j.1471-4159.2006.03728.x. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Davidson M, Kanof PD, Perl DP, Powchik P, Losonczy M, McCrystal J, Purohit DP, Bierer LM, Davis KL. Cortical cholinergic markers in schizophrenia. Schizophr Res. 1994;12:137–144. doi: 10.1016/0920-9964(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 5. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. What is the function of hippocampal theta rhythm? —Linking behavioral data to phasic properties of field potential and unit recording data. Hippocampus. 2005;15:936–949. doi: 10.1002/hipo.20116. [DOI] [PubMed] [Google Scholar]
- Hohmann CF. A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neurosci Biobehav Rev. 2003;27:351–363. doi: 10.1016/s0149-7634(03)00066-6. [DOI] [PubMed] [Google Scholar]
- Hohmann CF, Berger-Sweeney J. Cholinergic regulation of cortical development and plasticity. New twists to an old story. Perspect Dev Neurobiol. 1998;5:401–425. [PubMed] [Google Scholar]
- Holt DJ, Herman MM, Hyde TM, Kleinman JE, Sinton CM, German DC, Hersh LB, Graybiel AM, Saper CB. Evidence for a deficit in cholinergic interneurons in the striatum in schizophrenia. Neuroscience. 1999;94:21–31. doi: 10.1016/s0306-4522(99)00279-1. [DOI] [PubMed] [Google Scholar]
- Holt DJ, Bachus SE, Hyde TM, Wittie M, Herman MM, Vangel M, Saper CB, Kleinman JE. Reduced density of cholinergic interneurons in the ventral striatum in schizophrenia: An in situ hybridization study. Biol Psychiatry. 2005;58:408–416. doi: 10.1016/j.biopsych.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Crook JM. Cholinergic systems and schizophrenia: Primary pathology or epiphenomena? J Chem Neuroanat. 2001;22:53–63. doi: 10.1016/s0891-0618(01)00101-6. [DOI] [PubMed] [Google Scholar]
- Ichikawa J, Dai J, O’Laughlin IA, Fowler WL, Meltzer HY. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacology. 2002;26:325–339. doi: 10.1016/S0893-133X(01)00312-8. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, D’Souza DC, Mencl WE, Pugh KR, Skudlarski P, Krystal JH. Nicotine effects on brain function and functional connectivity in schizophrenia. Biol Psychiatry. 2004;55:850–888. doi: 10.1016/j.biopsych.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Jann MW. Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer’s disease. Pharmacotherapy. 2000;20:1–12. doi: 10.1592/phco.20.1.1.34664. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Nedza FM, Spracklin DK, Ward KM, Schmidt AW, Iredale PA, Godek DM, Rollema H. The role of muscarinic receptor antagonism in antipsychotic-induced hippocampal acetylcholine release. Eur J Pharmacol. 2005;506:209–219. doi: 10.1016/j.ejphar.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Rutkowski JL, Wainer BH, Long JB, Mobley WC. NGF effects on developing forebrain cholinergic neurons are regionally specific. Neurochem Res. 1987;12:985–994. doi: 10.1007/BF00970927. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Ferrier IN, Frith CD, Owens DG, Bourne RC, Gamble SJ. Adverse effects of anticholinergic medication on positive schizophrenic symptoms. Psychol Med. 1983;13:513–527. doi: 10.1017/s0033291700047942. [DOI] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504(Pt 3):603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- Kane JM, Marder SR, Schooler NR, Wirshing WC, Umbricht D, Baker RW, Wirshing DA, Safferman A, Ganguli R, McMeniman M, Borenstein M. Clozapine and haloperidol in moderately refractory schizophrenia: A 6-month randomized and double-blind comparison. Arch Gen Psychiatry. 2001;58:965–972. doi: 10.1001/archpsyc.58.10.965. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: Regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante F, Srivastava LK, Quirion R. Alterations in dopaminergic modulation of prefrontal cortical acetylcholine release in post-pubertal rats with neonatal ventral hippocampal lesions. J Neurochem. 2004;89:314–323. doi: 10.1111/j.1471-4159.2004.02351.x. [DOI] [PubMed] [Google Scholar]
- Larrison-Faucher AL, Matorin AA, Sereno AB. Nicotine reduces antisaccade errors in task impaired schizophrenic subjects. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:505–516. doi: 10.1016/j.pnpbp.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: Bridging the gap from molecules to behaviour. Nat Rev Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci. 2005;25:4365–4369. doi: 10.1523/JNEUROSCI.0178-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi A, Maltinti E, Poggi E, Fabrizio L, Coli E. Effects of rivastigmine on cognitive function and quality of life in patients with schizophrenia. Clin Neuropharmacol. 2003;26:317–321. doi: 10.1097/00002826-200311000-00011. [DOI] [PubMed] [Google Scholar]
- Leonard S, Adams C, Breese CR, Adler LE, Bickford P, Byerley W, Coon H, Griffth JM, Miller C, Myles-Worsley M, Nagamoto HT, Rollins Y, et al. Nicotinic receptor function in schizophrenia. Schizophr Bull. 1996;22:431–445. doi: 10.1093/schbul/22.3.431. [DOI] [PubMed] [Google Scholar]
- Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, et al. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry. 2002;59:1085–1096. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- Levin ED, Wilson W, Rose JE, McEvoy J. Nicotine-haloperidol interactions and cognitive performance in schizophrenics. Neuropsychopharmacology. 1996;15:429–436. doi: 10.1016/S0893-133X(96)00018-8. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: Behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Lewis DA. The organization of chemically-identified neural systems in monkey prefrontal cortex: Afferent systems. Prog Neuropsychopharmacol Biol Psychiatry. 1990;14:371–377. doi: 10.1016/0278-5846(90)90025-c. [DOI] [PubMed] [Google Scholar]
- Li Z, Huang M, Ichikawa J, Dai J, Meltzer HY. N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology. 2005;30:1986–1995. doi: 10.1038/sj.npp.1300768. [DOI] [PubMed] [Google Scholar]
- Liao DL, Hong CJ, Chen HM, Chen YE, Lee SM, Chang CY, Chen H, Tsai SJ. Association of muscarinic m1 receptor genetic polymorphisms with psychiatric symptoms and cognitive function in schizophrenic patients. Neuropsychobiology. 2003;48:72–76. doi: 10.1159/000072880. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Phillips M, Gu H, Stroup S, Zhang P, Kong L, Ji Z, Koch G, Hamer RM. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: A 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28:995–1003. doi: 10.1038/sj.npp.1300157. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ford B, Mann MA, Fischbach GD. Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J Neurosci. 2001;21:5660–5669. doi: 10.1523/JNEUROSCI.21-15-05660.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Cautinat A, Sanchez JA, Bielle F, Flames N, Garratt AN, Talmage DA, Role LW, Charnay P, Marin O, Garel S. Tangential neuronal migration controls axon guidance: A role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network. Pflugers Arch. 2003;446:17–29. doi: 10.1007/s00424-002-0999-2. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB. Nicotinic modulation of neuronal networks: From receptors to cognition. Psychopharmacology (Berl) 2006;184:292–305. doi: 10.1007/s00213-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazeh D, Zemishlani H, Barak Y, Mirecki I, Paleacu D. Donepezil for negative signs in elderly patients with schizophrenia: An add-on, double-blind, crossover, placebo-controlled study. Int Psychogeriatr. 2006;18(3):429–436. doi: 10.1017/S1041610205003017. [DOI] [PubMed] [Google Scholar]
- McEvoy JP, Freter S. The dose-response relationship for memory impairment by anticholinergic drugs. Compr Psychiatry. 1989;30:135–138. doi: 10.1016/0010-440x(89)90065-5. [DOI] [PubMed] [Google Scholar]
- Mechawar N, Descarries L. The cholinergic innervation develops early and rapidly in the rat cerebral cortex: A quantitative immunocytochemical study. Neuroscience. 2001;108:555–567. doi: 10.1016/s0306-4522(01)00389-x. [DOI] [PubMed] [Google Scholar]
- Mendelsohn E, Rosenthal M, Bohiri Y, Werber E, Kotler M, Strous RD. Rivastigmine augmentation in the management of chronic schizophrenia with comorbid dementia: An open-label study investigating effects on cognition, behaviour and activities of daily living. Int Clin Psychopharmacol. 2004;19:319–324. doi: 10.1097/00004850-200411000-00001. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Spatial attention and neglect: Parietal, frontal and cingulate contributions to the mental representation and attentional targeting of salient extrapersonal events. Philos Trans R Soc Lond B Biol Sci. 1999;354:1325–1346. doi: 10.1098/rstb.1999.0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. The cholinergic innervation of the human cerebral cortex. Prog Brain Res. 2004;145:67–78. doi: 10.1016/S0079-6123(03)45004-8. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Atlas of cholinergic neurons in the forebrain and upper brainstem of the macaque based on monoclonal choline acetyltransferase immunohistochemistry and acetylcholinesterase histochemistry. Neuroscience. 1984;12:669–686. doi: 10.1016/0306-4522(84)90163-5. [DOI] [PubMed] [Google Scholar]
- Michal P, Lysikova M, El-Fakahany EE, Tucek S. Clozapine interaction with the M2 and M4 subtypes of muscarinic receptors. Eur J Pharmacol. 1999;376:119–125. doi: 10.1016/s0014-2999(99)00341-6. [DOI] [PubMed] [Google Scholar]
- Mirza NR, Peters D, Sparks RG. Xanomeline and the antipsychotic potential of muscarinic receptor subtype selective agonists. CNS Drug Rev. 2003;9:159–186. doi: 10.1111/j.1527-3458.2003.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 1986;387:53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- Moore H, Fadel J, Sarter M, Bruno JP. Role of accumbens and cortical dopamine receptors in the regulation of cortical acetylcholine release. Neuroscience. 1999;88:811–822. doi: 10.1016/s0306-4522(98)00261-9. [DOI] [PubMed] [Google Scholar]
- Mori T, Yuxing Z, Takaki H, Takeuchi M, Iseki K, Hagino S, Kitanaka J, Takemura M, Misawa H, Ikawa M, Okabe M, Wanaka A. The LIM homeobox gene, L3/Lhx8, is necessary for proper development of basal forebrain cholinergic neurons. Eur J Neurosci. 2004;19:3129–3141. doi: 10.1111/j.0953-816X.2004.03415.x. [DOI] [PubMed] [Google Scholar]
- Myers CS, Robles O, Kakoyannis AN, Sherr JD, Avila MT, Blaxton TA, Thaker GK. Nicotine improves delayed recognition in schizophrenic patients. Psychopharmacology (Berl) 2004;174:334–340. doi: 10.1007/s00213-003-1764-8. [DOI] [PubMed] [Google Scholar]
- Nahas Z, George MS, Horner MD, Markowitz JS, Li X, Lorberbaum JP, Owens SD, McGurk S, DeVane L, Risch SC. Augmenting atypical antipsychotics with a cognitive enhancer (donepezil) improves regional brain activity in schizophrenia patients: A pilot double-blind placebo controlled BOLD fMRI study. Neurocase. 2003;9:274–282. doi: 10.1076/neur.9.3.274.15563. [DOI] [PubMed] [Google Scholar]
- Newhouse PA, Potter A, Singh A. Effects of nicotinic stimulation on cognitive performance. Curr Opin Pharmacol. 2004;4:36–46. doi: 10.1016/j.coph.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Ninan I, Kulkarni SK. Clozapine-induced cognitive dysfunction in mice. Methods Find Exp Clin Pharmacol. 1996;18:367–372. [PubMed] [Google Scholar]
- Olsson M, Bjorklund A, Campbell K. Early specification of striatal projection neurons and interneuronal subtypes in the lateral and medial ganglionic eminence. Neuroscience. 1998;84:867–876. doi: 10.1016/s0306-4522(97)00532-0. [DOI] [PubMed] [Google Scholar]
- Parikh V, Evans DR, Khan MM, Mahadik SP. Nerve growth factor in never-medicated first-episode psychotic and medicated chronic schizophrenic patients: Possible implications for treatment outcome. Schizophr Res. 2003;60:117–123. doi: 10.1016/s0920-9964(02)00434-6. [DOI] [PubMed] [Google Scholar]
- Park S, Knopick C, McGurk S, Meltzer HY. Nicotine impairs spatial working memory while leaving spatial attention intact. Neuropsychopharmacology. 2000;22:200–209. doi: 10.1016/S0893-133X(99)00098-6. [DOI] [PubMed] [Google Scholar]
- Partridge JG, Apparsundaram S, Gerhardt GA, Ronesi J, Lovinger DM. Nicotinic acetylcholine receptors interact with dopamine in induction of striatal long term depression. J Neurosci. 2002;22:2541–2549. doi: 10.1523/JNEUROSCI.22-07-02541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Nishimura M, Armanini MP, Chen K, Albers KM, Davis BM. Rescue of NGF-deficient mice II: Basal forebrain cholinergic projections require NGF for target innervation but not guidance. Brain Res Mol Brain Res. 2004;124:1–11. doi: 10.1016/j.molbrainres.2003.01.001. [DOI] [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of behavior: Beyond the inverted U. Trends Pharmacol Sci. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, Egan MF, Coppola R, Weinberger DR. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am J Psychiatry. 2003;160:118–127. doi: 10.1176/appi.ajp.160.1.118. [DOI] [PubMed] [Google Scholar]
- Reddy GR, Basha MR, Devi CB, Suresh A, Baker JL, Shafeek A, Heinz J, Chetty CS. Lead induced effects on acetylcholinesterase activity in cerebellum and hippocampus of developing rat. Int J Dev Neurosci. 2003;21:347–352. doi: 10.1016/s0736-5748(03)00071-6. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Robinson SE. Effects of perinatal buprenorphine and methadone exposures on striatal cholinergic ontogeny. Neurotoxicol Teratol. 2002;24:137–142. doi: 10.1016/s0892-0362(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Rosse RB, Deutsch SI. Adjuvant galantamine administration improves negative symptoms in a patient with treatment-refractory schizophrenia. Clin Neuropharmacol. 2002;25:272–275. doi: 10.1097/00002826-200209000-00010. [DOI] [PubMed] [Google Scholar]
- Sacco KA, Bannon KL, George TP. Nicotinic receptor mechanisms and cognition in normal states and neuropsychiatric disorders. J Psychopharmacol. 2004;18:457–474. doi: 10.1177/0269881104047273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci. 2005;6:48–56. doi: 10.1038/nrn1588. [DOI] [PubMed] [Google Scholar]
- Sarter M, Nelson CL, Bruno JP. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophr Bull. 2005;31:117–138. doi: 10.1093/schbul/sbi006. [DOI] [PubMed] [Google Scholar]
- Scarr E, Keriakous D, Crossland N, Dean B. No change in cortical muscarinic M2, M3 receptors or [35S]GTPgammaS binding in schizophrenia. Life Sci. 2006;78:1231–1237. doi: 10.1016/j.lfs.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Semba K, Vincent SR, Fibiger HC. Different times of origin of choline acetyltransferase- and somatostatin-immunoreactive neurons in the rat striatum. J Neurosci. 1988;8:3937–3944. doi: 10.1523/JNEUROSCI.08-10-03937.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr JD, Myers C, Avila MT, Elliott A, Blaxton TA, Thaker GK. The effects of nicotine on specific eye tracking measures in schizophrenia. Biol Psychiatry. 2002;52:721–728. doi: 10.1016/s0006-3223(02)01342-2. [DOI] [PubMed] [Google Scholar]
- Shirazi-Southall S, Rodriguez DE, Nomikos GG. Effects of typical and atypical antipsychotics and receptor selective compounds on acetylcholine efflux in the hippocampus of the rat. Neuropsychopharmacology. 2002;26:583–594. doi: 10.1016/S0893-133X(01)00400-6. [DOI] [PubMed] [Google Scholar]
- Singh MM, Kay SR, Opler LA. Anticholinergic-neuroleptic antagonism in terms of positive and negative symptoms of schizophrenia: Implications for psychobiological subtyping. Psychol Med. 1987;17:39–48. doi: 10.1017/s0033291700012964. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Morrel F, Mesulam MM. Cholinergic synapses in human cerebral cortex: An ultra structural study in serial sections. Exp Neurol. 1997;144(2):361–368. doi: 10.1006/exnr.1997.6413. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Subramanian M, Mesulam MM. Monoaminergic-cholinergic interactions in the primate basal forebrain. Neuroscience. 1999;93:817–829. doi: 10.1016/s0306-4522(99)00116-5. [DOI] [PubMed] [Google Scholar]
- Smith RC, Warner-Cohen J, Matute M, Butler E, Kelly E, Vaidhyanathaswamy S, Khan A. Effects of nicotine nasal spray on cognitive function in schizophrenia. Neuropsychopharmacology. 2006;31:637–643. doi: 10.1038/sj.npp.1300881. [DOI] [PubMed] [Google Scholar]
- Smythies J. Section I. The cholinergic system. Int Rev Neurobiol. 2005;64:1–122. doi: 10.1016/S0074-7742(05)64001-9. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Strauss ME, Reynolds KS, Jayaram G, Tune LE. Effects of anticholinergic medication on memory in schizophrenia. Schizophr Res. 1990;3:127–129. doi: 10.1016/0920-9964(90)90045-9. [DOI] [PubMed] [Google Scholar]
- Stryjer R, Strous RD, Bar F, Werber E, Shaked G, Buhiri Y, Kotler M, Weizman A, Rabey JM. Beneficial effect of donepezil augmentation for the management of comorbid schizophrenia and dementia. Clin Neuropharmacol. 2003;26:12–17. doi: 10.1097/00002826-200301000-00004. [DOI] [PubMed] [Google Scholar]
- Stryjer R, Strous R, Bar F, Shaked G, Shiloh R, Rozencwaig S, Grupper D, Buchman N, Kotler M, Rabey JM, Weizman A. Donepezil augmentation of clozapine monotherapy in schizophrenia patients: A double blind cross-over study. Hum Psychopharmacol. 2004;19:343–346. doi: 10.1002/hup.595. [DOI] [PubMed] [Google Scholar]
- Takahashi LK. Prenatal stress: Consequences of glucocorticoids on hippocampal development and function. Int J Dev Neurosci. 1998;16:199–207. doi: 10.1016/s0736-5748(98)00020-3. [DOI] [PubMed] [Google Scholar]
- Takahashi LK, Goh CS. Glucocorticoid facilitation of cholinergic development in the rat hippocampus. Neuroscience. 1998;83:1145–1153. doi: 10.1016/s0306-4522(97)00472-7. [DOI] [PubMed] [Google Scholar]
- Tanabe J, Tregellas JR, Martin LF, Freedman R. Effects of nicotine on hippocampal and cingulate activity during smooth pursuit eye movement in schizophrenia. Biol Psychiatry. 2006;59:754–761. doi: 10.1016/j.biopsych.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Tandon R, Shipley JE, Greden JF, Mann NA, Eisner WH, Goodson JA. Muscarinic cholinergic hyperactivity in schizophrenia. Relationship to positive and negative symptoms. Schizophr Res. 1991;4:23–30. doi: 10.1016/0920-9964(91)90006-d. [DOI] [PubMed] [Google Scholar]
- Tandon R, DeQuardo JR, Goodson J, Mann NA, Greden JF. Effect of anticholinergics on positive and negative symptoms in schizophrenia. Psychopharmacol Bull. 1992;28:297–302. [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4 receptors: Sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Thomas JD, La Fiette MH, Quinn VR, Riley EP. Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicol Teratol. 2000;22:703–711. doi: 10.1016/s0892-0362(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Tugal O, Yazici KM, Anil Yagcioglu AE, Gogus A. A double-blind, placebo controlled, cross-over trial of adjunctive donepezil for cognitive impairment in schizophrenia. Int J Neuropsychopharmacol. 2004;7:117–123. doi: 10.1017/S1461145703004024. [DOI] [PubMed] [Google Scholar]
- Tune LE, Strauss ME, Lew MF, Breitlinger E, Coyle JT. Serum levels of anticholinergic drugs and impaired recent memory in chronic schizophrenic patients. Am J Psychiatry. 1982;139:1460–1462. doi: 10.1176/ajp.139.11.1460. [DOI] [PubMed] [Google Scholar]
- van Vulpen EH, van der Kooy D. Striatal cholinergic interneurons: Birthdates predict compartmental localization. Brain Res Dev Brain Res. 1998;109:51–58. doi: 10.1016/s0165-3806(98)00012-1. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Kiss JP. Neurochemistry and pharmacology of the major hippocampal transmitter systems: Synaptic and nonsynaptic interactions. Hippocampus. 1998;8:566–607. doi: 10.1002/(SICI)1098-1063(1998)8:6<566::AID-HIPO2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Ward NL, Hagg T. BDNF is needed for postnatal maturation of basal forebrain and neostriatum cholinergic neurons in vivo. Exp Neurol. 2000;162:297–310. doi: 10.1006/exnr.1999.7346. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2003;8:592–610. doi: 10.1038/sj.mp.4001308. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2005;10:637–650. doi: 10.1038/sj.mp.4001678. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. Striatal D2 receptors and LTD: Yes, but not where you thought they were. Neuron. 2006;50:347–348. doi: 10.1016/j.neuron.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Sidhpura N, Balfour DJ. Nicotine: From molecular mechanisms to behaviour. Curr Opin Pharmacol. 2005;5:53–59. doi: 10.1016/j.coph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Woolf N. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Yang X, Kuo Y, Devay P, Yu C, Role L. A cysteine-rich isoform of neuregulin controls the level of expression of neuronal nicotinic receptor channels during synaptogenesis. Neuron. 1998;20:255–270. doi: 10.1016/s0896-6273(00)80454-7. [DOI] [PubMed] [Google Scholar]
- Zemishlany Z, Aizenberg D, Weiner Z, Weizman A. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11:199–202. doi: 10.1097/00004850-199609000-00007. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Marin O, Hermesz E, Powell A, Flames N, Palkovits M, Rubenstein JL, Westphal H. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zwart R, Van Kleef RG, Vijverberg HP. Physostigmine and atropine potentiate and inhibit neuronal alpha 4 beta 4 nicotinic receptors. Ann NY Acad Sci. 1999;868:636–639. doi: 10.1111/j.1749-6632.1999.tb11339.x. [DOI] [PubMed] [Google Scholar]