

Abstract
When choosing a recombinant P450 enzyme system for in vitro studies, it is critical to understand the strengths, limitations, and applicability of the enzyme system to the study design. While literature kinetic data may be available to assist in enzyme system selection, comparison of data from separate laboratories is often confounded by differences in experimental conditions and bioanalytical techniques. We measured the Michaelis-Menten kinetic parameters for four CYP2C9 substrates (diclofenac, (S)-warfarin, tolbutamide, and (S)-flurbiprofen) using four recombinant CYP2C9 enzyme systems (Supersomes™, Baculosomes®, RECO® system, and in-house purified, reconstituted enzyme) to determine if the enzyme systems exhibited kinetic differences in metabolic product formation rates under uniform experimental conditions. The purified, reconstituted enzyme systems exhibited higher Km values, reduced substrate affinity, and lower calculated intrinsic clearance values compared to baculovirus microsomal preparations. Six to twenty five-fold differences in predicted intrinsic clearance values were calculated for each substrate depending on the enzyme system-substrate combination. Results suggest that P450 reductase interactions with the CYP2C9 protein and varying ratios of CYP2C9/P450 reductase in the enzyme preparations may play a role in these observed differences. Additionally, when (S)-flurbiprofen was used as a substrate probe to determine CYP2C9 inhibition with a set of twelve inhibitors, decreased inhibition potency was observed across eleven of those inhibitors in the RECO® purified, reconstituted enzyme as compared to the Supersomes™ baculovirus microsomal preparation and pooled human liver microsomes. Considering these differences, consistent use of enzyme source is an important component in producing comparable and reproducible kinetics and inhibition data with CYP2C9.
Introduction
The cytochrome P450 super-family of enzymes is responsible for the metabolism of a majority of new chemical entities (NCEs) and currently marketed drugs. The CYP2C sub-family is a significant contributor to overall P450 activity, accounting for up to 15 percent of P450-mediated metabolism (Williams et al., 2004). Of the CYP2C isoforms, CYP2C9 plays a role in the metabolism of NSAIDs, endogenous steroids, and is the major clearance pathway for the low therapeutic index drugs warfarin and phenytoin (Rettie and Jones, 2005). Due to the role of CYP2C9 in drug metabolism, it is important to evaluate the kinetic behavior of CYP2C9 substrates and their potential to undergo inhibition upon concomitant drug administration in a pre-clinical setting. A combination of recombinant expressed enzyme and human liver tissue fractions (human liver microsomes, S9 fraction, or hepatocytes) are typically used to evaluate the activity of P450s. When evaluating certain enzyme characteristics, such as kinetic differences between CYP2C9 polymorphic variants, use of a recombinant enzyme system is often a necessity due to the scarcity of donor material available from individuals having the genetic variation of interest. However, it is still unclear as to how directly comparable a recombinant enzyme system is to the human situation.
Recombinant CYP2C9 enzyme is commonly obtained by expression using baculovirus or E. Coli expression systems and may be membrane bound or made soluble by the action of detergents and purified. Purified CYP2C9 may be full length or truncated to increase solubility, but generally requires reconstitution with P450 reductase and possibly cytochrome b5 in a liposome environment to attain human liver microsome-like activity (Shaw et al., 1997). The kinetic parameters obtained using recombinant P450 enzyme systems are sensitive to experimental conditions such as the presence of magnesium ion, ionic strength, pH, and membrane constituents, and differences in kinetic parameters due to changes in the conditions are often substrate dependent (Schrag and Wienkers, 2000). While comparison of literature kinetics data for the different enzyme systems can be made, true quantitative comparisons require that experiments be conducted using a uniform set of experimental conditions.
Before proceeding with a large number of inhibition potency (Ki) experiments for inhibitors of CYP2C9, we sought to determine whether kinetic and inhibition profile differences existed between commonly used recombinant systems studied under a uniform set of experimental conditions. To this end, metabolite formation rates of four commonly used CYP2C9 substrates (diclofenac, (S)-flurbiprofen, (S)-warfarin, and tolbutamide) were measured in four recombinant enzyme systems to determine whether kinetic differences in Vmax, Km, or CLint existed among the enzyme systems. Based on the kinetic results, the inhibitor potency of twelve CYP2C9 inhibitors was evaluated in Supersomes™ (a baculovirus expressed, microsomal preparation), human liver microsomes (HLMs) and RECO® (a purified, reconstituted preparation) enzyme systems to determine if systematic differences in inhibition potential were present as well.
Materials and Methods
Chemicals
CYP2C9.1 + b5 BD Supersomes™, human liver microsomes (16.1 mg/ml protein, 20 pooled donors), 4'-hydroxydiclofenac, 4-hydroxytolbutamide, and 7-hydroxywarfarin were purchased from BD Gentest (Woburn, MA). CYP2C9.1 Baculosomes®, cytochrome b5, and RECO® CYP2C9 purified, reconstituted enzyme were purchased from Invitrogen (Madison, WI). Purified CYP2C9.1 was expressed according to previously published methods (Hummel et al., 2005). The metabolite 4'-hydroxyflurbiprofen was obtained internally from the Pfizer compound storage library. All other chemicals used were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest purity available.
Incubation Conditions
For kinetic studies performed using Supersomes™, Baculosomes®, RECO® purified reconstituted enzyme, and in-house purified reconstituted enzyme, incubation times (10 min for diclofenac and (S)-flurbiprofen, and 20 minutes for (S)-warfarin and tolbutamide) and protein concentrations (2-5 pmol enzyme/incubation) were within the linear range for each substrate. Incubations using Supersomes™ and Baculosomes® reagents contained 2 pmol of CYP2C9 reagent suspended in phosphate buffer (50 mM, pH 7.4), and incubations using RECO® system and in-house purified reconstituted reagents contained 5 pmol of CYP2C9 reagent suspended in phosphate buffer. Reconstitution conditions for the in-house purified reconstituted system have been published previously (Hutzler et al., 2003). Samples in a 96-well plate format were pre-incubated in an incubator-shaker at 37 °C for 3 min and each reaction was initiated by the addition of NADPH (1 mM final concentration) in a final volume of 100 μl. Organic solvent amounts were kept constant and final concentrations did not exceed 1% (v/v). Eight substrate concentrations ranged from 1 to 600 μM (diclofenac and (S)-flurbiprofen), 2 to 1000 μM ((S)-warfarin), and 4 to 2000 μM (tolbutamide) were used to determine Vmax and Km. Reactions were terminated by the addition of 50 μl of acetonitrile containing 1 μM tenoxicam as internal standard except for diclofenac in which 100 μl was used. Samples were centrifuged at 4,000 rpm for 5 min and analyzed by liquid chromatography/tandem mass spectrometry. Data for the kinetics experiments were collected in duplicate on three successive days.
Inhibitory studies with (S)-flurbiprofen as the probe substrate were conducted under identical incubation conditions, except for the addition of inhibitor. Incubations with HLMs contained 0.05 mg/ml protein suspended in a final volume of 100 μl. For each Ki determination, a matrix of three substrate ((S)-flurbiprofen) and five inhibitor concentrations were run in duplicate. The concentration values of (S)-flurbiprofen (0.5Km, Km, and 2Km) used were 10, 20, and 40 μM for the Supersome™-containing incubations, 2, 4, and 8 μM for the HLM-containing incubations and 60, 120, and 240 μM for the RECO®-containing incubations. Inhibitor concentrations were chosen to span a ten-fold range of the expected Ki estimate.
Liquid Chromatography/Tandem Mass Spectral Analysis
The LC/MS system was comprised of an API 4000 triple quadrapole mass spectrometer with an atmospheric pressure electrospray ionization source (MDS SCIEX, Concord, Ontario, Canada), and two LC-10ADvp pumps with a SCL-10ADvp controller (Shimadzu, Columbia, MD). A Thermo Electron Aquasil-C18 column (2.1 × 20 mm, 3.0 μm, Waltham, MA) was used for separation with initial conditions of 10% B, followed by a gradient of 10% B to 90% B over 1 min (solvent A 0.1% formic acid, solvent B 100% acetonitrile) followed by an immediate return to initial conditions that were maintained for 1 min with a flow rate of 0.5 ml/min.
The system was operated in negative ion mode, and the deprotonated molecular ions were formed using an ion spray voltage of −3500 V, curtain gas of 10 V, collision gas of 8 eV, and source temperature of 600 °C for all compounds. Product ions formed at collision energies of −16 eV (4'-hydroxydiclofenac, m/z 309.8→265.8), −12 eV (4-hydroxyflurbiprofen, m/z 258.9→214.9), −28 eV (7-hydroxywarfarin, m/z 322.9→176.6), −26 eV (4-hydroxytolbutamide, m/z 285.0→185.9), and −14 eV (tenoxicam, m/z 335.9→271.8).
Spectral Binding
Spectral binding studies were conducted as previously reported (Hummel et al., 2005). Briefly, 200 pmol of enzyme was placed in the sample and reference cuvettes. For determination of spectral changes, 5 μl aliquots of (S)-flurbiprofen were added to the sample cuvette, while 5 μl of 50 mM potassium phosphate buffer, pH 7.4, was added to the reference cuvette. The sample and reference cuvettes were allowed to equilibrate for 3 min before spectral analysis. Spectra were recorded on an Aminco DW-2000 UV-visible spectrophotometer with Olis modifications (Olis, Inc., Bogart, GA). The spectrophotometer was set to record data between 350 and 500-nm wavelengths with a slit-width of 6.0 nm and a scan rate of 100 nm/min. The temperature was held constant at 28 °C. The difference in absorbance between the peak (∼390 nm) and trough (∼420 nm) of the type I-binding spectrum was calculated and plotted against (S)-flurbiprofen concentration. A binding constant (KS) was determined by fitting the resulting data to the following equation:
Data Analysis
Kinetic parameters (Vmax and Km) for each substrate were obtained from the untransformed data by non-linear least square regression of the Michaelis-Menten equation using Prism4 (GraphPad Software, San Diego, CA), except for the RECO®, Baculosome® and purified reconstituted CYP2C9 diclofenac kinetic parameters, which were obtained by non-linear least square regression using the following substrate inhibition equation:
For inhibition potency experiments, data were visualized and inspected using Dixon plots. Inhibition potency (Ki) values were obtained from the untransformed data by non-linear least square regression of the Michaelis-Menten competitive inhibition equation using Prism4.
Measurement of P450 Concentrations
The concentration of CYP2C9 for each enzyme preparation was determined according to published methods (Omura and Sato, 1964). Briefly, 100 μl of enzyme preparation was mixed with 500 μl of 50 mM potassium phosphate buffer, pH 7.4, and dithionite was added. Carbon monoxide was gently bubbled through the solution and the spectrum was recorded from 400 to 500 nm using a Cary 4000 UV-Vis spectrophotometer (Varian, Inc, Palo Alto, CA). P450 concentration was determined by the absorbance difference between the 450 nm and 490 nm peak using an extinction coefficient of 91 mM−1cm−1.
Measurement of P450 Reductase Concentrations
The concentration of P450 reductase for each enzyme preparation was determined by in-gel immunochemical detection. A standard curve for P450 reductase ranging from 0.1-10 μg based on commercially available P450 reductase standard (Invitrogen, Madison, WI) was prepared by adding sequential dilutions of P450 reductase solution to 5 μl of Tris-glycine SDS sample buffer (Invitrogen, Madison, WI) and 1 μl of NuPage® reducing buffer (Invitrogen, Madison, WI) and loading the standard solutions onto a Tris-glycine gel. Duplicate samples from each CYP2C9 enzyme system were prepared in a manner identical to the standard curve, heated at 70 °C for 10 minutes, loaded onto the gel and subjected to electrophoresis at 125 V for 90 min. The gel was fixed in 50% v/v isopropanol / 5% v/v acetic acid, washed using ultrapure water and incubated overnight with rabbit anti-P450 reductase primary antibody (Research Diagnostics, Inc, Concord, MA) diluted 1:2000 in 50 mM phosphate buffered saline, pH 7.1, with 0.1% v/v Tween®-20. The gel was rinsed three times with 50 mM phosphate buffered saline, pH 7.1, with 0.1% v/v Tween®-20 and incubated overnight with Alexa Fluor® 680 goat anti-rabbit IgG secondary antibody (Invitrogen, Madison, WI) diluted 1:2000 in 50 mM phosphate buffered saline, pH 7.1, with 0.1% v/v Tween®-20. The gel was rinsed with ultrapure water and analyzed at 680 nm using a LI-COR Odyssey infrared analyzing system (Lincoln, NE). P450 reductase levels for the enzyme preparations were within the standard curve limits.
Measurement of Cytochrome b5 Concentations
The cytochrome b5 concentration for each enzyme preparation was detemined using a previously published method (Venkatakrishnan et al., 2000). Briefly, 200 μl of enzyme preparation was added to 1000 μl of 100 mM potassium phosphate buffer, pH 7.4, and split between two cuvettes. After determining a baseline, 5 μl of 20 mM β-NADH was added to the sample cuvette and the difference spectrum was measured. An extinction coefficient of 185 mM−1cm−1 was used to determine the amount of cytochrome b5 present using the absorbance difference between the peak (425 nm) and trough (410 nm) values using a Cary 4000 UV-Vis spectrophotometer (Varian, Inc, Palo Alto, CA).
Results
Kinetic profiles for each of the four probe substrates and enzyme systems are depicted in Figure 1 and the resulting kinetic parameter estimates are presented in Table 1. Differences in Vmax estimates for each of the substrates were noted among various preparations, and the Baculosomes® system consistently exhibited the highest Vmax estimates when compared to the other three enzyme preparations. No consistent pattern for Vmax estimates emerged as to rank order of preparations across substrates for the Supersomes™, RECO®, or the in-house purified preparations. Literature values for Vmax estimates using human liver microsomes were obtained, though comparison of these estimates to the expressed preparations is difficult due to the lack of specific quantitation of CYP2C9 in the liver microsomes. However, incubations carried out using the purified reconstituted enzyme systems, either RECO® or the in house enzyme preparation, exhibited higher Km values than observed using the baculovirus microsomal enzyme preparations, regardless of CYP2C9 substrate tested. When compared to literature Km values obtained using human liver microsomes, the baculovirus preparations generally exhibited Km values within 2-3 fold of the human liver microsome estimates, while the reconstituted preparations varied generally from 2-20 fold.
Figure 1.
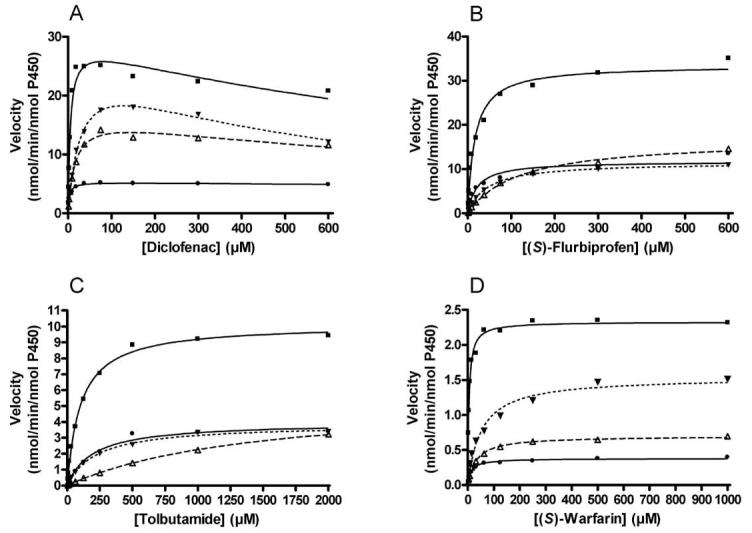
Substrate concentration versus velocity graphs for the four CYP2C9 substrates in the following enzyme systems: RECO® (---Δ), Supersomes™ (—●), Baculosomes® (—■), and a purified, reconstituted system (…▼) using diclofenac (A), (S)-flurbiprofen (B), tolbutamide (C), and (S)-warfarin (D) as substrates.
Table 1.
Kinetic parameters for each enzyme/substrate combination.
| Substrate | Enzyme Source |
Km | Vmax | CLint |
|---|---|---|---|---|
| μM |
nmol/min/nmol P450 |
ml/min/nmol P450 |
||
| Diclofenac | Baculosomes® | 5.1 ± 0.9 | 29 ± 2 | 5.7 |
| Supersomes™ | 2.6 ± 0.3 | 14.8 ± 0.4 | 5.7 | |
| RECO® | 16 ± 2 | 16.8 ± 0.9 | 1.1 | |
| Reconstituted | 30 ± 3 | 27 ± 1 | 0.9 | |
| HLMc | 4.0 | 1670a | 420b | |
| (S)-Flurbiprofen | Baculosomes® | 16.2 ± 0.9 | 34 ± 1 | 2.1 |
| Supersomes™ | 21.6 ± 0.8 | 11.7 ± 0.8 | 0.54 | |
| RECO® | 120 ± 10 | 16.8 ± 0.5 | 0.14 | |
| Reconstituted | 45 ± 2 | 11.5 ± 0.2 | 0.25 | |
| HLMd | 1.9 | 343 a | 180 b | |
| Tolbutamide | Baculosomes® | 103 ± 6 | 10.1 ± 0.2 | 0.098 |
| Supersomes™ | 194 ± 29 | 4.0 ± 0.2 | 0.021 | |
| RECO® | 1462 ± 50 | 5.6 ± 0.1 | 0.0038 | |
| Reconstituted | 220 ± 19 | 3.9 ± 0.1 | 0.018 | |
| HLMc | 147 | 276 a | 1.88 b | |
| (S)-Warfarin | Baculosomes® | 4.6 ± 0.3 | 2.33 ± 0.03 | 0.51 |
| Supersomes™ | 13 ± 2 | 0.38 ± 0.01 | 0.029 | |
| RECO® | 32 ± 2 | 0.698 ± 0.009 | 0.022 | |
| Reconstituted | 52 ± 9 | 1.54 ± 0.07 | 0.030 | |
| HLMe | 3.7 | 10.5a | 2.8 b |
Units of pmol/min/mg protein
Units of μl/min/mg protein
Since the Km parameter is reflective of both affinity of the substrate for the enzyme and the rate at which bound substrate is converted to product, cause(s) of these differences was not obvious (Northrup, 1998). Thus, spectral binding studies were carried out to determine whether substrate affinity differed between the reconstituted and membrane bound systems (Figure and Table 2), using (S)-flurbiprofen as the substrate. The spectral binding constants (KS) for the two reconstituted enzyme systems (RECO® and in-house) were higher than observed in the membrane bound systems. Since reported literature spectral binding constants for (S)-flurbiprofen were similar to those found for membrane bound CYP2C9 (Hummel et al., 2005), additional spectral binding studies were carried out to determine which incubation constituent was responsible for the increase in KS. Interestingly, omission of human P450 reductase from the spectral binding study incubations resulted in KS values similar to those obtained with either of the membrane bound systems or purified P450 in the absence of P450 reductase and cytochrome b5. Omission of cytochrome b5 had no effect on the binding constant estimate, confirming that P450 reductase in the reconstituted preparations was responsible for the increased KS values. In order to better determine the effects of differences in content of redox partner proteins on the observed kinetic parameters and binding, amounts of CYP2C9, P450 reductase and cytochrome b5 in each enzyme preparation were quantitated (Table 3).
Table 2.
Spectral binding constants for each enzyme system with (S)-flurbiprofen as substrate.
| Enzyme Source | KS |
|---|---|
| μM | |
| Baculosomes® | 5.7 |
| Supersomes™ | 8.0 |
| RECO® | 445 |
| Reconstituted (enzyme + P450 reductase + b5) |
304 |
| Reconstituted (enzyme + P450 reductase) |
363 |
| Reconstituted (enzyme + b5) |
8.9 |
| Reconstituted (enzyme alone) |
7.5 |
Table 3.
CYP2C9 stock enzyme preparation concentrations and constituent/CYP2C9 ratios
| Enzyme Preparation |
CYP2C9 Concentration (μM) |
Ratio of P450 reductase/CYP2C9 |
Ratio of Cytochrome b5/CYP2C9 |
Phospholipid Concentration (μM) |
|---|---|---|---|---|
| Baculosomes® | 0.48 | 1.3 | Does not contain |
NA |
| Supersomes™ | 0.48 | 0.4 | 1.3 | NA |
| RECO® | 0.43 | 3.7 | 0.16 | 0.1 μg/ml total liposomesa (DLPC, DOPC, DLPS) |
| Reconstituted | 21.9 | 2.0 | 1.0 | 1.0 μg/μl DLPC |
DLPC = Dilauroyl phosphatidylcholine; DOPC = Dioleoyl phosphatidylcholine; DLPS = Dilauroyl phosphatidylserine
To assess whether enzyme preparation type (membrane bound versus reconstituted) also affects inhibition kinetics, the probe substrate (S)-flurbiprofen was tested against twelve model inhibitors in the RECO® and Supersomes™ systems, as well as human liver microsomes. Results from these inhibition experiments are summarized in Table 4. Seven out of the eleven inhibitors exhibited a greater than three-fold increase in inhibition potency, while half displayed a greater than five-fold increase in inhibition potency in the Supersomes™ system, when compared to results with these same inhibitors in the RECO® enzyme system. Seven of the twelve inhibitors for each recombinant system exhibited Ki estimates within three-fold of human liver microsomal values.
Table 4.
Ki values determined with (S)-flurbiprofen as the substrate probe
| Inhibitor | Supersomes™ Ki (μM) |
HLM Ki (μM) |
RECO® Ki (μM) |
|---|---|---|---|
| Benzbromarone | 0.004 | 0.011 | 0.11 |
| Diclofenac | 1.6 | 2.9 | 9.3 |
| Fluvoxamine | 0.63 | 5.7 | 6.6 |
| Gemfibrozil | 12.4 | 0.98 | 13.9 |
| (S)-Ibuprofen | 4.3 | 3.2 | 17.1 |
| Indomethacin | 53.4 | 12.8 | 48.7 |
| Ketoconazole | 0.38 | 2.1 | 2.4 |
| Miconazole | 0.028 | 0.038 | 0.11 |
| Phenytoin | 5.6 | 10.2 | 11.1 |
| Piroxicam | 0.47 | 9.8 | 6.7 |
| Sulphaphenazole | 0.06 | 0.15 | 1.3 |
| Tolbutamide | 1.5 | 3.5 | 2.0 |
Discussion
Comparison of the kinetic parameters for the substrates using the different enzyme systems revealed two systematic trends. The Baculosomes® system consistently exhibited the highest Vmax estimates, and the purified reconstituted systems consistently exhibited higher Km estimates than the baculovirus microsomal preparations. Because differences in both Vmax and Km across enzyme preparations were observed with the four substrates, the distribution of calculated intrinsic clearance (CLint) values for a given substrate varied widely depending upon the enzyme system used. A six-fold difference in CLint for diclofenac hydroxylation was observed across enzyme preparations, whereas the substrates (S)-flurbiprofen, (S)-warfarin, and tolbutamide exhibited 15, 23, and 26-fold differences between the highest and lowest CLint values, respectively. Increases in Km due in part to reduced affinity for substrates noted in the reconstituted enzyme preparations explains some of this variation, as higher Km values result in reduced calculated CLint values. Additional factors influencing the distribution of the intrinsic clearance values were the consistently higher Vmax values seen with the Baculosmes® preparation and the inconsistent rankings of the other enzyme preparations for a given substrate.
Many factors can influence the rates and efficiency of P450 catalyzed reactions, including the ratios of P450 reductase or cytochrome b5 to P450 and differences in model membrane composition. Spectral determination of CYP2C9 concentration in each of the enzyme preparations did not differ significantly from vendor provided values (Table 3). The ratio of P450 reductase/CYP2C9 in the preparations varied from 0.4 to 3.7, while the amount of cytochrome b5 in the preparations varied from none to a cytochrome b5/CYP2C9 ratio of 2. Comparing the microsomal and reconstituted preparations, the Baculosomes® preparation contained three times more P450 reductase than did the Supersomes™ preparation, while the RECO® preparation contained nearly twice the amount in the in-house reconstituted preparation. Recent experiments indicate that the highest activity for CYP2C9 using reconstituted systems is generally obtained with a P450 reductase/CYP2C9 ratio near 4 and that varying reductase concentrations generally has a grater effect on CYP2C9 activity than does the addition of cytochrome b5 (unpublished data). This bias will influence estimations of intrinsic clearance toward higher values with the Baculosomes® and RECO® preparations. In order to account for the activity differences between recombinant preparations based on levels of accessory proteins when extrapolating to values expected from human liver microsomes, a relative activity factor (RAF) is often used (Crespi and Penman, 1997). The RAF compares activity rates (Vmax estimate) for a specific substrate reaction in human liver microsomes to the activity of the same substrate using the reconstituted system. Extrapolation of kinetic parameters from recombinant P450 systems to human liver microsomes based on relative protein abundance alone may lead to incorrect prediction of activity or relative contribution of individual isoforms (Venkatakrishnan et al., 2000).
The spectral binding results indicated that the presence of human P450 reductase in the reconstituted system increased the KS of (S)-flurbiprofen when compared to preparations containing P450 or a mixture P450 and cytochrome b5. These results were unexpected considering recent reports indicating that the addition of E. coli expressed rat P450 reductase does not alter the KS of alpha-napthoflavone, coumarin, quinidine, and testosterone in CYP1A2, CYP2A6, CYP2D6 and CYP3A4, respectively (Shimada et al., 2005). It is unclear at this time whether factors such as the P450 reductase expression system, species differences in P450 reductase (rat versus human), the ratio of P450 reductase to cytochrome P450 or species-dependent P450 reductase differences may be contributing to the shift in the spectral binding constant. Regardless, differences in substrate affinity may need to be accounted for when using commercially available premixes or in-house reconstituted systems containing human P450 reductase. Although outside of the scope of the current work, further studies are underway to determine the source of these P450 reductase-dependent alterations in substrate affinity.
Differences were also noted with regard to diclofenac kinetic profile observed among the different enzyme preparations. The Baculosomes®, RECO®, and purified reconstituted enzyme systems exhibited substrate inhibition kinetics with diclofenac as probe, but the Supersomes™ preparation did not. Differences in atypical kinetics profiles observed have been reported for membrane bound, baculovirus expressed CYP3A4 when compared to both lymphoblast expressed and human liver microsome preparations employing diclofenac as a substrate and quinidine as effector (Zhang et al., 2004). With human liver microsomes and lymphoblast-expressed enzyme, diclofenac and quinidine exhibited a high degree of positive cooperativity. However, in a baculovirus expressed enzyme, no cooperativity was observed. Solubilization of the baculovirus CYP3A4 enzyme preparation with CHAPS detergent resulted in quinidine-mediated enhancement (cooperativity) of diclofenac turnover equivalent to the lymphoblast and HLM systems. It was postulated that differential positioning of CYP3A4 in the insect cell membrane might mask a putative effector-binding site or not allow an enzymatic conformational change induced by effector binding. While the mechanism of substrate inhibition is not known, positioning of the enzyme in the baculovirus membrane or reduction in the ability of the enzyme to undergo conformational change provides a potential explanation why diclofenac hydroxylation did not exhibit substrate inhibition kinetics in the Supersomes™ preparation
Inhibitory studies were conducted to determine if differences in inhibition potency would also be exhibited between the enzyme preparations. Because general kinetic differences were exhibited between the purified, reconstituted and the baculovirus microsomal preparations, one baculovirus microsomal system (Supersomes™) and one purified, reconstituted system (RECO®) were used for the inhibition studies, along with human liver microsomes. When compared to the results from Supersomes™, the RECO® system exhibited higher Ki values for all but one of the inhibitors and a greater than five fold reduction in inhibition potency for half of the inhibitors. When compared to the results from human liver microsomes, the Supersomes™ exhibited lower Ki values for nine of the inhibitors while the RECO® system exhibited higher Ki values for all of the inhibitors, although values for seven of the twelve inhibitors were within three fold of the human liver microsome data for both recombinant systems. It should be noted that to ensure that the reduction in inhibition potency was due to enzymatic differences and not to non-specific protein binding, equilibrium dialysis protein binding experiments were performed and indicated similar unbound fractions of the inhibitors in both the Supersome™ and RECO® systems (data not shown). The CYP2C9-P450 reductase interactions mentioned previously may alter inhibitor affinity as well as substrate affinity, and provide a potential explanation for these findings
In conclusion, differences in kinetic parameters and the resulting intrinsic clearance values are observed among commonly available CYP2C9 enzyme preparations that may have profound implications for in vitro-in vivo correlations and prediction of in vivo doses and pharmacokinetics. Substrate affinity was reduced in the reconstituted systems likely due to protein-protein interactions. In addition, differences in kinetic profile (typical versus atypical) may be observed among enzyme preparations, though it is unclear whether this is due to changes in enzyme active site conformation, access to putative effector binding sites or other reasons. Finally, choices of enzyme preparation may have dramatic effects on estimations of inhibition potency of a compound, confounding predictions of potential drug-drug interactions. Thus, when comparing literature CYP2C9 kinetic data or conducting screening assays for metabolism kinetics or inhibition, attention must be given to the enzyme source under investigation. Studies are still needed to assess which in vitro system affords the most accurate prediction of in vivo results.
Figure 2.

(S)-Flurbiprofen spectral binding difference plots used in the estimation of binding constants using Baculosomes® (A), RECO® (B), a purified, reconstituted system containing P450 + P450 reductase + b5 (C), and a purified reconstituted system containing P450 + b5 (D).
Acknowledgments
This study was supported by Pharmacokinetics, Dynamics and Metabolism at Pfizer and by National Institutes of Health grants GM063215 and GM069753 to T.S.T.
Non-Standard Abbreviations
- P450
Cytochrome P450
- P450 reductase
Cytochrome P450 reductase
References
- Crespi CL, Penman BW. Use of cDNA-expressed human cytochrome P450 enzymes to study potential drug-drug interactions. Adv Pharmacol. 1997;43:171–188. doi: 10.1016/s1054-3589(08)60205-7. [DOI] [PubMed] [Google Scholar]
- Hemeryck A, De Vriendt C, Belpaire FM. Inhibition of CYP2C9 by selective serotonin reuptake inhibitors: in vitro studies with tolbutamide and (S)-warfarin using human liver microsomes. Eur J Clin Pharmacol. 1999;54:947–951. doi: 10.1007/s002280050580. [DOI] [PubMed] [Google Scholar]
- Hummel MA, Locuson CW, Gannett PM, Rock DA, Mosher CM, Rettie AE, Tracy TS. CYP2C9 genotype-dependent effects on in vitro drug-drug interactions: switching of benzbromarone effect from inhibition to activation in the CYP2C9.3 variant. Mol Pharmacol. 2005;68:644–651. doi: 10.1124/mol.105.013763. [DOI] [PubMed] [Google Scholar]
- Hutzler JM, Wienkers LC, Wahlstrom JL, Carlson TJ, Tracy TS. Activation of cytochrome P450 2C9-mediated metabolism: mechanistic evidence in support of kinetic observations. Arch Biochem Biophys. 2003;410:16–24. doi: 10.1016/s0003-9861(02)00665-3. [DOI] [PubMed] [Google Scholar]
- Northrop DB. On the meaning of Km and Vmax/Km in Enzyme Kinetics. Journal of Chemical Education. 1998;75:1153–1157. [Google Scholar]
- Omura T, Sato R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- Rettie AE, Jones JP. Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu Rev Pharmacol Toxicol. 2005;45:477–494. doi: 10.1146/annurev.pharmtox.45.120403.095821. [DOI] [PubMed] [Google Scholar]
- Schrag ML, Wienkers LC. Topological alteration of the CYP3A4 active site by the divalent cation Mg(2+) Drug Metab Dispos. 2000;28:1198–1201. [PubMed] [Google Scholar]
- Shaw PM, Hosea NA, Thompson DV, Lenius JM, Guengerich FP. Reconstitution premixes for assays using purified recombinant human cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b5. Arch Biochem Biophys. 1997;348:107–115. doi: 10.1006/abbi.1997.0378. [DOI] [PubMed] [Google Scholar]
- Shimada T, Mernaugh RL, Guengerich FP. Interactions of mammalian cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b(5) enzymes. Arch Biochem Biophys. 2005;435:207–216. doi: 10.1016/j.abb.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Tracy TS, Rosenbluth BW, Wrighton SA, Gonzalez FJ, Korzekwa KR. Role of cytochrome P450 2C9 and an allelic variant in the 4′-hydroxylation of (R)- and (S)-flurbiprofen. Biochem Pharmacol. 1995;49:1269–1275. doi: 10.1016/0006-2952(95)00048-5. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, von Moltke LL, Court MH, Harmatz JS, Crespi CL, Greenblatt DJ. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab Dispos. 2000;28:1493–1504. [PubMed] [Google Scholar]
- Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32:647–660. doi: 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32:1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Li Y, Shou M, Zhang Y, Ngui JS, Stearns RA, Evans DC, Baillie TA, Tang W. Influence of different recombinant systems on the cooperativity exhibited by cytochrome P4503A4. Xenobiotica. 2004;34:473–486. doi: 10.1080/00498250410001691271. [DOI] [PubMed] [Google Scholar]