

Abstract
Huntington’s disease (HD), a dominantly inherited neurodegenerative disorder characterized by relatively selective degeneration of striatal neurons, is caused by an expanded polyglutamine tract of the huntingtin protein. The huntingtin mutation reduces levels of brain-derived neurotrophic factor (BDNF) in the striatum, likely by inhibiting cortical BDNF gene expression and anterograde transport of BDNF from cortex to striatum. However, roles of the BDNF reduction in HD pathogenesis have not been established conclusively. We reasoned that increasing striatal BDNF through overexpression would slow progression of the disease if BDNF reduction plays a pivotal role in HD pathogenesis. We employed a Bdnf transgene driven by the promoter for the alpha subunit of Ca2+/calmodulin-dependent kinase II to overexpress BDNF in the forebrain of R6/1 mice which express a fragment of mutant huntingtin with a 116-glutamine tract. The Bdnf transgene increased BDNF levels and TrkB signaling activity in the striatum, ameliorated motor dysfunction, and reversed brain weight loss in R6/1 mice. Furthermore, it normalized DARPP-32 expression, increased the number of enkephalin-containing boutons, and reduced formation of neuronal intranuclear inclusions in the striatum of R6/1 mice. These results demonstrate crucial roles of reduced striatal BDNF in HD pathogenesis and suggest potential therapeutic values of BDNF to HD.
Keywords: brain-derived neurotrophic factor, Huntington’s disease, motor coordination, brain atrophy, neuronal intranuclear inclusions, gene expression
Introduction
Huntington’s disease (HD), a dominantly inherited neurodegenerative disorder characterized by abnormalities of movement and cognition along with changes in psychiatric symptoms, is caused by expansion of a polyglutamine tract at the N-terminus of huntingtin (htt). The clinical signs and symptoms result from relatively selective degeneration of striatal neurons (Vonsattel and DiFiglia 1998). It remains unknown how the mutation in ubiquitously expressed htt leads to relatively selective striatal degeneration. Discovery of neuronal intranuclear inclusions containing mutant htt in HD patients and mouse models led to suggestions that these protein aggregates might cause neuronal death (Davies et al. 1997; DiFiglia et al. 1997). However, studies in mice and cultured neurons indicate that formation of nuclear inclusions does not correlate with neuronal death, suggesting that soluble mutant htt could be toxic to neurons (Saudou et al. 1998; Kim et al. 1999). Since many proteins containing polyglutamine tracts function as transcription factors (Alba and Guigo 2004), one possible harmful effect of soluble mutant htt is disruption of transcription (Sugars and Rubinsztein 2003).
Recent studies suggest that loss of beneficial activities of normal htt might also contribute to HD pathogenesis. Htt is important for neuronal survival in the central nervous system (Dragatsis et al. 2000). Moreover, in cortical neurons normal htt has been shown to induce BDNF gene expression while mutant htt has been shown to suppress BDNF gene expression (Zuccato et al. 2001). Since striatal neurons express TrkB but very little BDNF and BDNF in the striatum arrives by anterograde transport from cell bodies primarily in the cerebral cortex and secondarily in the substantia nigra (Altar et al. 1997; Conner et al. 1997), inhibition of BDNF gene expression in the cerebral cortex by the htt mutation should reduce striatal BDNF. Loss of normal htt further reduces striatal BDNF by decreasing BDNF axonal transport (Gauthier et al. 2004). Consistent with these observations, levels of striatal BDNF are reduced in HD patients and mice (Ferrer et al. 2000; Spires et al. 2004). Furthermore, intrastriatal injection of BDNF-expressing adenovirus or grafting of BDNF-expressing cells protects striatal neurons from loss in excitotoxic rat models of HD (Bemelmans et al. 1999; Perez-Navarro et al. 2000), and progression of disease phenotypes in R6/1 HD mice is accelerated in bdnf +/− mice (Canals et al. 2004). However, since BDNF is only one of many proteins affected in HD (Cha et al. 1999; Luthi-Carter et al. 2000; Zuccato et al. 2001) and since deficiency in BDNF-mediated signaling alone is sufficient to cause dendritic abnormalities and neuronal loss in the cerebral cortex and striatum (Xu et al. 2000; Baquet et al. 2004), it remains unclear whether the reduction in striatal BDNF seen in HD is such a crucial part of disease pathogenesis that restoration of striatal BDNF would normalize many aspects of HD pathology. Here we report that BDNF overexpression in the forebrain significantly ameliorates many disease phenotypes in R6/1 mice.
Materials and Methods
Animals
R6/1 mice were obtained from the Jackson Laboratory via recovery of frozen embryos. They were on the genetic background of B6CBA. Generation of Bdnf transgenic (BTg) mice under the control of the CaMKIIα promoter was reported previously (Huang et al. 1999b). BTg mice were maintained on the genetic background of C57BL/6. R6/1 mice were crossed to BTg mice to produce WT, BTg, R6/1, and BTg;R6/1 mice. Each mouse was marked with an ear tag. We used polymerase chain reactions to identify the two transgenes. All animal procedures were approved by the Georgetown University Animal Care and Use Committee.
Antibodies
Antibodies were purchased from Santa Cruz Biotechnology (BDNF, 1:1000), Cell Signaling Technology (DARPP-32, 1:1,000 for Western blots and 1:500 for immunohistochemistry; Akt, 1:1,000; phospho-Akt, 1:1,000), Chemicon International (EM48, 1:500), and Sigma (α-tubulin, 1:7,500). Antibodies to TrkB (1:1000) and phospho-TrkB (1:1000) were kindly provided by Dr. Louis Reichardt (University of California, San Francisco, CA) and Dr. Moses Chao (New York University, New York, NY), respectively.
In situ hybridization
In situ hybridization was performed as described previously (Xu et al. 2003). In brief, mouse brains were dissected and frozen immediately in an isopentane-dry ice bath. BDNF in situ hybridization was performed on cryostat coronal sections at 10 μm using 35S–labeled antisense cRNA probes complementary to the coding region of the mouse BDNF cDNA. After hybridization and washes, sections were exposed to Beta–Max Hyperfilm (Amersham). Images from three sections separated by 100 μm were scanned at 600 dpi for each mouse, and the optical density of in situ signals in the striatum and the cortical area dorsal to the striatum was determined using NIH Image J. After subtraction from the background, the mean optical density of a brain region was used for comparisons.
Western blotting
Dissected cortical and striatal tissues were homogenized in lysis buffer (80 mM Tris-Cl, 2% SDS, 10% glycerol, pH 6.8) and centrifuged at 12,500 rpm for 30 min. Protein concentrations of extracts (supernatants) were measured using the Dc protein assay kit (Bio-Rad Laboratories, Hercules, CA). Extracts (25 μg) in SDS loading buffer were denatured for 5 minutes at 100 °C, separated on an SDS-PAGE gel, and transferred onto a PVDF membrane. The membrane was blocked with 5% nonfat milk in TBST (10 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween-20) for one hour and then incubated with primary antibodies overnight at 4°C. After two washes with TBST, the membrane was incubated with the appropriate secondary antibody conjugated to horseradish peroxidase (Pierce, Rockford, IL) for 1 hour at room temperature. The membrane was then washed three times with TBST, and proteins were visualized using the chemiluminescence (ECL) plus system (Amersham Biosciences, Piscataway, NJ). The membrane was then stripped and blotted with an antibody to α-tubulin. To quantify Western blots, X-ray films were scanned into digital images and analyzed with the NIH image J software. Intensity of each band was normalized to the level of α-tubulin on the same lane.
Rotarod test
Motor coordination was tested using a Rotarod apparatus (UGO Basile, Italy). The observer was unaware of the genotype of each mouse during tests. Mice were trained for three consecutive days and each day had three trials, each with a 1-hr interval. A mouse was placed on a rotating rod which accelerated from 4 to 40 rpm in 5 min. The time the mouse stayed on the rotating rod was recorded. If the mouse still stayed on the rotating rod after 5 min, a score of 300 seconds was recorded. The best score on the third day was used for comparisons among genotypes.
Immunohistochemistry
Mice were anaesthetized with avertin and transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde sequentially. Their brains were removed from the skull, postfixed in 4% paraformaldehyde overnight, and soaked in 30% sucrose. Coronal brain sections (50 μm) were obtained with a sliding microtome, rinsed once with Tris buffered saline (TBS; 10 mM Tris–HCl, 150 mM sodium chloride, pH 7.5), and incubated with 10% methanol-3% hydrogen peroxide in TBS to quench endogenous peroxidase. After incubating with a blocking buffer (0.4% Triton X-100, 2.5% bovine serum albumin, and 10% horse serum in TBS) for 1 hour, the sections were incubated with a primary antibody diluted in the blocking buffer overnight at room temperature. After three washes in the blocking buffer, the sections were incubated with an appropriate biotinylated secondary antibody followed by the avidin–biotin–peroxidase complex (Vector Laboratories, Burlingame, CA) according to the instructions of the manufacturer. Sections were developed in 0.05% 3-3′-diaminobenzidine tetrahydrochloride and 0.003% hydrogen peroxide in 0.1 M Tris–HCl (pH 7.5), mounted onto slides, dehydrated, and coverslipped with DPX.
Cortical volume, striatal volume, and ventricle size
Perfused brains were cut coronally into 50 μm sections throughout the striatum. Every 12th coronal brain section was stained with cresyl violet. Stereo Investigator software and Neuroexplorer software (MicroBrightField Inc, Williston, VT, USA) were used to trace areas of interest and calculate volume, respectively. Cortical volume was estimated in the region with the largest percentage of striatal volume (bregma 1.18 mm − 0.22 mm), as described by Slow et al (Slow et al. 2003). Size of the lateral ventricle and dorsal 3rd ventricle was estimated on all sections with the striatum.
Htt aggregates
Color digital images of htt aggregates were obtained with a 100 × objective lens from the dorsal lateral striatum and converted to gray-scale images using the Image J software. The threshold of the gray-scale images were then adjusted until staining of soluble htt disappeared. The same parameters were used to take and adjust all images. We used the particle analysis function of Image J to measure the number and size of htt aggregates larger than 0.1 μm2. Four to five images from each mouse were analyzed.
Statistical analysis
All data are expressed as mean ± S.E.M. Results on protein levels, rotarod tests, weights, volumes, aggregate number, and aggregate size were compared using an unpaired Student’s t-test.
Results
BDNF overexpression in the forebrain increases striatal BDNF in R6/1 mice
We employed a Bdnf transgene (BTg) under the control of the promoter for the alpha subunit of Ca2+/calmodulin-dependent kinase II (CaMKIIα) to overexpress BDNF in the forebrain. In agreement with the previous observation (Huang et al. 1999b), in situ hybridization showed that the transgene led to an ~3-fold increase in levels of BDNF mRNA in the cerebral cortex (Fig. 1A, B). The Bdnf transgene was also expressed in the striatum where the activity of the endogenous Bdnf gene is very low (Fig. 1A).
Figure 1.
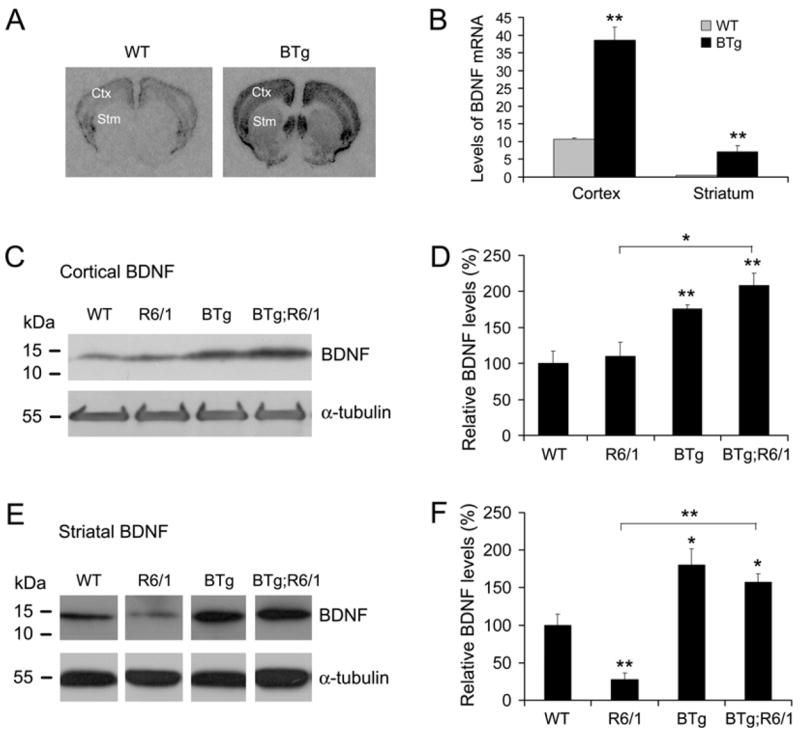
Cortical and striatal levels of BDNF protein in R6/1 and Bdnf transgenic mice. A, Representative brain sections of BDNF in situ hybridization show that levels of BDNF mRNA are increased in the cerebral cortex (Ctx) and striatum (Stm) of BTg mice. The in situ probe was 35S-labeled antisense RNA complementary to the coding region of BDNF transcripts. B, The optical density of BDNF in situ signals was measured with Image J software on three sections for each mouse. Four adult mice were used for each genotype. C, Representative Western blots show that levels of mature BDNF protein in the cerebral cortex. D, Relative levels of cortical BDNF were determined from 4 WT mice, 4 R6/1 mice, 4 BTg mice, and 3 BTg;R6/1 mice. E, Representative Western blots show that levels of mature BDNF protein in the striatum are reduced in R6/1 mice and elevated in BTg mice and BTg;R6/1 double transgenic mice. F, Relative levels of striatal BDNF were determined from 4 WT mice, 5 R6/1 mice, 4 BTg mice, and 3 BTg;R6/1 mice. Student’s t test: *, P<0.05; **, P<0.01.
In order to determine whether the Bdnf transgene was able to increase levels of striatal BDNF protein in HD mice, we crossed BTg mice to R6/1 mice to generate WT, BTg, R6/1, and BTg;R6/1 mice. R6/1 mice express exon 1 of the human huntingtin (htt) with a tract of 116 glutamine in the brain as well as in the peripheral tissues (Mangiarini et al. 1996). We prepared protein extracts from striatal and cortical tissues dissected from the four genotypes of mice at 6 months of age and examined levels of mature BDNF with Western blots. In the cerebral cortex, the BDNF level in R6/1 mice was similar to that in WT mice (Fig. 1C, D). The Bdnf transgene increased the cortical BDNF level by more than 76% in both BTg mice and BTg;R6/1 mice (Fig. 1C, D). In contrast, the level of mature BDNF in the striatum was dramatically reduced in R6/1 mice, compared to WT mice (Fig. 1E). Transgenic BDNF overexpression greatly increased levels of striatal BDNF in BTg and BTg;R6/1 mice (Fig. 1E, F). Levels of striatal BDNF protein in R6/1 mice were only 28% of that in WT mice (P<0.01), while those in BTg mice and BTg;R6/1 mice were increased by 80% (P<0.05) and 57% (P<0.05), respectively, in comparison with WT mice (Fig. 1F). Importantly, the striatal BDNF level in BTg;R6/1 mice was nearly 5 fold higher than that in R6/1 mice (Fig. 1F, P<0.001). There was not a significant difference in striatal BDNF levels between BTg mice and BTg;R6/1 mice (Fig. 1F, P=0.44), suggesting that the htt N-terminal fragment with expanded polyglutamine does not significantly interfere with expression of the Bdnf transgene and that overexpressed BDNF in the cerebral cortex is still transported to the striatum in R6/1 mice. These results demonstrate that the Bdnf transgene can be used to increase striatal BDNF supply in HD mice.
Activation of the TrkB receptor in R6/1 and BTg mice
To investigate whether TrkB activation is altered in R6/1 mice and whether BDNF overexpression in the forebrain enhances TrkB activation in the striatum, we first examined striatal levels of the TrkB receptor in the four genotypes of mice at 6 months of age. There are two isoforms of TrkB receptors: the full-length TrkB receptor tyrosine kinase and the truncated TrkB receptor that lacks the tyrosine kinase domain (Klein et al. 1990). Western blot analyses with an antibody against the TrkB extracellular domain revealed that levels of either isoform of TrkB receptors were not altered in R6/1 mice or by BDNF overexpression (Fig. 2A, B). We then examined TrkB activation with an antibody specific to the activated TrkB receptor in which the tyrosine residue at the PLCγ1 docking site is phosphorylated. Levels of the activated TrkB receptor were reduced by 49% in R6/1 mice and normalized in BTg;R6/1 mice (Fig. 2C, D). This is consistent with the observed alterations in striatal BDNF levels in these two genotypes of mice (Fig. 1E, F). To further confirm that TrkB signaling is diminished in R6/1 mice and enhanced in BTg;R6/1 mice, we investigated activation of Akt, one of the downstream targets of activated TrkB receptors in the striatum. Although the amount of total Akt protein in striatal extracts was only slightly lower in R6/1 mice than in WT mice (Fig. 2C, E), levels of phospho-Akt (Ser473) were dramatically reduced in R6/1 mice, compared to WT mice (Fig. 2C, F). Furthermore, striatal levels of phospho-Akt (Ser473) in BTg mice and BTg;R6/1 mice were nearly twice as high as those in WT mice (Fig. 2C, F), which correlates well with striatal levels of BDNF in these mice (Fig. 1F). These results indicate that R6/1 mice have diminished TrkB signaling in the striatum due to a reduced striatal BDNF level, which can be reversed by BDNF overexpression in the forebrain.
Figure 2.
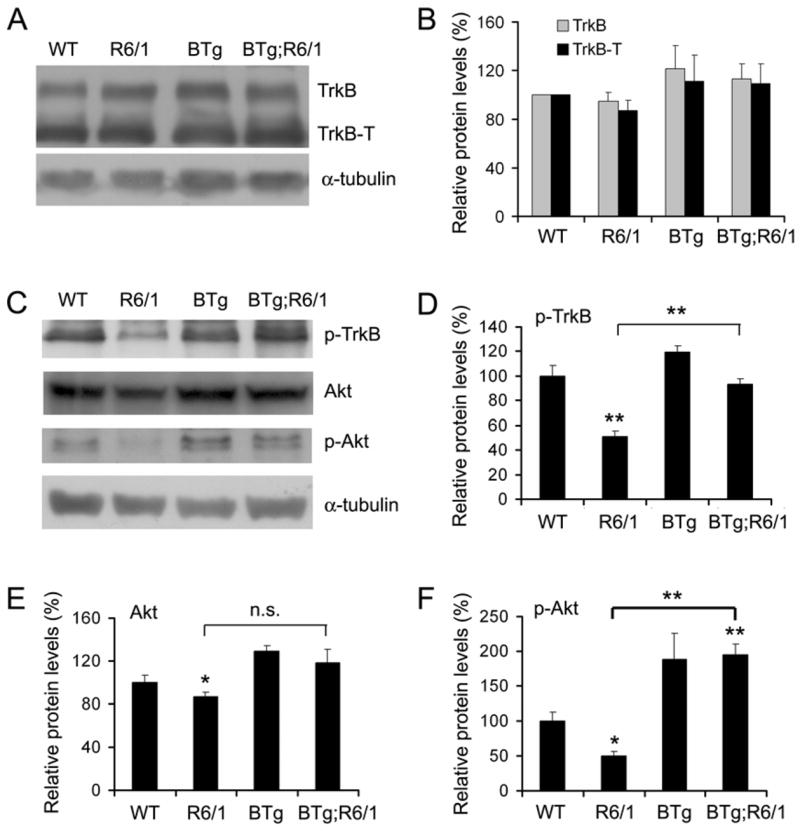
Expression and activation of the TrkB receptor in R6/1 and Bdnf transgenic mice. A, Representative Western blots show expression of the full-length TrkB receptor (TrkB) and the truncated TrkB receptor (TrkB-T) in the striatum. B, Levels of TrkB and TrkB-T are similar in striata of WT (n=4), R6/1 (n=4), BTg (n=4), and BTg;R6/1 (n=3) mice. The graph represents the average of three independent experiments. C, Representative Western blots show levels of activated TrkB (p-TrkB), Akt, and activated Akt (p-Akt) in the striatum. D–F, Striatal levels of p-TrkB, Akt, and p-Akt in WT (n=4), R6/1 (n=3), BTg (n=4), and BTg;R6/1 (n=3) mice. Student’s t test: *, P<0.05; **, P<0.01.
BDNF overexpression ameliorates disease phenotypes in R6/1 mice
R6/1 mice display several HD-related phenotypes, including poor motor coordination, brain atrophy, and loss of body weight (Mangiarini et al. 1996; Spires et al. 2004). We performed rotarod tests to monitor motor coordination when mice were at 2, 3, 4, 5, and 6 months of age. WT mice and BTg mice performed similarly well on the test and stayed on the rotating rod for the maximum amount of allowed time in most trials (Fig. 3A). R6/1 mice stayed on an accelerating rotating rod for a significantly shorter time, compared to WT mice (Fig. 3A, P<0.01 at all time points). Although double transgenic mice (BTg;R6/1) did not perform as well as WT mice (Fig. 3A, P=0.017 at 3 months of age and P<0.01 at other time points), they performed significantly better on the test than R6/1 mice at 4 months of age and afterward (Fig. 3A, P=0.019 at 4 months of age and P<0.01 at 5 and 6 months of age). These results indicate that overexpression of BDNF in the forebrain slows progression of motor dysfunction in R6/1 mice.
Figure 3.
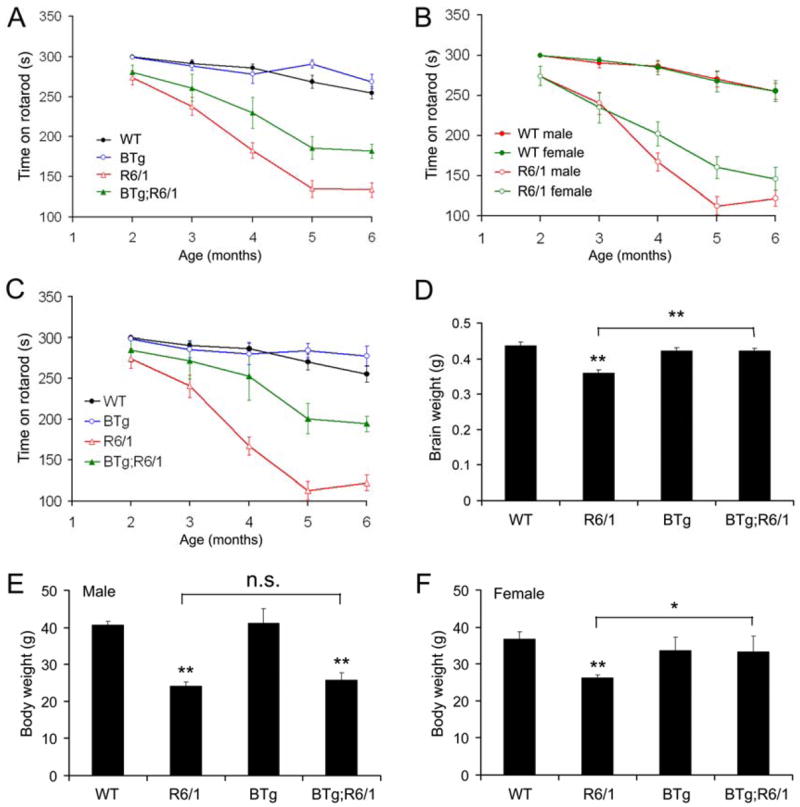
BDNF overexpression improves motor coordination and reverses loss of brain weight in R6/1 mice. A, Performance on rotarod tests. Not every mouse was tested at each time point. The mouse number at each time point was as follows: 30 (2 months), 31 (3 months), 30 (4 months), 30 (5 months), and 24 (6 months) for WT mice; 31, 32, 32, 34, and 24 for R6/1 mice; 13, 16, 15, 21, and 16 for BTg mice; 12, 12, 12, 13, and 11 for BTg;R6/1 mice. B, Differential rotarod performance in male and female R6/1 mice. The mouse number at each time point was as follows: 18 (2 months), 18 (3 months), 18 (4 months), 18 (5 months), and 12 (6 months) for male WT mice; 12, 13, 12, 12, and 12 for female WT mice; 18, 18, 17, 18, and 12 for male R6/1 mice; 13, 14, 15, 16, and 11 for female R6/1 mice. C, Performance of male mice on rotarod tests. The mouse number at each time point was as follows: 18 (2 months), 18 (3 months), 18 (4 months), 18 (5 months), and 12 (6 months) for male WT mice; 18, 18, 17, 18, and 12 for male R6/1 mice; 8, 10, 9, 11, and 7 for male BTg mice; 4, 6, 6, 8, and 7 for male BTg;R6/1 mice. D, Brain weights of WT (6 mice), R6/1 (8 mice), BTg (4 mice) and BTg;R6/1 (4 mice) at 6 months of age. E, Body weight of male WT (10 mice), R6/1 (9 mice), BTg (6 mice) and BTg;R6/1 (6 mice) at 6 months of age. F, Body weight of female WT (10 mice), R6/1 (9 mice), BTg (5 mice) and BTg;R6/1 (4 mice) at 6 months of age. Student’s t test: *, P<0.05; **, P<0.01.
Male and female R6/1 mice displayed differential progression in motor dysfunction. Both male and female R6/1 mice stayed on an accelerating rotating rod for a significantly shorter time than sex-matched WT mice (Fig. 3B, P<0.01 at 3 months of age and later time points). The motor dysfunction progressed faster in male R6/1 mice than in female R6/1 mice, so that male R6/1 mice performed significantly worse on rotarod tests than female R6/1 mice at 5 months of age (Fig. 3B, P=0.011). Despite of the accelerated disease progression, BDNF overexpression still dramatically improved motor coordination in male R6/1 mice (Fig. 3C, P<0.01 between male R6/1 mice and male BTg;R6/1 mice at ages of 4, 5, and 6 months).
Body weights were measured for some mice at 6 months of age. BTg mice had a similar body weight to WT mice for both genders (Fig. 3E, F). Although both male and female R6/1 mice had significantly lower body weights than their sex-matched WT littermates, the reduction of body weight was more severe in male R6/1 mice (Fig. 3E and F, 40% in male vs. 29% in female), which is consistent with the gender difference in motor dysfunction in R6/1 mice (Fig. 3B). BDNF overexpression had little effect on the body weight of male R6/1 mice (P=0.478), but significantly increased the body weight of female R6/1 mice (P=0.027).
We perfused the four genotypes of mice at 6 months of age, and dissected and weighed their brains. Brain weight of R6/1 mice was 18% lighter than that of WT mice (Fig. 3D, P<0.001). Brain weight of BTg;R6/1 mice was significantly higher than that of R6/1 mice and similar to that of WT and BTg mice (Fig. 3D). Thus, BDNF overexpression in the forebrain completely reverses the loss of brain weight in R6/1 mice.
To determine if the change in brain weight reflects an alteration in brain size, we sectioned fixed brains, performed Nissl staining, and measured volumes of the cerebral cortex, striatum, and ventricles. An examination of comparable coronal sections suggests that R6/1 mice have smaller brains (Fig. 4A). Using the Nissl-stained coronal sections, we employed Stereo Investigator software to measure the striatal volume, cortical volume, and size of the lateral and dorsal third ventricles. In agreement with the previous observation (Canals et al. 2004), cortical and striatal volumes were significantly smaller by 12% and 18% in R6/1 mice than in WT mice, respectively (Fig. 4B, C). Importantly, BDNF overexpression in the forebrain normalized both cortical volume and striatal volume in R6/1 mice (Fig. 4B, C). BDNF overexpression also significantly increased the striatal volume in BTg mice by 13% (Fig. 4C), which is consistent with the observation that BTg mice have significantly smaller ventricles (Fig. 4D). There was not a significant difference in ventricle size among WT, R6/1, and BTg;R6/1 mice (Fig. 4D). Taken together, these results demonstrate that BDNF overexpression under the control of the CaMKIIα promoter at least partially reverses many aspects of disease phenotypes in R6/1 mice.
Figure 4.
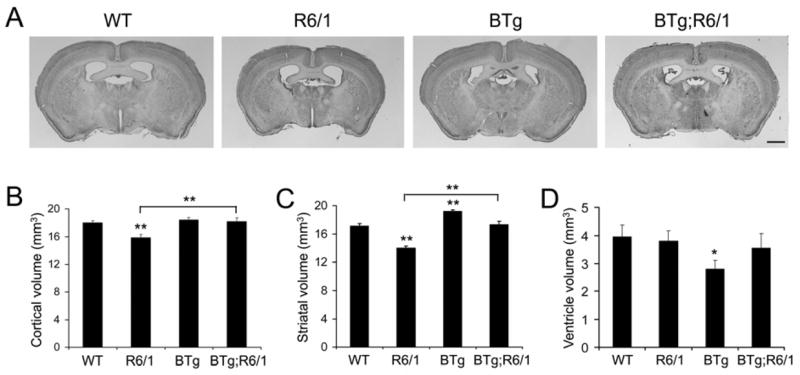
BDNF overexpression in the forebrain increases volumes of the cerebral cortex and striatum in R6/1 mice. A, Representative coronal brain sections of four genotypes of mice. Note that R6/1 mice have a smaller brain. Scale bar, 1 mm. B, Cortical volumes of WT (n=7), R6/1 (n=6), BTg (n=7), and BTg;R6/1 (n=5) mice at 6 months of age. C, Striatal volumes of WT (n=8), R6/1 (n=6), BTg (n=5), and BTg;R6/1 (n=5) mice at 6 months of age. D, Ventricle volumes of WT (n=9), R6/1 (n=10), BTg (n=7), and BTg;R6/1 (n=5) mice at 6 months of age. The volume of the lateral ventricle and dorsal 3rd ventricle was calculated on coronal sections containing the striatum. Student’s t test: *, P<0.05; **, P<0.01.
Effect of BDNF overexpression on levels of DARPP-32 and enkephalin in the R6/1 striatum
DARPP-32 is a 32 kDa dopamine- and cAMP-regulated phosphoprotein important for dopamine neurotransmission (Svenningsson et al. 2004), while enkephalin serves as a peptide neurotransmitter for striatal neurons at the origin of the indirect pathway (Kawaguchi 1997). Expression of DARPP-32 and enkephalin in the striatum is severely impaired in HD mice (Luthi-Carter et al. 2000; Spires et al. 2004) and may be regulated by BDNF (Ivkovic and Ehrlich 1999; Canals et al. 2004). To determine if BDNF overexpression slows disease progression of R6/1 mice in part by normalizing expression of DARPP-32 and enkephalin, we examined striatal levels of these two polypeptides in the four genotypes of mice at 6 months of age using Western blots and immunohistochemistry.
Immunohistochemistry showed that the striatal level of DARPP-32 was diminished in R6/1 mice, as reported (Spires et al. 2004), and that BDNF overexpression reversed the reduction (Fig. 5A–D). To quantify the change we performed Western blots with striatal protein extracts prepared from mice at 6 months of age. As shown in Fig. 5E and F, the level of DARPP-32 in R6/1 mice was only 21% of that in WT mice. Although the Bdnf transgene did not significantly alter the level of DARPP-32 on the WT genetic background, it increased the striatal level of DARPP-32 in BTg;R6/1 mice by ~5 fold (Fig. 5E, F). This result suggests that downregulation of DARPP-32 in the striatum of R6/1 mice likely results from BDNF deficiencies.
Figure 5.

BDNF overexpression normalizes levels of DARPP-32 in the striatum of R6/1 mice. A–D, Immunohistochemistry shows striatal expression of DARPP-32 in four genotypes of mice. Scale bar, 50 μm. E, Representative blots show levels of striatal DARPP-32 in four genotypes of mice. F, Levels of DARPP-32 protein were determined in the striatal extracts from WT (n=4), R6/1 (n=3), BTg (n=4), and BTg;R6/1 (n=3) mice. Student’s t test: *, P<0.05 and **, P<0.01.
As expected, enkephalin immunoreactivity in the striatum was reduced in R6/1 mice as compared to WT mice (Fig. 6A, B). Introduction of the Bdnf transgene to either WT or R6/1 mice did not appear to significantly increase striatal enkephalin immunoreactivity (Fig. 6C, D). However, BDNF overexpression in the forebrain did increase the number of enkephalinergic boutons in the R6/1 striatum. These boutons are the presynaptic terminals of collaterals from enkephalinergic medium-sized spiny neurons to innervate other striatal neurons (Kawaguchi 1997). Very few enkephalin-immunoreactive boutons were observed in the striatum of R6/1 mice and, if present, they were small, compared to those in WT mice (Fig. 6E, F). BDNF overexpression increased the number of enkephalin-immunoreactive boutons in the striatum of BTg;R6/1 mice (Fig. 6F, H), although it had a minimal effect on these boutons in the striatum of BTg mice (Fig. 6E, G). However, BDNF overexpression does not completely reverses the deficit in enkephalin-immunoreactive boutons since these boutons were still fewer and smaller in BTg;R6/1 mice than in WT mice (Fig. 6E, H). This result indicates that an elevated BDNF level in the striatum partially rescues alterations in enkephalin synthesis and/or formation of enkephalinergic synapses on striatal neurons in R6/1 mice.
Figure 6.
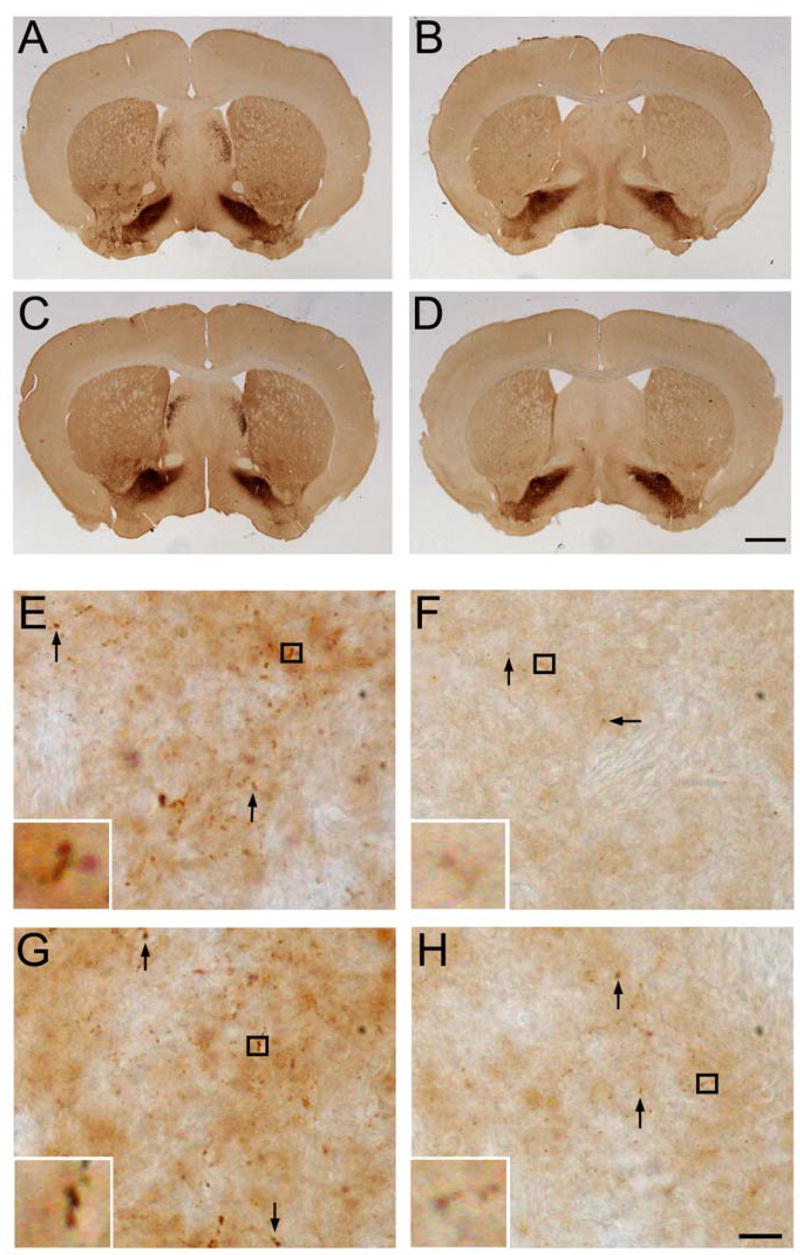
Effect of BDNF overexpression on enkephalergic medium-sized spiny neurons. A–D, Immunohistochemistry shows that striatal levels of enkephalin in R6/1 mice (B) are reduced in comparison with those in WT mice (A). BDNF overexpression does not appear to alter significantly striatal levels of enkephalin on either the wild-type (C) or the R6/1 (D) genetic background. Scale bar, 1 mm. E–H, High-magnification images show that the number and size of striatal enkephalin-containing boutons (arrows) in four genotypes of mice. Inserts are enlarged images of the boxed areas to show representative boutons. Scale bar, 10 μm.
Reduced formation of htt aggregates in double transgenic mice
To examine the effect of BDNF overexpression on formation of htt aggregates, we analyzed brain samples from R6/1 and BTg;R6/1 mice at 6 months of age with EM48 antibodies against polyglutamine tracts (DiFiglia et al. 1997). Striata of R6/1 mice showed extensive EM48-positive aggregates (Fig. 7A). Fewer and smaller htt aggregates were observed in striata of BTg;R6/1 mice (Fig. 7A). To quantify the difference between htt aggregates in the two genotypes of mice, we measured the density and size of the aggregates in the striatum. The average size of htt aggregates and the number of the aggregates in a unit of striatal area were significantly reduced by 35% and 31% respectively in BTg;R6/1 mice compared to R6/1 mice (Fig. 7B and C). A further analysis based on the cross area of an aggregate showed that the reductions resulted from the presence of fewer large htt aggregates in BTg;R6/1 mice (Fig. 7D). Thus, an elevation in striatal BDNF reduces formation of neuronal intranuclear htt aggregates in R6/1 mice.
Figure 7.
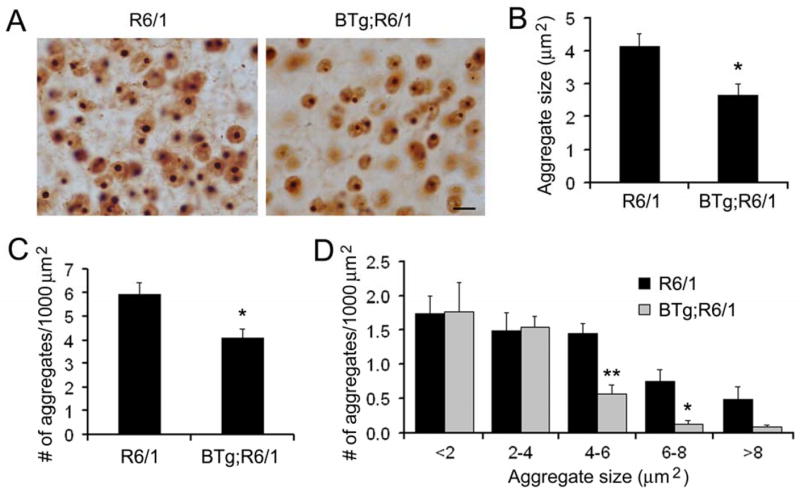
Formation of neuronal intranuclear inclusion is reduced in BTg;R6/1 mice. A, Immunohistochemistry revealed htt aggregates in striata of R6/1 and BTg;R6/1 mice. Scale bar, 10 μm. B, The average size of htt aggregates was smaller in BTg;R6/1 mice than in R6/1 mice. C, The number of htt aggregates on one area unit was reduced in BTg;R6/1 mice in comparison with R6/1 mice. D, BTg;R6/1 mice had fewer large htt aggregates than R6/1 mice. Five R6/1 mice and four BTg;R6/1 mice at 6 months of age were used for the analysis of neuronal intranuclear inclusions. Student’s t test: *, P<0.05 and **, P<0.01.
Discussion
It has been shown that intrastriatal injection BDNF-expressing adenovirus or grafting of BDNF-expressing cells protects striatal neurons from loss in excitotoxic rat models of HD (Bemelmans et al. 1999; Perez-Navarro et al. 2000). However these types of studies have several limitations in evaluating the role of BDNF deficiency in HD pathogenesis and the value of BDNF or its mimetics as HD therapeutic agents. First, the excitotoxic models do not allow for examination of motor dysfunction or alterations in gene expression related to HD. Second, it is unclear if the excitotoxic rat model is physiologically relevant to HD since R6/1 and R6/2 mice were observed to be resistant to intrastriatal injections of the NMDA-receptor agonist quinolinic acid (Hansson et al. 1999). Third, it is difficult to perform similar studies in transgenic HD mice, as these mice show no or only a small loss of striatal neurons (Rubinsztein 2002). Finally, it is not feasible to use viral injection or cell grafting to examine the effect of BDNF on a whole brain region. Here we examined roles of BDNF deficiency in HD pathogenesis through transgenic overexpression of BDNF in the R6/1 forebrain. In this system, the Bdnf transgene likely increases levels of striatal BDNF by overexpressing BDNF in the striatum and cerebral cortex. Our results provide three lines of evidence to support a crucial role of BDNF deficiency in HD pathogenesis. First, striatal BDNF levels were sharply reduced in R6/1 mice and increased by BDNF overexpression in the forebrain. Consistent with this observation, our results show that TrkB signaling in the striatum is decreased in R6/1 mice and enhanced in BTg;R6/1 mice. Second, BDNF overexpression ameliorated motor dysfunction and normalized brain weight in R6/1 mice. Third, BDNF overexpression normalized expression of DARPP-32. These observations suggest that BDNF or its mimetics should have therapeutic values to HD.
Previous studies obtained conflicting results with regard to levels of striatal BDNF in R6/1 mice. Spires et al observed significantly reduced levels of mature BDNF protein in the R6/1 striatum but not in the cerebral cortex by using Western blots (Spires et al. 2004). However, two other studies reported no changes in levels of striatal BDNF in R6/1 mice by using ELISA assays (Canals et al. 2004; Pang et al. 2006). The discrepancy likely results from the different detection methods employed in these studies. If BDNF antibodies used in ELISA assays also bind to other proteins, the nonspecific binding will mask a reduction in BDNF which is expressed at low levels. Furthermore, ELISA assays do not distinguish mature BDNF from pro-BDNF. In light of these considerations, Western blots offer some advantages over ELISA assays. We were able to identify a specific band for mature BDNF on the basis of its molecular weight. In agreement with the observation made by Spires et al, we found that levels of mature BDNF protein were dramatically reduced in the striatum but not in the cerebral cortex in R6/1 mice at 6 months of age. This observation supports the notion that both production of BDNF in the cortex and its anterograde transport from cortex to striatum are impaired in R6/1 mice.
The activity of the BDNF-to-TrkB pathway can be further diminished if levels of the TrkB receptor are also reduced in the R6/1 striatum. In fact, one recent study reported that levels of striatal TrkB are reduced in HD mice (Gines et al. 2006). We revisited this issue by using highly specific antibodies raised against the TrkB extracellular domain (Huang et al. 1999a; Paredes et al. 2004). We did not observe a change in levels of either the full-length TrkB receptor or the truncated receptor in the striatum of R6/1 mice at 6 months of age. Consistent with the results on levels of striatal BDNF and TrkB, we found that levels of activated TrkB and Akt were reduced by ~ 50% in the R6/1 striatum. Therefore, our results suggest that reduced TrkB signaling in the R6/1 striatum results from decreased BDNF availability.
It has been reported that the htt mutation reduces axonal transport of BDNF in cultured neurons (Gauthier et al. 2004). However, it is unlikely that the htt mutation will completely block BDNF axonal transport because a significant amount of striatal BDNF is still present in HD mouse models and in human HD patients (Gauthier et al. 2004; Spires et al. 2004). Therefore, overexpression of BDNF in the cerebral cortex should be able to increase striatal levels of BDNF in HD mice. Our data on levels of BDNF in the striatum and cortex support this argument. First, levels of cortical and striatal BDNF were not significantly different between BTg mice and BTg;R6/1 mice. Second, levels of striatal BDNF in BTg;R6/1 mice were significantly higher than those in WT mice, although levels of BDNF mRNA produced from the Bdnf transgene in the striatum were not as high as those in WT cortex. Therefore, both locally expressed BDNF and anterogradely transported BDNF contribute to elevated levels of striatal BDNF in BTg;R6/1 mice. However, increased cortical BDNF levels and decreased striatal BDNF levels in BTg;R6/1 mice in comparison with BTg mice, despite statistically insignificant, do suggest that mutant htt inhibits BDNF anterograde transport.
How might BDNF overexpression in the forebrain normalize brain weight in R6/1 mice? The cerebral cortex, hippocampus, and striatum are atrophic in HD patients as well as in R6/1 mice (Vonsattel et al. 1985; Mangiarini et al. 1996). The Bdnf transgene should increase BDNF levels in these brain regions (Huang et al. 1999b) and thus might protect these regions from atrophy in R6/1 mice. Indeed, introduction of the Bdnf transgene into R6/1 mice normalizes cortical and striatal volumes. BDNF has been shown to be essential for dendritic growth and/or maintenance and long-term survival of neurons in the cortex and striatum (Xu et al. 2000; Baquet et al. 2004). Since loss of striatal medium-sized spiny neurons in R6/1 mice is not significant (Mangiarini et al. 1996), BDNF overexpression likely restores size and weight of the brain by overcoming detrimental effects of the mutant htt N-terminal fragment on dendritic growth and maintenance.
BDNF overexpression in the forebrain significantly, but not fully, reverses motor dysfunction in R6/1 mice, even though it normalizes brain weight, striatal volume, and expression of some genes. This could be due to the fact that R6/1 mice develop early onset and severe motor dysfunction in association with pathology in peripheral tissues such as skeletal muscle (Mangiarini et al. 1996). It is unlikely that BDNF overexpression in the forebrain of R6/1 mice would ameliorate dysfunction of their skeletal muscle and therefore be able to completely restore rotarod performance.
BDNF overexpression significantly increases body weight in female R6/1 mice but not in male R6/1 mice. One possibility for this differential effect is that BDNF overexpression disturbs the balance of TrkB signaling in different subsets of hypothalamic neurons, which leads to more weight gain in females because female mice develop more severe obesity when TrkB signaling is deficient (Xu et al. 2003). However, this does not appear likely since BTg mice had similar body weights to WT mice. A more plausible possibility is that loss of body weight in male R6/1 mice is too severe to be overcome by BDNF overexpression in the brain.
The number and size of neuronal intranuclear inclusions are reduced in BTg;R6/1 mice, compared to R6/1 mice. This is the first demonstration that signaling cascades activated by receptor tyrosine kinases regulate aggregation of mutant htt. This is a somewhat surprising finding considering the previous observation that deletion of one copy of the Bdnf gene does not affect htt inclusions in R6/1 mice (Canals et al. 2004). One explanation for the apparent discrepancy is that TrkB signaling has already been so reduced in R6/1 mice that a further reduction in levels of striatal BDNF will not increase formation of htt aggregates anymore. A reduction in formation of htt aggregates could result from either decreased levels of the mutant htt or reduced propensity of soluble mutant htt to form aggregates. The latter mechanism should increase levels of soluble mutant htt in neurons. However, EM48 immunoreactivity of soluble mutant huntingtin appeared to be lower in BTg;R6/1 mice than in R6/1 mice (Fig. 7A). Therefore, it is likely that increased TrkB signaling somehow reduces levels of the mutant htt in neurons. It is an intriguing open question whether TrkB signaling decreases cellular content of the mutant htt by stimulating protein degradation pathways, by keeping striatal neurons healthier so that they are more efficient in clearing misfolded proteins, or by other mechanisms. Since the Bdnf transgene completely restored brain weight in BTg;R6/1 mice while many intranuclear inclusions were still formed in these mice, BDNF overexpression should provide direct beneficial effects on gene expression and dendritic morphology independent of its potential role in degradation of mutant htt.
Acknowledgments
We thank Drs. Moses Chao and Louis Reichardt for antibodies to phospho-TrkB and TrkB. This work was supported by the grant from the National Institutes of Health (NS050596).
References
- Alba MM, Guigo R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004;14:549–554. doi: 10.1101/gr.1925704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemelmans AP, Horellou P, Pradier L, Brunet I, Colin P, Mallet J. Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington’s disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther. 1999;10:2987–2997. doi: 10.1089/10430349950016393. [DOI] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Frey AS, Alsdorf SA, Kerner JA, Kosinski CM, Mangiarini L, Penney JB, Jr, Davies SW, Bates GP, Young AB. Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:981–989. doi: 10.1098/rstb.1999.0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gines S, Bosch M, Marco S, Gavalda N, Diaz-Hernandez M, Lucas JJ, Canals JM, Alberch J. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur J Neurosci. 2006;23:649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci U S A. 1999;96:8727–8732. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Wilkinson GA, Farinas I, Backus C, Zang K, Wong SL, Reichardt LF. Expression of Trk receptors in the developing mouse trigeminal ganglion: in vivo evidence for NT-3 activation of TrkA and TrkB in addition to TrkC. Development. 1999a;126:2191–2203. doi: 10.1242/dev.126.10.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999b;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Neostriatal cell subtypes and their functional roles. Neurosci Res. 1997;27:1–8. doi: 10.1016/s0168-0102(96)01134-0. [DOI] [PubMed] [Google Scholar]
- Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci. 1999;19:964–973. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Pang TY, Stam NC, Nithianantharajah J, Howard ML, Hannan AJ. Differential effects of voluntary physical exercise on behavioral and brain-derived neurotrophic factor expression deficits in Huntington’s disease transgenic mice. Neuroscience. 2006;141:569–584. doi: 10.1016/j.neuroscience.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Paredes A, Romero C, Dissen GA, DeChiara TM, Reichardt L, Cornea A, Ojeda SR, Xu B. TrkB receptors are required for follicular growth and oocyte survival in the mammalian ovary. Dev Biol. 2004;267:430–449. doi: 10.1016/j.ydbio.2003.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Navarro E, Canudas AM, Akerund P, Alberch J, Arenas E. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington’s disease. J Neurochem. 2000;75:2190–2199. doi: 10.1046/j.1471-4159.2000.0752190.x. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. Lessons from animal models of Huntington’s disease. Trends Genet. 2002;18:202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Varshney NK, Cordery PM, van Dellen A, Blakemore C, Hannan AJ. Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J Neurosci. 2004;24:2270–2276. doi: 10.1523/JNEUROSCI.1658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]