

Abstract
The oxidation of six derivatives of terfenadone by recombinant human CYP2J2 was studied by HPLC coupled to mass spectrometry (MS) using tandem MS techniques and by 1H NMR spectroscopy. CYP2J2 exhibited a surprising regioselectivity in favor of the hydroxylation of the substrate terminal chain at the weakly reactive homobenzylic position. In contrast, hydroxylation of the same substrates by CYP3A4 mainly occurred on the most chemically reactive sites of the substrates (N-oxidation and benzylic hydroxylation). A 3D homology model of CYP2J2 was constructed using recently published structures of CYP2A6, 2B4, 2C8, 2C9 and 2D6 as templates. In contrast with other CYP2 structures, it revealed an active site cavity with a severely restricted access of substrates to the heme through a narrow hydrophobic channel. Dynamic docking of terfenadone derivatives in the CYP2J2 active site allowed one to interpret the unexpected regioselectivity of the hydroxylation of these substrates by CYP2J2, which is mainly based on this restricted access to the iron. The structural features that have been found to be important for recognition of substrates or inhibitors by CYP2J2 were also interpreted on the basis of CYP2J2-substrate interactions in this model.
Keywords: Monooxygenases, drug metabolism, terfenadine, ebastine, hemeproteins
Cytochromes P450 (CYPs) constitute a superfamily of hemoproteins that play key roles in the metabolism of a large variety of xenobiotics and endogenous compounds (1). In the human genome, 57 genes have been found to code for CYPs (2). Many studies on the major isoforms implicated in hepatic drug metabolism and those responsible for the biosynthesis of steroid hormones have been carried out. Specific inhibitors and substrates have been extensively studied (2). Moreover, several X-ray structures of human CYPs, including CYP2A6, 2C8, 2C9, 2D6 and 3A4, which together metabolize a large majority of drugs clinically used, have been recently published (3-12).
Fewer data are available for the other human P450s that have been more recently characterized. Among them, CYP2J2 is the only cytochrome that is mainly expressed in the cardiovascular system (13). Its presence was also detected in kidney, lung and the gastrointestinal tract, and to a lower extent in liver (14, 15). CYP2J2 is assumed to be the main arachidonic acid epoxygenase in the heart as the regio- and stereoselectivity of cis-epoxyeicosatrienoic acids (EETs) formation by CYP2J2 match that in human heart (13). EETs are important intracellular messengers in vascular tissues, as they play important roles in the regulation of vascular tone (16, 17), have anti-inflammatory (18) and anti-fibrinolytic properties (19), and protect endothelial cells from ischemic or hypoxic injuries (20, 21). Moreover, it has been shown that CYP2J2-derived metabolites are involved in the recovery of heart function following ischemia in mice (20), that the risk of coronary artery disease is associated with polymorphisms in CYP2J2 gene in humans (22), and that CYP2J2-derived products can affect cardiac electrophysiology (23, 24). EETs and EETs-derived metabolites are also involved in a host of processes related to cancer cell behavior, angiogenesis and tumor pathogenesis (25, 26). Very recent data suggest that CYP2J2 promotes the neoplastic phenotype of carcinoma cells and may represent a novel biomarker and potential target for therapy of human cancers (27).
Besides these roles in the metabolism of endogenous compounds, CYP2J2 could be involved in the metabolism of some drugs, especially in the intestine. So far, CYP2J2 has been shown to contribute to the metabolism of three drugs, ebastine (28, 29), astemizole (30) and terfenadine (31).
Very little data are presently available on the active site topology and substrate specificity of CYP2J2 (2). Quite recently, we have shown that it was possible to obtain high-affinity inhibitors for CYP2J2 by chemical modification of terfenadone, 1 (see Figure 1), that is an isomer of ebastine and a derivative of the drug terfenadine (32, 33). Thus, compound 4 was found to be a selective, competitive inhibitor of CYP2J2 with a Ki value as low as 0.16 μM, and compound 5 was found to be an efficient mechanism-based inhibitor of CYP2J2 (33) (see Figure 1 for the formulas of 4 and 5).
Figure 1. Structure of terfenadone derivatives 1-6 and of their products formed after oxidation by CYP2J2.

This article shows that terfenadone derivatives, such as 4, are good CYP2J2 substrates which are hydroxylated by this P450 with an unexpected regioselectivity favoring the homobenzylic position of their R chain (Figure 1). Construction of a 3D homology model of CYP2J2 and dynamic docking of these terfenadone derivatives in its active site allowed us to interpret the particular affinity of some of these derivatives, as well as the peculiar regioselectivity of their hydroxylation by CYP2J2.
EXPERIMENTAL PROCEDURES
Materials
Terfenadone, 4-[4(hydroxydiphenylmethyl)piperidin-1-yl]-1-(4-terbutylphenyl)butan-1-one, 1, 4-[(4(hydroxydiphenylmethyl)piperidin-1-yl]-1-(4-methylphenyl)-butan-1-one, 2, 4-[(4(hydroxydiphenylmethyl)piperidin-1-yl]-1-(4-ethylphenyl)butan-1-one, 3, 4-[(4(hydroxydiphenyl-methyl)piperidin-1-yl]-1-(4-propylphenyl)butan-1-one, 4, 1-(4-allylphenyl)-4-(4-(hydroxydiphenylmethyl)piperidin-1-yl)butan-1-one, 5, 4-[(4(hydroxydiphenyl-methyl)piperidin-1-yl]-1-(4-butylphenyl)butan-1-one, 6, and diphenyl(1-(4-(4-propylphenyl)butyl)piperidin-4-yl)methanol, 7, were synthesized as described previously (33). Reference compounds for products 2a, 3b, and 4c were synthesized as reported recently (33). Ebastine was provided by Pharmafarm (Paris, France). All organic solvents were purchased from SDS (Peypin, France) and were of the highest purity available. NADP+ and NADPH-generating system (glucose 6-phosphate and glucose 6-phosphate dehydrogenase) were purchased from Boehringer-Mannheim (Mannheim, Germany).
Origins of recombinant cytochromes P450
CYP2J2 was co-expressed with human P450 reductase in baculovirus-infected Spodoptera frugiperda insect cells (Sf9) and microsomes of these cells were prepared as described previously (13). Microsomes from insect cells expressing CYP3A4 (Supersomes) were purchased from BD Discovery Labware (Woburn, MA, USA).
Oxidation of compounds 1-7 by CYP2J2 and CYP3A4. Incubation procedure and HPLC-MS-UV analysis
Substrate (0.1-20 μM) and microsomes of insect cells expressing CYP2J2 or CYP3A4 (1-5 nM P450) were preincubated for temperature equilibration at 37 °C in a shaking bath for 2-3 min in 0.1 M phosphate buffer, pH 7.4, containing 0.1 mM EDTA. Incubation was started (t0=0 min) with the addition of a NADPH-generating system (1mM NADP+, 10mM glucose 6-phosphate and 2 units of glucose 6-phosphate dehydrogenase per mL) preincubated at 37°C for 2 min. Usual incubation times were 2 to 5 min for kinetic constants determination and up to 30 min for products identification. At t0 and regularly thereafter, aliquots (200 μL) were taken and the reaction was terminated by treatment with 100 μL of a cold CH3CN/CH3COOH (10:1) mixture. Proteins were precipitated by centrifugation for 10 min at 10,000 rpm, and the supernatant was analyzed by reversed-phase HPLC. The apparatus for HPLC/MS-UV analysis was composed of a Surveyor HPLC system and LCQ Advantage-ion trap mass spectrometer (Thermo Finnigan, Les Ulis, France). The column used was a Kromasil C18 column (150 × 2.1 mm, 3.5 μ) (AIT, Marly, France). The mobile phase consisted of water/acetonitrile/formic acid (80/20/1) (solvent A) and acetonitrile/formic acid (99/1) (solvent B), at a flow rate of 200 μL/min. Elution was performed with a linear gradient from 0% of B to 70 % in 13 min, followed by a gradient to 100% of B in 4 min, and ended by 4 min at 100 % B. Elution was monitored at 254 nm for quantification. The MS ionisation was carried out using an ESI source in positive mode, with a capillary temperature at 275°C, a capillary voltage of 41V and a spray voltage of 5 kV. Amounts of products were quantified by using UV detection at 254 nm. KM and kcat parameters were determined using Kaleidagraph software (Synergy, PA, USA).
Identification of oxidation products by tandem mass spectrometry (MS2 and MS3)
MS2 and MS3 analyses were performed with activated broadband and a fragmentation power set to 40-50 %, depending on the compound analyzed.
1: MS2: m/z 470 (M + H+), 452, 268, 250, 203, 161; 1a: MS2: m/z 486 (M + H+), 468, 450, 268, 250, 219, 177; 2: MS2: m/z 428 (M + H+), 410, 268, 250, 161, 119; 2a: MS2: m/z 444 (M + H+), 426, 408, 268, 250, 177, 135; 3: MS2: m/z 442 (M + H+), 424, 268, 250, 175, 133; 3a: MS2: m/z 458 (M + H+), 440, 422, 268, 250, 191, 176, 149, 131; MS3 for m/z = 191: 176 (m/z - CH3); 3b: MS2: m/z 458 (M + H+), 440, 422, 268, 250, 191, 149, 131; 4: MS2: m/z 456 (M + H+), 438, 268, 250, 189, 147; 4a: MS2: m/z 472 (M + H+), 454, 436, 268, 250, 205, 163; 4b: MS2: m/z 472 (M + H+), 454, 436, 268, 250, 205, 161, 163; MS3 for m/z = 205: 161(m/z - CH3CHO); 5: MS2: m/z 454 (M + H+), 436, 268, 250, 187, 145; 5a: MS2: m/z 470 (M + H+), 452, 434, 250, 203, 176, 161; MS3 for m/z = 203: 176 (m/z - CH=CH2); 5b: MS2: m/z 470 (M + H+), 452, 434, 268, 250, 203, 161, 131; MS3 for m/z = 161: 131 (m/z - CH2O); 5c: MS2: m/z 488 (M + H+), 452, 268, 250, 221, 203, 190, 179, 161, 131; MS3 for m/z = 221: 190 (m/z - CH2OH).
Identification of oxidation products by 1H NMR spectroscopy
Amounts of 3a, 4a, 4b, 5a, 5b, and 5c that were sufficient for 1H NMR structure determination were obtained from incubations of 3, 4, and 5 with rat liver microsomes. Each incubation involved 40mL 0.1 M phosphate buffer, pH 7.4, containing 0.1 mM EDTA and 35 nmol P450 (25 mg protein from liver microsomes from dexamethasone-pretreated rats). In the particular case of the isolation of products 5a, 5b, and 5c, incubations were done in the presence of an inhibitor of microsomal epoxide hydrolases, 3,3,3-trichloropropene oxide (1 mM) (34), in order to accumulate sufficient amounts of 5b. Reaction was started with the addition of a NADPH-generating system (1 mM NADP+, 10 mM glucose 6-phosphate and 2 units of glucose 6-phosphate dehydrogenase per mL), lasted 1h at 37 °C, and was terminated by treatment with 20 μL CH3COOH per mL of incubate. Proteins were precipitated by centrifugation at 10,000 rpm for 10 min. After filtration on SepPak C18 cartridge (Waters, MA, USA), washing of the column with 5 mL H2O, and elution with 2 mL CH3OH, the reaction products were separated by reversed-phase HPLC using the equipment described above. The fractions containing a single product were evaporated; the residue was dissolved in 200 μL D2O, evaporated to dryness, redissolved in 100 μL D2O saturated with Na2CO3, and extracted with 0.75 mL CD2Cl2 (except in the case of 5a for which CDCl3 was used). The final CD2Cl2 (or CDCl3) solution was studied by 1H NMR spectroscopy using a 250 MHz and, for some products, a 500 MHz Bruker instrument. Structure determination was done on the basis of one-dimension 1H NMR and COSY experiments. The signals observed for all the hydrogens of 3a, 4a, 4b, 5a, 5b, and 5c, except for those of the R group, were almost identical to those of the starting substrate (3, 4, and 5), that are described in ref (33). The 1H NMR signals of the R group, that are characteristic of the indicated structure (Figure 1), are the following ones (δ in ppm relative to tetramethylsilane, s, d, t, q, m, and dd used for singulet, doublet, triplet, quadruplet, massif and doublet of doublet, respectively):
3a: δ = 4.81 (1H, q, J = 6.3 Hz, CHOH), 1.97 (3H, d, J = 6.3 Hz, CH3); 4a: δ = 4.64 (1H, t, J = 7.3 Hz, CHOH), 1.73 (2H, m, CH2), 0.88 (3H, t, J = 6.8 Hz, CH3); 4b: δ = 4.02 (1H, m, CHOH), 2.80 (1H, dd, J = 5.1, 13.3 Hz, CHaHb), 2.73 (1H, dd, J = 7.7, 13.3 Hz, CHaHb), 1.20 (3H, d, J = 6.2 Hz, CH3); 5a: δ = 6.00 (1H, m, CH), 5.35 (1H, d, J = 17.3 Hz, CHaHb), 5.25 (1H, d, J = 10.3 Hz, CHOH), 5.21 (1H, d, J = 5.9 Hz, CHaHb); 5b: 3.10 (1H, m, CHO), 2.94 (1H, dd, J = 5.0, 14.8 Hz, CHaHb), 2.85 (1H, dd, J = 6.2, 14.8 Hz, CHaHb), 2.74 (1H, dd, J = 3.9, 5.0 Hz, CHcHdO), 2.50 (1H, dd, J = 2.7, 5.0 Hz, CHcHdO); 5c: δ = 3.91 (1H, m, CHOH), 3.63 (1H, m, CHaHbOH), 3.45 (1H, m, CHaHbOH), 2.81 (2H, m, CH2).
Homology modeling and model refinement
Construction of a CYP2J2 model was done as follows:
The amino acid sequence of CYP2J2 was submitted to SWISS-MODEL v3.5 server in automatic mode (35). Templates used for homology modeling were the x-ray structures reported for CYP2A6 (9), CYP2B4 (36), CYP2C5 (37), CYP2C8 (4) and CYP2D6 (8) (pdb codes 1z11, 2bdm, 1nr6, 1pq2 and 2f9q, respectively). Validation of the protein structure (devoid of heme) was made at this early stage using ANOLEA (38) and WHATCHECK (39), as proposed by SWISS-MODEL.
Iron-protoporphyrin IX was then added to the protein using the X-ray structure data of one of the templates (CYP2C8).
The complete structure was then optimized by 15 cycles of minimizations (1000 steps, Powell method (40)), and short molecular dynamics (MD) runs (500 ps at 50 K) for equilibration. Then, a minimization of 2000 steps (Powell method) was performed to obtain the final model. All computations were performed using Sybyl software (Tripos, Courtaboeuf, France) on an Octane 2 Silicon Graphics workstation (Mountain view, CA, USA), using Tripos force field parameters. MD simulations and minimizations were carried out in vacuo. Temperature for all MD simulations was held to 50 or 100 K. The cutoff for the computation of non-bonded interactions was set to 12 Å.
Validation of the final optimized model was performed using PROCHECK tools (41), yielding an overall score for the model. Stability of the model was also tested by running a 1ns MD simulation at 300 K in vacuo, during which no partial unfolding of the protein secondary and tertiary structures was observed.
Solvent accessible molecular surfaces were calculated using VOIDOO software from Uppsala Software Factory (Uppsala University, Sweden) (42), with a probe solvent radius of 1.4 Å and a grid size set to 0.33 Å. PYMOL software was used for structure rendering (http://www.pymol.org) in figures 5 to 9.
Figure 5. 3D model of the substrate binding site of CYP2J2 and cavity contours.

The active site is viewed perpendicular (top) and parallel (bottom) to the heme. The heme is represented by red sticks with the iron atom shown as a Van der Waals sphere. The active site cavity surface is rendered with a green mesh calculated using VOIDOO (42) with a probe size of 1.4 Å. Portions of the structural elements of the protein surrounding the active site (helices A, B’, F and I; β1 and β4 sheets) are rendered as green (helices) or yellow (sheets) cartoons. Portions of B’-C and K-β1 loops are rendered as grey ribbons. The residues bordering the cavity are shown (sticks), with side chain atoms colored in grey for carbon, red for oxygen, blue for nitrogen and orange for sulfur.
Figure 9. Comparison of the positioning of 4 (A) and 7 (B) in the CYP2J2 active site.

H-bonding between the keto group of 4 and Arg117 is indicated in dash lines.
Docking procedure
The docking protocol used was based on a soft-restrained MD approach previously described (43) and applied to CYP2C8 (44). Briefly, in this protocol, the substrate was placed outside the protein structure in front of the entrance of a possible substrate access channel, its hydroxylation site being in front of the entrance about 20 Å from the iron. The three initial orientations corresponded to different angles (between -15° and +15°) between the mean axis of the substrate molecule and of the substrate access channel entrance, globally considered as cylinders. The choice of the substrate access channel is described in the following paragraph. Two ps MD simulations were performed in vacuo at 50 K to thermally equilibrate the substrate and the protein without restraints applied to the system. Then, a distance-dependent constraint whose force constant values ranged from 3 to 9 kcal/mol/Å2, was applied between the heme iron and the substrate hydroxylation site, and MD simulations were performed at 50 K for 200 ps. Equilibration of docked ligand in the active site was done by releasing the constraint in a final MD run of 200 ps at 100 K. Final minimization (1000 steps, conjugate gradient) was performed to obtain the CYP2J2 - substrates complexes.
Choice of access channel
The docking protocol needs a well-defined access channel through which the substrate will be driven. After thorough examination of the CYP2J2 model and consideration of previous published work on substrate access channels of mammalian P450s (37, 45-50), three possible access channels were found for CYP2J2. Channel 1 was delineated by helices B’, G and I, and the B’-C loop; it corresponds to the substrate access channel previously proposed in CYP2C5 (37, 45, 46). Channel 2 was found to be located between helices F and I and β5-sheet; it is described as the solvent access channel of several P450s (48). Channel 3 was located on the opposite side of the cleft bordered by the B-C loop utilized by channel 1, between helices B’ and G’ and β1-sheet. It should correspond to the cleft between helices B’ and G’ visible in the X-ray structure of an open conformation of CYP2B4 (47). In a more general manner, it corresponds to the access channel identified as “pathway 2b” by Cojocaru et al. in several mammalian P450s (50). Calculation of potential energy profiles of the CYP2J2-terfenadone complex after entrance of the substrate through each of these channels showed that channel 3 presented the lowest energy barrier for this entry. Moreover, when decreasing the constraint force constant value from 9 to 3 kcal/mol/Å2, the distance between the iron and the substrate hydroxylated carbon remained larger than 7 Å in the case of channels 1 and 2. In the case of channel 3, the substrate came closer to the iron, with an iron-hydroxylated carbon distance of about 4 Å, even with the lowest constraint applied. Considering these preliminary docking results, channel 3 was chosen for docking the terfenadone derivatives. Residues used to define the channel entrance were Phe52, Gly84, Ile86, Val113, Pro115, and Asn231.
RESULTS AND DISCUSSION
Oxidation of terfenadone derivatives by CYP2J2
Oxidation of terfenadone derivatives 1-6 by microsomes of insect Sf9 cells expressing CYP2J2, in the presence of a NADPH-generating system, was studied by HPLC-MS. Compounds 1 and 2 led to only one oxidation product, the mass spectrum of which exhibited a molecular ion at m/z = m/z of the molecular ion of the starting compound + 16. A study of the fragments of these molecular ions by MS2 showed that the oxygen atom introduced into 1 and 2 was inserted at the level of the R substituent (Figure 1). Actually, oxidation products of 1 and 2 exhibited HPLC and MS characteristics identical to those of authentic samples of the alcohols derived from an hydroxylation of the terminal methyl group of 1 and 2, 1a and 2a, respectively (Figure 1). These results are in agreement with those of a recent study (Krausz K.W., Lafite P. et al., submitted for publication), that compared the oxidations of terfenadone, 1, by 15 recombinant human P450s including CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2J2, CYP3A4, CYP3A5, CYP4A11 and CYP4F12. The latter results showed that only three of those P450s, CYP2J2, CYP3A4 and CYP3A5, were efficient catalysts of the oxidation of 1. However, contrary to CYP3A4 and CYP3A5, that mainly oxidized 1 at the level of its amine function with formation of dealkylation products, CYP2J2 catalyzed the hydroxylation of the R group of 1 (R = t-butyl in that case), in a highly regioselective manner (Figure 2).
Figure 2. Oxidation of terfenadone, 1, by CYP2J2 and CYP3A4.

Oxidation of compounds 3, 4 and 6 respectively led to 2, 3 and 4 products that exhibited molecular ions at m/z = m/z of the molecular ion of the starting compound + 16, and fragments consistent with an hydroxylation at the level of the R substituent.
Actually, structural determination of the oxidation products in this terfenadone series was possible in general because of the existence of three major fragments in the MS2 spectra of these compounds (Figure 3). Two fragments resulted from the breaking of the N-CH2 (exocyclic) bond - i.e. one corresponding to the HOCPh2-piperidinyl fragment at m/z = 268 and the other corresponding to the (CH2)3COC6H4R fragment. A third fragment corresponding to COC6H4R was also formed by cleavage of the CH2-CO bond. In all products derived from CYP2J2-catalyzed oxidations of compounds 1-6, the two latter fragments exhibited a m/z value equal to m/z of the corresponding starting compound fragment + 16. This clearly showed that the oxygen atom introduced into the substrate was systematically inserted at the level of the C6H4R group. A further detailed analysis of the (CH2)3COC6H4R[O] fragment using MS2 and MS3 techniques showed the formation of fragments resulting from the breaking of C-C bonds whose at least one carbon bore the introduced oxygen atom. This allowed us to determine, in most cases, the site of R in which an oxygen atom was inserted (see Materials and Methods).
Figure 3. Mass spectra of compound 3 and of its products formed upon oxidation by CYP2J2.

Mass spectra were obtained from HPLC-MS2 and MS3 analysis of incubations of 20 μM 3 with 5 nM CYP2J2 and a NADPH-generating system for 30 min, as described in Materials and Methods. The m/z values corresponding to 3 and its oxidation products (3a and 3b) are indicated into squares. Fragments containing the hydroxylated carbon are indicated in boldface. The m/z value of the characteristic fragment for identification of compound 3a is circled.
This is illustrated in the case of CYP2J2-catalyzed hydroxylation of 3 that led to two products. The major product was easily identified as the primary alcohol 3b (33), on the basis of its HPLC and MS characteristics which were identical to those of an authentic sample synthesized previously (33). MS2 and MS3 analyses of the minor product clearly showed that it was a regioisomer of 3b resulting from a benzylic hydroxylation of 3 (fragments at m/z = 149 and 191 corresponding to COC6H4CHOHCH3 and (CH2)3COC6H4CHOHCH3, and fragmentation of the latter leading to a loss of a methyl group, m/z = 176) (Figure 3).
One of the two minor oxidation products of 4 was also identified to a previously described authentic compound (33), the alcohol 4c, by using the same HPLC-MS characterization. One of the other products, 4b, was characterized on the basis of MS2 and MS3 analyses as indicated above (presence of a fragment corresponding to the loss of CH3CHOH).
Compound 5 led to two kinds of oxidation products. The first ones, which comprise 5a and 5b, exhibited a MS molecular ion at m/z = m/z of 5 + 16, whereas the second one (5c) exhibited a MS molecular ion at m/z = m/z of 5 + 34. A more detailed analysis of their MS2 characteristics was in favor of the structures shown in Figure 1. These HPLC-MS data showed that oxidation of 5 by CYP2J2 mainly occurred at the level of its double bond with formation of the corresponding epoxide 5b, whose hydrolysis by insect cell microsomes, that should contain epoxide hydrolases (51), led to the diol 5c. Product 5a should result from an hydroxylation of the benzylic-allylic CH2 group of 5.
Finally, the structures of all these oxidation products of compounds 3, 4, and 5 were completely confirmed on the basis of their 1H NMR spectra, after separation and isolation by preparative HPLC. In order to obtain sufficient amounts of each product for 1H NMR studies, compounds 3, 4, and 5 were incubated with liver microsomes from rats pretreated with dexamethasone. Such preparative experiments required relatively large amounts of cytochrome P450 (up to 35 nmol P450 per incubation), and we had not enough recombinant CYP2J2 for that purpose. Fortunately, oxidation of 3, 4, and 5 by liver microsomes led to the expected products 3a, 4a, 4b, 5a, 5b, and 5c in sufficient amounts for isolation by preparative HPLC and 1H NMR structure determination. These products were found to be identical to those derived from CYP2J2-catalyzed oxidations of 3, 4, and 5, on the basis of their HPLC and MS (including MS2 and MS3) characteristics. The 1H NMR characteristics of 3a, 4a, 4b, 5a, 5b, and 5c completely confirmed the structures indicated in Figure 1. The signals corresponding to the hydrogens of the R substituent clearly showed the position of the oxygen atom introduced into the substrate (see Experimental Procedures for a detailed description of these signals). As mentioned above, the other oxidation products 3b and 4c were identified to previously described authentic compounds (33) on the basis of their identical HPLC and MS characteristics.
As mentioned above, oxidation of 6 by CYP2J2 led to four products that should result from the hydroxylation of the four carbons of the butyl chain of 6. The analysis of the mass spectra of these four products did not allow us to make definitive conclusions about their respective structures.
Table 1 compares the kinetic constants that were calculated for the oxidation (based on the amounts of the major product detected by HPLC-UV) of compounds 1, 3, 4, 5 and 6 by CYP2J2. The amounts of product formed after oxidation of substrate 2 were too low (for 2 concentrations below 5 μM) to calculate the kinetic constants of this oxidation. The best CYP2J2 substrate in this series of compounds was 4 both in terms of KM (lowest value of 0.14 ± 0.02 μM) and of kcat/KM (highest value of 141 min-1.μM-1). Interestingly, the variation of the KM value as a function of the R structure well corresponded to that of the IC50 value previously found for the inhibition of CYP2J2-catalyzed hydroxylation of ebastine by the same compounds (33). Actually, the KM value calculated for 4 (0.14 ± 0.02 μM) was in very good agreement with the Ki (0.16 ± 0.05 μM) and IC50 (0.4 ± 0.1 μM) values previously found (33) for the inhibition of CYP2J2-dependent hydroxylation of ebastine by 4, assuming that Ki = IC50/2 for a competitive inhibitor (52). In a more general manner the KM values found for compounds 1 and 3-6 were roughly equal to IC50/2 (Table 1).
Table 1.
Kinetic constants for the oxidation of compounds 1 - 6 by CYP2J2 (a)
| Compound | KM (μM) | kcat (min-1) | kcat/KM (μM-1.min-1) | IC50(b) (μM) |
|---|---|---|---|---|
| 1 | 0.39 ± 0.01 | 36 ± 2 | 90 | 0.7 ± 0.1 |
| 2 | n.d. | n.d. | n.d. | 0.7 ± 0.2 |
| 3 | 0.24 ± 0.07 | 5 ± 0.6 | 23 | 0.6 ± 0.1 |
| 4 | 0.14 ± 0.02 | 19 ± 1 | 141 | 0.4 ± 0.1 |
| 5 | 0.21 ± 0.02 | 23 ± 2 | 110 | 0.4 ± 0.2 |
| 6 | 0.32 ± 0.02 | 42 ± 5 | 132 | 0.7 ± 0.2 |
Kinetic constants were calculated for the formation of the major product upon oxidation of each compound by microsomes of insect cells expressing recombinant CYP2J2 (conditions described in Materials and Methods). Values are means ± SD from three independent experiments.
Values previously reported (33) for the inhibition of CYP2J2-catalyzed hydroxylation of ebastine by the indicated compound.
n.d.: not determined because of the formation of too low amounts of product.
Regioselectivity of CYP2J2-catalyzed oxidations of compounds 3, 4, and 5. Comparison with the corresponding CYP3A4-catalyzed reactions
Table 2 shows the regioselectivity of CYP2J2-dependent oxidation of 3 that led almost exclusively to the homobenzylic alcohol 3b (98 % hydroxylation on the β-carbon relative to the aryl ring). This result could only be explained by a strict positioning of 3 in the CYP2J2 active site, as, from a chemical point of view, benzylic C-H bonds are much more reactive towards oxidants than unactivated C-H bonds. This was illustrated by a comparative study of the regioselectivity of the oxidation of the R group of 3 by CYP3A4, which is well known to possess a large substrate binding site, that might allow free substrate reorientation, and thus multiple positioning of substrates in the active site (11, 53). CYP3A4-catalyzed hydroxylation of the R group of 3 almost exclusively occurred at the most reactive position, i.e. the benzylic position (Table 2). Such completely different regioselectivities of CYP2J2 and CYP3A4 in the hydroxylation of the R group of 3 were also observed for compounds 4 and 5, as the CYP3A4-dependent oxidation of the R group of 4 and 5 mainly occurred on the benzylic position (88 and 81 % regioselectivity, respectively), whereas the CYP2J2 oxidation of these substrates mainly occurred on the homobenzylic (β) position (85 % and 99 % regioselectivity, respectively; Table 2). Actually, CYP3A4-catalyzed oxidation of terfenadone and its derivatives 3-5 mainly led to N-dealkylation products resulting from an oxidation occurring at the level of their amino function (Figure 2). In the following, we will only discuss the regioselectivity of the minor CYP3A4-dependent oxidation that occurs at the level of the substrate R group, for comparison with oxidations by CYP2J2.
Table 2.
Regioselectivity of the oxidation of the R group of compounds 3, 4 and 5 by CYP2J2 and CYP3A4 (a)
| Compound | Regioselectivity (%) (b) | |||||
|---|---|---|---|---|---|---|
| CYP2J2 | CYP3A4 | |||||
| α | β | γ | α | β | γ | |
| 3 | 2 | 98 | - | 98 | 2 | - |
| 4 | 5 | 85 | 10 | 88 | 9 | 3 |
| 5 | 1 | 99 | 81 | 19 | ||
Oxidation conditions as described in Materials and Methods.
α, β, γ indicate the position of oxidation on the R chain (fig. 1) relative to the aryl ring, i.e. the α- and β- positions refer to the benzylic and homobenzylic positions, respectively. In the case of compound 5, oxidation at positions and corresponds to the epoxidation of the double bond. Values are means from three independent experiments.
The above data revealed two main differences between CYP2J2 and CYP3A4 as catalysts for the oxidation of terfenadone derivatives. The first difference had been described already in the case of terfenadone itself and was also observed in the case of its derivatives 3, 4 and 5. It is related to the ability of CYP2J2 to only oxidize the R substituent of the terfenadone derivatives, whereas CYP3A4 is much less selective as it oxidizes both the amine function (major pathway) and the R group of the same compounds. The second difference between CYP2J2 and CYP3A4 concerned their very different regioselectivities in the oxidation of the R group of terfenadone derivatives, as CYP2J2 mainly oxidizes the homobenzylic position, whereas CYP3A4 favors benzylic oxidation.
The regioselectivity of CYP3A4 seems to be dictated by the intrinsic chemical reactivity of the different parts of the terfenadone-derived molecules, that varies as follows: tertiary amine function > benzylic C-H bonds > unactivated C-H bonds (including homobenzylic C-H bonds). This is presumably due to the wide substrate binding site of CYP3A4 that permits several positionings of the substrates relative to the iron-oxo hydroxylating species. At the opposite, the highly regioselective oxidation of terfenadone derivatives by CYP2J2, that occurs on the poorly reactive homobenzylic C-H bonds, can be attributed to a unique, very strict positioning of these substrates in its active site.
Influence of the presence of the keto group of terfenadone derivatives on the regioselectivity of their oxidation by CYP2J2
Previous studies on the inhibitory effects of a series of terfenadone derivatives towards CYP2J2 have shown the importance of the presence of a keto group para to the R substituent for a good recognition by CYP2J2 (33). For instance, compound 7 (Figure 4) that results from a complete reduction of the keto function of 4, exhibited an IC50 value for CYP2J2-dependent ebastine hydroxylation 10-times higher than 4 (33). An HPLC-MS study of the oxidation of 7 by microsomes of insect cells expressing CYP2J2, under conditions identical to those previously used for the oxidation of compounds 1-6, showed that CYP2J2 catalyzed the hydroxylation of the R group of 7 with a regioselectivity different from the one found in the case of 4 (Figure 4). The loss of regioselectivity observed in the case of 7 corresponds to a marked increase of the benzylic hydroxylation.
Figure 4. Regioselectivity of the hydroxylation of compounds 4 and 7 by CYP2J2.
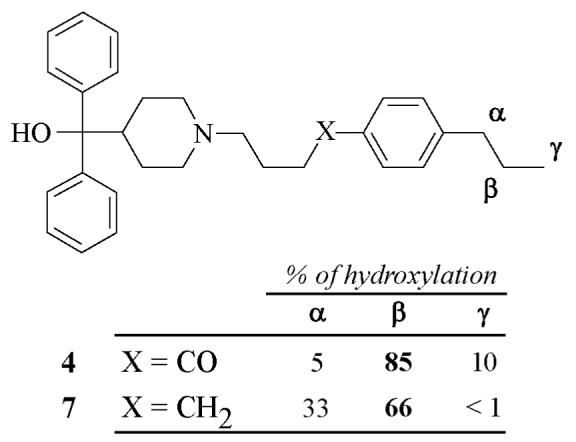
These data indicate that the very strict positioning of terfenadone derivatives in the CYP2J2 active site, which would be at the origin of the high regioselectivity of this enzyme in favor of homobenzylic C-H bonds, implies, at least in part, a binding of the keto group in the CYP2J2 active site.
Construction of a 3D model of CYP2J2 by homology modeling
In a preliminary step, the SWISS-MODEL software, available at the address http://swissmodel.expasy.org, was used to build 3D models of CYP2 proteins whose x-ray crystal structures had been recently published (CYP2C8, CYP2B4 and CYP2C9). This was done to further validate the method which had been previously used to build models of CYP2B6 (54), CYP2E1 (55, 56) and CYP11A1 (57), and to find the most appropriate parameters for producing accurate models. For instance, a model of CYP2B4 was constructed from a combination of 3 CYP2s templates (CYP2A6, CYP2C8 and CYP2C9; pdb codes 1z10, 1pq2 and 1og2, respectively). The root mean square (RMS) deviation of the peptide backbone atoms between the resulting model and the X-ray structure published for CYP2B4 in a closed conformation (58) (pdb code 1suo) was found to be 1.05 Å, whereas the sequence identities between CYP2B4 and X-ray templates were 53, 54 and 51 %, for CYP2A6, CYP2C8 and CYP2C9, respectively.
A 3D model of CYP2J2 was then built using a combination of the X-ray structures of CYP2A6, CYP2B4, CYP2C5, CYP2C8 and CYP2D6 (pdb codes: 1z11, 2bdm, 1nr6, 1pq2 and 2f9q, respectively) as templates. The choice of these templates was made in order to start from the greatest possible diversity of subfamilies in CYP family 2 (CYP2A, CYP2B, CYP2C and CYP2D) and from structures of CYP2s with (CYP2A6-methoxalene, CYP2B4-bifonazole, CYP2C5-diclofenac) and without (CYP2C8, CYP2D6) substrate. Sequence identities of the templates with CYP2J2 varied from 41 to 44 %. Final optimization of the model was done by several cycles of MD simulations in vacuo and energy minimizations. Optimization of the model was followed by validation of the geometry of the model using PROCHECK tools (41) that give an overall score for the model. A structure is considered to be of sufficient quality if its PROCHECK score is below the limit of 0.5. The score obtained for the CYP2J2 model was 0.29 which was within the score range calculated for the templates (X-ray structures) with the same method (from 0.17 to 0.39). Ramachandran plots also gave a good reliability of this model with 81% of residues in the most favored region (to be compared with 84 % to 90 % for the templates).
The global folding of the protein in the final model was very similar to that found in previously described X-ray structures of CYP2s (59), as expected if one considers the construction method based on CYP2s templates. The substrate binding site appeared as a truncated cone between the I, B’, F and F’ helices with an extension up to the β1-sheet (Figure 5). The volume of the cavity corresponding to the solvent-accessible molecular surface was calculated using VOIDOO and found to be 945 Å3. It is larger than that reported for CYP2D6 (540 Å3, pdb code 2f9q (8)) but smaller than that calculated for CYP2C8 (1438 Å3, pdb code 1pq2 (4)). The part of this cavity that is close to the heme and leads to a possible access to the iron is markedly narrower than in other CYP2s, such as CYP2C5 (37), CYP2C8 (4) or CYP2C9 (3). Figure 6 illustrates this particular characteristic of CYP2J2 by comparing the access to the heme in the active site cavity in CYP2J2 and CYP2C9. This restricted access to the heme is due to the presence of a crown of bulky amino acid residues I127, F310, A311, T315, I375, I376 and V380 that are in close proximity to the heme (Figure 7). They form a small, hydrophobic tunnel which is the only possible access to CYP2J2 iron for substrates.
Figure 6. Comparison of the active sites of CYP2J2 and CYP2C9.

The active site cavities for CYP2C9 (pdb 1r9o) and CYP2J2 model were calculated using VOIDOO (42) and are represented as grey mesh. The heme is rendered in sticks and F and I helices as ribbons. The arrows indicate the width of the active site cavity available to the substrate in close proximity to the heme.
Figure 7. Detailed distal view of the amino acid residues determining the access to the heme in the CYP2J2 active site.

The amino acid residues that delineate the active site near the heme are rendered in Van der Waals spheres.
Docking of terfenadone derivatives in the CYP2J2 active site
Docking of compounds 1-4 and 7 in the CYP2J2 active site model was performed using the Soft-restrained MD docking method described previously (43) and applied to CYP2C8 (44). The interest of this method which follows the substrate from its entrance into the substrate access channel to its final positioning close to the heme, is to take into account possible conformational changes of the protein and of substrates that may occur after the entrance of the substrate into the access channel. The different steps of this protocol and the method used for choosing the most appropriate substrate access channel are described in Materials and Methods. The channel used for dynamic docking was delineated by helices B’, G’ and β1-sheet, and corresponded to the access channel identified as pathway 2b by Cojocaru et al. observed in several mammalian P450 X-ray structures (50). Each substrate was docked three times by changing the initial substrate orientation. Using this protocol, the substrate positioning most often found (80 % of the docking experiments) for all the studied compounds is shown in Figure 8 in the case of terfenadone. In this model of CYP2J2-terfenadone complex, the hydrophobic terminal part of 1, that involves the t-butyl group, is in contact with some of the amino acid residues constituting the hydrophobic, narrow tunnel of access to the heme - i.e. I127, F310, I376 and V380. The keto function of 1 is well positioned to establish one or two hydrogen bonds with the guanidine moiety of R117. The (CH2)3 chain of 1 is in contact with M116. Finally, the two phenyl groups of the terminal Ph2COH moiety are in a hydrophobic region formed by several leucine residues (L378, L402, and L83). The terminal OH group of 1 is located in a small, less hydrophobic pocket of this region containing T488, even though it seems to be too far away to establish an hydrogen bond with the oxygen atom of this threonine (O-O distance of 6 Å).
Figure 8. Schematic positioning of terfenadone, 1, in the active site of CYP2J2.

Hydrophobic interactions are shown in red, the hydrogen bond between the keto function of 1 and Arg117 is drawn in blue, and putative polar interaction with Thr488 is represented in green.
Docking of compounds 2, 3, and 4 led to positionings of these substrates in the CYP2J2 active site highly similar to that of terfenadone. The distances between the iron and the carbon atoms of the R group are compared in Table 3. Their absolute values must be considered cautiously as they derive from a model. However, they are in the range of distances (4 to 5 Å) expected for an hydroxylation of the corresponding C-H bonds by the P450 iron-oxo species (48). More interestingly, a comparison of the Fe-C distances for a given substrate allowed one to explain the surprising CYP2J2 regioselectivity mentioned above (see Table 3). Thus, the Fe-C β distance found in the case of 3 is more than 1 Å shorter than the Fe-Cα distance, in agreement with the high regioselectivity that was observed in favor of homobenzylic hydroxylation. In the case of 4, the order of the Fe-C distances completely corresponds to the hydroxylation regioselectivity: Fe-Cβ < Fe-Cγ < Fe-Cα for a 85/10/5 regioselectivity.
Table 3.
Distances calculated between iron and the carbon atoms of R in CYP2J2-terfenadone derivative complex models (a)
| Distances (Å) | |||
|---|---|---|---|
| Substrate | Cα | Cβ | Cγ |
| 3 | 5.1 ± 0.1 | 3.9 ± 0.1 | - |
| (2%) | (98 %) | ||
| 4 | 4.9 ± 0.2 | 3.8 ± 0.1 | 4.6 ± 0.1 |
| (5 %) | (85 %) | (10 %) | |
| 7 | 4.2 ± 0.5 | 4.4 ± 0.5 | 4.6 ± 0.5 |
| (33 %) | (66 %) | (1 %) | |
α, β and γ positions relative to the phenyl ring. Values are means ± SD from 3 structural models obtained from various docking procedures where the initial position of the substrate was changed. Values in parentheses, drawn from Table 2, refer to the regioselectivity of CYP2J2-catalyzed hydroxylation of the carbons of the R chain for each substrate.
Docking of compound 7 in the CYP2J2 model led to a slightly different positioning of the propyl chain relative to the heme, when compared to 4 (Figure 9). The loss of the interaction with R117, that was important in the CYP2J2 - 4 complex, leads to a much greater flexibility of the terminal part of 7 in the active site, and allows the benzylic carbon (Cα) to be closer to the iron, as shown by the Fe-Cα distance which is shorter in the CYP2J2 - 7 complex than in the CYP2J2 complexes with either 3 or 4 (Table 3). This should be at the origin of the lower regioselectivity of CYP2J2-dependent hydroxylation of 7. Actually, in the case of 7, the order of the Fe-C distances does not fit to the observed hydroxylation regioselectivity as well as in the case of 3 and 4. However, presumably because of the greater flexibility of the aryl-R part of 7 in CYP2J2 active site, the error on Fe-C distances determination is much higher than in the case of 3 and 4.
CONCLUSION
The specific behavior of CYP2J2, in terms of its recognition of terfenadone derivatives as inhibitors (33), and its regioselective oxidation of several of those derivatives (shown in Figure 1), can be explained by considering the CYP2J2 3D model described above.
The surprising regioselectivity of CYP2J2-catalyzed oxidation of compounds 3-5, which is strongly in favor of the weakly reactive homobenzylic position of the R chain, can be explained by the shape of the CYP2J2 active site leading to a severely restricted access to the iron. The presence of a small channel constituted by several bulky amino acid residues (I127, A311, I375, I376 and V380) just above the heme would only permit the access to iron of the terminal part of the R chain. This access would also be controlled by a hydrogen bond between the keto function of the terfenadone derivatives and arginine 117. This would explain the loss of regioselectivity observed in CYP2J2-catalyzed hydroxylation of compound 7.
The main structural features that appear to be important for recognition of terfenadone derivatives, as inhibitors (33) or as substrates (this work) of CYP2J2, are: (i) the existence of a small hydrophobic terminal chain (R = propyl leads to the highest affinity), (ii) the presence of a keto substituent on the phenyl ring, para to the R group, and (iii) the existence of an hydrophobic moiety at the other extremity of the substrate. The present 3D model of CYP2J2 explains the high affinity of compounds such as 4 (KM = 0.14 μM, this work; KI = 0.16 μM (33)), as they have the proper length and shape to fill the CYP2J2 active site cavity. The R = propyl chain of 4 is particularly well adapted to establish favorable hydrophobic interactions with the narrow hydrophobic protein channel close to the heme. Moreover, the keto group of 4 is well positioned to establish hydrogen bonds with R117 (Figure 8). Removal of this keto group (as in 7) leads to a 10-fold increase of the IC50 value (33). Introduction of polar alcohol, ether or amide functions into R was also shown to lead to a dramatic decrease of the affinity (33), presumably because of steric hindrance and loss of hydrophobic interactions in the narrow hydrophobic channel. Finally, removal of the HOCPh2 terminal moiety of the molecule was found to cause a 19-fold decrease of affinity (33). Based on the model of figure 8, this would be explained by the loss of several, favorable, hydrophobic interactions of the CPh2 group with a series of leucines of the active site (Leu 83, 378 and 402).
Supplementary experiments, using site-directed mutants, are required to confirm this molecular description of the interactions between CYP2J2 and its substrates and inhibitors. However, the aforementioned results, particularly the 3D model proposed for CYP2J2-substrate interactions, should be important tools (i) to find even more selective and high-affinity substrates and inhibitors, (ii) to interpret or to predict the metabolism of xenobiotics, including drugs, by CYP2J2, and (iii) to discover new possible endogenous substrates of this enzyme.
ACKNOWLEDGMENTS
We thank Dr. Didier Buisson (UMR 8601) for a gift of compound 2, and Dr. Gildas Bertho for his help in 1H NMR structure determination.
ABBREVIATIONS
- COSY
correlation spectroscopy
- CYP or P450
cytochrome P450
- EDTA
ethylenediaminetetraacetic acid
- EET
epoxyeicosatrienoic acid
- ESI
electrospray ionization
- HPLC
high performance liquid chromatography
- MS
mass spectrometry
- MD
molecular dynamics
- MS2/MS3
tandem mass spectrometry
- NMR
nuclear magnetic resonance
- UV
ultraviolet
Footnotes
This work was supported by the C.N.R.S. (Centre National de la Recherche Scientifique) and Ministère de la Recherche (France), and by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (USA).
REFERENCES
- 1.Ortiz de Montellano PR. Cytochrome P450: structure, mechanism, and biochemistry. 3d ed. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 2.Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: structure, mechanism, and biochemistry. 3d ed. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 377–530. [Google Scholar]
- 3.Williams PA, Cosme J, Ward A, Angove HC, MatakVinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 4.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Structure of human microsomal cytochrome P450 2C8: evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 5.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J. Biol. Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 6.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 7.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 8.Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, Lewis CJ, Tennant MM, Modi S, Eggleston DS, Chenery RJ, Bridges AM. Crystal structure of human cytochrome P450 2D6. J. Biol. Chem. 2005;281:7614–7622. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]
- 9.Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 2005;12:822–823. doi: 10.1038/nsmb971. [DOI] [PubMed] [Google Scholar]
- 10.Yano JK, Denton TT, Cerny MA, Zhang X, Johnson EF, Cashman JR. Synthetic inhibitors of cytochrome P-450 2A6: inhibitory activity, difference spectra, mechanism of inhibition, and protein cocrystallization. J. Med. Chem. 2006;49:6987–7001. doi: 10.1021/jm060519r. [DOI] [PubMed] [Google Scholar]
- 11.Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang CW, Yeh HC, Wang LH, Chan NL. Crystal structure of the human prostacyclin synthase. J. Mol. Biol. 2006 doi: 10.1016/j.jmb.2006.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- 14.Zeldin DC, Foley J, Ma J, Boyle JE, Pascual JM, Moomaw CR, Tomer KB, Steenbergen C, Wu S. CYP2J subfamily P450s in the lung: expression, localization, and potential functional significance. Mol. Pharmacol. 1996;50:1111–1117. [PubMed] [Google Scholar]
- 15.Zeldin DC, Foley J, Goldsworthy SM, Cook ME, Boyle JE, Ma J, Moomaw CR, Tomer KB, Steenbergen C, Wu S. CYP2J subfamily cytochrome P450s in the gastrointestinal tract: expression, localization, and potential functional significance. Mol. Pharmacol. 1997;51:931–943. doi: 10.1124/mol.51.6.931. [DOI] [PubMed] [Google Scholar]
- 16.Fleming I, Busse R. Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hypertension. 2006;47:629–633. doi: 10.1161/01.HYP.0000208597.87957.89. [DOI] [PubMed] [Google Scholar]
- 17.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 18.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Ga s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 20.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ. Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 21.Yang B, Graham L, Dikalov S, Mason RP, Falck JR, Liao JK, Zeldin DC. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol. Pharmacol. 2001;60:310–320. doi: 10.1124/mol.60.2.310. [DOI] [PubMed] [Google Scholar]
- 22.Spiecker M, Darius H, Hankeln T, Soufi M, Sattler AM, Schaefer JR, Node K, Borgel J, Mugge A, Lindpaintner K, Huesing A, Maisch B, Zeldin DC, Liao JK. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110:2132–2136. doi: 10.1161/01.CIR.0000143832.91812.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao YF, Ke Q, Seubert JM, Bradbury JA, Graves J, Degraff LM, Falck JR, Krausz K, Gelboin HV, Morgan JP, Zeldin DC. Enhancement of cardiac L-type Ca2+ currents in transgenic mice with cardiac-specific overexpression of CYP2J2. Mol. Pharmacol. 2004;66:1607–1616. doi: 10.1124/mol.104.004150. [DOI] [PubMed] [Google Scholar]
- 24.Lu T, Ye D, Wang X, Seubert JM, Graves JP, Bradbury JA, Zeldin DC, Lee HC. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J. Physiol. 2006;575:627–644. doi: 10.1113/jphysiol.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J. Pharmacol. Exp. Ther. 2005;314:522–532. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- 26.Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol. Ther. 2006;111:584–595. doi: 10.1016/j.pharmthera.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005;65:4707–4715. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- 28.Hashizume T, Imaoka S, Mise M, Terauchi Y, Fujii T, Miyazaki H, Kamataki T, Funae Y. Involvement of CYP2J2 and CYP4F12 in the metabolism of ebastine in human intestinal microsomes. J. Pharmacol. Exp. Ther. 2002;300:298–304. doi: 10.1124/jpet.300.1.298. [DOI] [PubMed] [Google Scholar]
- 29.Liu KH, Kim MG, Lee DJ, Yoon YJ, Kim MJ, Shon JH, Choi CS, Choi YK, Desta Z, Shin JG. Characterization of ebastine, hydroxyebastine, and carebastine metabolism by human liver microsomes and expressed cytochrome P450 enzymes: major roles for CYP2J2 and CYP3A. Drug Metab. Dispos. 2006;34:1793–1797. doi: 10.1124/dmd.106.010488. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto S, Hirama T, Matsubara T, Nagata K, Yamazoe Y. Involvement of CYP2J2 on the intestinal first-pass metabolism of antihistamine drug, astemizole. Drug Metab. Dispos. 2002;30:1240–1245. doi: 10.1124/dmd.30.11.1240. [DOI] [PubMed] [Google Scholar]
- 31.Parikh S, Gagne P, Miller V, Crespi C, Thummel K, Patten C. CYP2J2 and CYP4F12 are active for the metabolism of non-sedating antihistamines: Terfenadine and astemizole. Drug Metab. Rev. 2003;35:190–190. [Google Scholar]
- 32.Lafite P, Dijols S, Buisson D, Macherey AC, Zeldin DC, Dansette PM, Mansuy D. Design and synthesis of selective, high-affinity inhibitors of human cytochrome P450 2J2. Bioorg. Med. Chem. Lett. 2006;16:2777–2780. doi: 10.1016/j.bmcl.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lafite P, Dijols S, Zeldin DC, Dansette PM, Mansuy D. Selective, competitive and mechanism-based inhibitors of human cytochrome P450 2J2. Arch. Biochem. Biophys. 2007 doi: 10.1016/j.abb.2007.03.028. in press; doi:10.1016/j.abb.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oesch F, Kaubisch N, Jerina DM, Daly JW. Hepatic epoxide hydrase. Structure-activity relations for substrates and inhibitors. Biochemistry. 1971;10:4858–4866. doi: 10.1021/bi00802a005. [DOI] [PubMed] [Google Scholar]
- 35.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, White MA, Muralidhara BK, Sun L, Halpert JR, Stout CD. Structure of microsomal cytochrome P450 2B4 complexed with the antifungal drug bifonazole: insight into P450 conformational plasticity and membrane interaction. J. Biol. Chem. 2006;281:5973–5981. doi: 10.1074/jbc.M511464200. [DOI] [PubMed] [Google Scholar]
- 37.Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD. Structure of mammalian cytochrome P450 2C5 complexed with diclofenac at 2.1 A resolution: evidence for an induced fit model of substrate binding. Biochemistry. 2003;42:9335–9345. doi: 10.1021/bi034556l. [DOI] [PubMed] [Google Scholar]
- 38.Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 39.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 40.Powell MJD. An efficient method for finding the minimum of a function of several variables without calculating derivatives. The Computer Journal. 1964;7:155–162. [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 42.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Cryst. 1994;D50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 43.André F, Delaforge M, Loiseau N. A method for performing restrained dynamics docking of one or multiple substrates on multi-specific enzymes. Patent. 2004
- 44.Delaforge M, Pruvost A, Perrin L, Andre F. Cytochrome P450-mediated oxidation of glucuronide derivatives: example of estradiol-17beta-glucuronide oxidation to 2-hydroxy-estradiol-17beta-glucuronide by CYP 2C8. Drug Metab. Dispos. 2005;33:466–473. doi: 10.1124/dmd.104.002097. [DOI] [PubMed] [Google Scholar]
- 45.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 46.Wester MR, Johnson EF, Marques-Soares C, Dansette PM, Mansuy D, Stout CD. Structure of a substrate complex of mammalian cytochrome P450 2C5 at 2.3 A resolution: evidence for multiple substrate binding modes. Biochemistry. 2003;42:6370–6379. doi: 10.1021/bi0273922. [DOI] [PubMed] [Google Scholar]
- 47.Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. An open conformation of mammalian cytochrome P450 2B4 at 1.6-A resolution. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poulos TL, Johnson EF. Structures of cytochromes P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: structure, mechanism, and biochemistry. 3d ed. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 87–114. [Google Scholar]
- 49.de Graaf C, Vermeulen NP, Feenstra KA. Cytochrome P450 in silico: an integrative modeling approach. J Med Chem. 2005;48:2725–2755. doi: 10.1021/jm040180d. [DOI] [PubMed] [Google Scholar]
- 50.Cojocaru V, Winn PJ, Wade RC. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Barth S, Fischer M, Schmid RD, Pleiss J. Sequence and structure of epoxide hydrolases: a systematic analysis. Proteins. 2004;55:846–855. doi: 10.1002/prot.20013. [DOI] [PubMed] [Google Scholar]
- 52.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 53.Scott EE, Halpert JR. Structures of cytochrome P450 3A4. Trends Biochem. Sci. 2005;30:5–7. doi: 10.1016/j.tibs.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 54.Bathelt C, Schmid RD, Pleiss J. Regioselectivity of CYP2B6: homology modeling, molecular dynamics simulation, docking. J. Mol. Model. 2002;8:327–335. doi: 10.1007/s00894-002-0104-y. [DOI] [PubMed] [Google Scholar]
- 55.Vidali M, Hidestrand M, Eliasson E, Mottaran E, Reale E, Rolla R, Occhino G, Albano E, Ingelman-Sundberg M. Use of molecular simulation for mapping conformational CYP2E1 epitopes. J. Biol. Chem. 2004;279:50949–50955. doi: 10.1074/jbc.M407329200. [DOI] [PubMed] [Google Scholar]
- 56.Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. CYP2E1 active site residues in substrate recognition sequence identified by photoaffinity labeling and homology modeling. Arch Biochem Biophys. 2007;459:59–69. doi: 10.1016/j.abb.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sivozhelezov V, Nicolini C. Homology modeling of cytochrome P450scc and the mutations for optimal amperometric sensor. J. Theor. Biol. 2005;234:479–485. doi: 10.1016/j.jtbi.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-Å resolution: insight into the range of P450 conformations and the coordination of redox partner binding. J. Biol. Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 59.Johnson EF, Stout CD. Structural diversity of human xenobiotic-metabolizing cytochrome P450 monooxygenases. Biochem. Biophys. Res. Commun. 2005;338:331–336. doi: 10.1016/j.bbrc.2005.08.190. [DOI] [PubMed] [Google Scholar]