

Abstract
Acetaminophen (APAP) overdose is the leading cause of drug related liver failure in many countries. N-acetyl-p-benzoquinone imine (NAPQI) is a reactive metabolite that is formed by the metabolism of APAP. NAPQI preferentially binds to glutathione and then cellular proteins. NAPQI binding is considered an upstream event in the pathophysiology, especially when binding to mitochondrial proteins and therefore leading to mitochondrial toxicity. APAP caused a significant increase in liver toxicity 3 h post APAP administration as measured by increased serum ALT levels. Using high-resolution mitochondrial proteomics techniques to measure thiol and protein changes, no significant change in global thiol levels was observed. However, 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 (HMG-CoA synthase) had significantly decreased levels of reduced thiols and activity after APAP treatment. HMG-CoA synthase is a key regulatory enzyme in ketogenesis and possesses a number of critical cysteines in the active site. Similarly, catalase, a key enzyme in hydrogen peroxide metabolism, also showed modification in protein thiol content. These data indicate post-translational modifications of a few selected proteins involved in mitochondrial and cellular regulation of metabolism during liver toxicity after APAP overdose. The pathophysiological relevance of these limited changes in protein thiols remains to be investigated.
Keywords: Acetaminophen, hepatotoxicity, mitochondria, proteomics, thiols, oxidative modification, HMG, CoA synthase
INTRODUCTION
Acetaminophen (APAP) is the leading cause of drug-induced liver failure in the US and the UK (Lee, 2004). A fraction of the dose of APAP is metabolized by cytochrome P450 enzymes to a reactive metabolite, presumably N-acetyl-p-benzoquinone imine (NAPQI) (Nelson, 1990). NAPQI can be conjugated with glutathione (GSH) and excreted into bile. However, after GSH stores are depleted, NAPQI covalently binds to cellular proteins (Mitchell et al., 1973). A substantial number of proteins adducted by NAPQI have been identified and include proteins of several sub-cellular compartments including cytosol, microsomes, and mitochondria (Pumford et al., 1990; Cohen et al., 1997; Dietze et al., 1997; Qiu et al., 1998). However, random protein binding alone is insufficient to severely impair the function of critical proteins and directly cause cell death. Therefore, protein binding of NAPQI is considered an important upstream event, which initiates toxicity (Jaeschke et al., 2003; Jaeschke and Bajt, 2006). In fact, the covalent binding of NAPQI to mitochondrial proteins (Tirmenstein and Nelson, 1989; Qiu et al., 2001) appears to trigger mitochondrial dysfunction resulting in the inhibition of the mitochondrial respiration (Meyers et al., 1988; Ramsey et al., 1989), ATP depletion (Jaeschke, 1990; Tirmenstein and Nelson, 1990), selective mitochondrial oxidant stress (Jaeschke, 1990; Knight et al., 2001) and peroxynitrite formation (Cover et al., 2005) and release of mitochondrial intermembrane proteins (Bajt et al., 2006). These events trigger the mitochondrial membrane permeability transition pore opening (MPT) and collapse of the mitochondrial membrane potential (Kon et al., 2004), which is ultimately responsible for the oncotic necrosis in this model (Gujral et al., 2002).
A possible consequence of the selective mitochondrial oxidant stress could be protein thiol oxidation, which has been discussed as a cause of the MPT (Kim et al., 2002; Lemasters et al., 2002) and mechanism of cell death (Nicotera et al., 1992). However, there are controversial reports on loss of protein thiols after APAP overdose in vivo. Smith and Mitchell (1985) did not find a significant change in hepatic protein thiols in a rat model at 3 and 6 h after APAP. On the other hand, Tirmenstein and Nelson (1990) observed 22% less protein thiols in livers of mice treated with APAP for 1 and 6 h. Limitations of both studies include the sensitivity of the assay and the fact that total liver homogenates rather than the mitochondrial compartments were evaluated. Based on the selective oxidation of GSH and detection of nitrotyrosine adducts only within mitochondria (Jaeschke, 1990; Knight et al., 2001; Cover et al., 2005), we hypothesized that mitochondrial protein thiol groups might be most susceptible to modifications in an experimental mouse model of APAP overdose. To test this hypothesis, we assessed mitochondrial protein thiol content using a highly sensitive thiol alkylating agent biotinylated iodoacetamide (BIAM) (Kim et al., 2000, Shiva et al., 2004), which essentially labels (i.e. “tags”) reduced thiols within proteins, but not those that have been oxidized and/or modified by reactive species. Changes in protein thiol content in response to APAP exposure were identified using high-resolution proteomics approaches (Venkatraman et al., 2004a; Bailey et al., 2005).
MATERIALS AND METHODS
Animals and Experimental Protocol
Male C3HeB/FeJ mice (8–10 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME). Animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”. The experimental protocol was approved by the institutional animal care and use committee of Kansas University Medical Center. The animals were fasted overnight, received 300 mg/kg APAP (Sigma Chemical Co., St. Louis, MO) dissolved in warm saline (15 mg/ml) (i.p.) and were killed by cervical dislocation under isoflurane anesthesia 1, 3, or 6 h after APAP administration. Blood was drawn from the caval vein into a heparinized syringe for measurement of plasma alanine aminotransferase (ALT) activities with test kit 68-B (Biotron Diagnostics, Inc., Hernet, CA). The liver was removed and mitochondria were isolated using standard isolation procedures with differential centrifugation as described previously (Cover et al., 2005). Purity of the mitochondrial fractions using this isolation procedure is routinely > 95% as measured by cytochrome c content (marker for mitochondria) and lactate dehydrogenase activities (marker for contamination with cytosolic proteins) (Cover et al., 2005).
BIAM Labeling of Mitochondrial Protein Thiols
Liver mitochondrial protein (1 mg) was re-suspended in 10 mM Tris buffer (pH 8) containing 1% (w/v) Triton X-100 and protease inhibitor cocktail (P8340, Sigma, St. Louis, Mo.). BIAM (biotinylated iodoacetamide, final concentration 0.5 μM) was added to the mitochondrial protein extract and allowed to incubate at room temperature for 15 min in the dark essentially as described in (Kim et al., 2000; Landar et al., 2006). The labeling reaction was quenched with β-mercaptoethanol (final concentration 20 mM) and kept on ice.
Two Dimensional Isoelectric focusing and SDS-PAGE (2D IEF/SDS-PAGE)
The protein concentration of BIAM-labeled samples was measured using the Bradford method (Bradford, 1976) and 100 μg of protein was added to rehydration buffer containing 7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 0.5% (w/v) lauryl maltoside, 0.002% (w/v) bromophenol blue, ampholine electrophoresis reagent (Sigma, St. Louis, Mo, range pH 3–10), 0.04 M DTT and 2 mM tributylphosphine. The mitochondrial sample in rehydration buffer was applied to the IEF gel strips (Invitrogen ZOOM Strips, pH 3–10, Carlsbad, CA) and rehydration of IEF gels was done overnight. IEF was performed using an Invitrogen IEF Runner with a high voltage protocol (175 V, 20 min; ramp up to 2000 V, 45 min; 2000 V, 30 min; ramp down to 500 V, 30 min). After IEF, gel strips were removed and frozen at −80°C until second dimension SDS-PAGE. For SDS-PAGE and the high-resolution separation of mitochondrial protein, IEF gel strips were placed horizontally on a 10% resolving gel with 4% stacking gel. Agarose solution (1% w/v) was added to hold the gel strips in place and gels were run at 100 V for 1 ½ h. After electrophoresis, gels were stained with Sypro Ruby™ for total protein or transferred to nitrocellulose where BIAM labeled proteins were detected using streptavidin conjugated to horseradish peroxidase (HRP). Protein stained gels were imaged using a Bio-Rad Fluor-S Imager.
Immunoblotting for BIAM-labeled proteins
Gels for immunoblotting were transferred to nitrocellulose membranes and standard western blotting techniques were used. BIAM-labeled proteins were detected by incubating membranes in a 1:20,000 streptavidin-HRP/BSA (1%, w/v) solution. Visualization of BIAM-labeled proteins was done using Pierce ECL reagents in combination with a BIO-Rad Chemi-Doc imager. Digital images were analyzed using PD-Quest Software (Bio-Rad).
Protein identification with matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry
Protein “spots” were excised from the gels and the “protein gel plugs” were subjected to processing via standard methods in the UAB mass spectrometry shared facility (http://www.uab.edu/proteomics). Samples were de-stained by three 30 min washes with a 50%, 50 mM NH4HCO3/50% acetonitrile solution. Samples were then treated with 10 mM dithiothreitol in 50 mM NH4HCO3 for 60 min at 60°C to reduce cysteine residues, which was then followed by alkylation of free cysteines with 55 mM iodoacetamide in 50 mM NH4HCO3 for 60 min at room temperature. Sixteen-hour incubations at 37°C with trypsin (12.5 ng/μL, Promega Gold Trypsin) were used to digest proteins. The resulting peptide solution was extracted by two, 30 min washes of a 50/50 solution of 5% formic acid and acetonitrile; supernatants were then collected, and dried using a Savant SpeedVac. Dried peptide samples were resuspended in 0.1% formic acid, desalted (C18 ZipTips, Millipore), and diluted 1:10 with a saturated solution of α-cyano-4-hydroxycinnamic acid matrix before application to MALDI-TOF target plates. After plating, samples were dried before analysis with a Voyager De-Pro mass spectrometer in the positive mode. Spectra were analyzed using Voyager Explorer software and peptide masses identified by mass spectrometry were submitted to the MASCOT database (see www.matrixscience.com) for protein identification.
2D Gel and Immunoblot Image Analysis
Gels were scanned using Bio-Rad Fluor-S imager and analyzed for differences in protein density using PD-Quest Image Analysis software (Bio-Rad). For image analysis of 2D gels, individual protein spots on each gel were identified by the software program and manually verified to generate a match-set of gels for control (untreated) and 1, 3, and 6 h APAP treatment gels. The protein spot density was compared across all gels and a reference gel was selected, which served as the master gel image. This reference gel was a control gel (i.e. untreated sample) and contained the highest abundance of detected protein spots. Using a built-in algorithm, automatic matching of protein spots in each gel to the corresponding protein spots in the master gel was performed and then manually verified to correct for any proteins that may have been incorrectly matched to proteins in the reference gel. To correct for any inter-gel protein loading differences, the density data for all protein spots in each gel was normalized to the total density in valid protein spots for that particular gel. Normalized protein spot densities were transferred into an Excel spreadsheet where the mean densities were calculated for statistical analyses. BIAM-labeled mitochondrial proteins were visualized using a Bio-Rad Chemi-Doc imager and analyzed for differences in protein thiol intensity using the same approaches described for gel analysis. Protein thiol blot intensities were normalized to total protein by dividing protein thiol blots density by protein gel densities. Similarly, the thiol intensity of individual proteins was normalized to density of its corresponding protein. Statistical analysis on both protein and protein thiol blot densities were performed essentially as described in (Venkatraman et al., 2004 a, b).
Measurement of HMG-CoA synthase activity
HMG-CoA synthase activity was determined in mitochondria isolated from controls and animals treated with 300 mg/kg APAP for 6 h according to a method described by Clinkenbeard et al. (1974) and Valera et al. (1994). After isolation, the mitochondria were resuspended in 20 mM KH2PO4 (pH 7.0) containing 0.1 mM EDTA and 0.1 mM dithiothreitol. Mitochondria were sonicated (Sonic Dismembrator, Fisher Scientific) and enzyme activity was determined using the following assay mixture, which contained in 1.0 ml volume: 100 μmol of Tris-HCl, pH 8.2; 0.1 μmol of EDTA; 0.2 μmol of acetyl-CoA; 50 nmol of acetoacetyl-CoA; and HMG-CoA synthase from mitochondria. The assay mixture minus acetyl-CoA was preincubated for 2 min at 30°C. The reaction was started by adding acetyl-CoA and the rate of HMG-CoA synthesis was monitored by following the acetyl-CoA-dependent consumption of acetoacetyl-CoA at 300 nm. The enzyme activity was calculated from the slope of substrate consumption and expressed as mU/mg mitochondrial protein.
Statistical Analysis
To determine differences between 3–5 animals per group, one-way ANOVA with Bonferroni post hoc test was performed. Statistical analysis was performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego CA).
RESULTS
Administration of 300 mg/kg APAP to C3Heb/FeJ mice resulted in an increase of plasma ALT activities as an indicator of cell injury at 3 h and a further increase at 6 h (Figure 1). These data are identical to previous observations in the same strain of mice using the same dose of APAP (Lawson et al., 1999; Knight et al., 2001; Gujral et al., 2002; Cover et al., 2005). The ALT values correlate well with the histological assessment of the area of necrosis (Knight et al., 2001; Gujral et al., 2002) (Table 1). Furthermore, it was previously shown that within 1 h after APAP, mitochondrial GSH levels were depleted and there was first evidence of nitrotyrosine protein adducts in mitochondria (Table 1). However, substantial mitochondrial oxidant stress and peroxynitrite formation was evident at 3 and 6 h, which correlated well with cell injury and loss of mitochondrial DNA (Table 1).
Figure 1. Increased serum ALT levels in response to Acetaminophen (APAP) exposure.
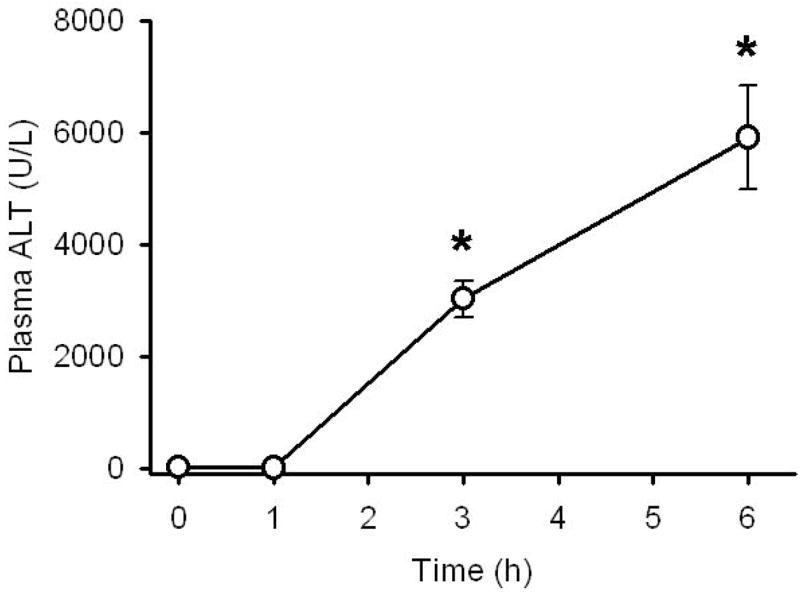
APAP treatment (300 mg/kg) causes liver injury as indicated by significant increases in plasma ALT levels at 3 and 6 h after treatment. Data represent mean ± SE of n = 5 animals per time point. *p < 0.05 (compared to time zero)
Table 1.
Summary of key findings on hepatic injury and mitochondrial oxidant stress/peroxynitrite formation in C3Heb/FeJ mice
| Control | 1 h APAP | 3 h APAP | 6 h APAP | REFERENCES | |
|---|---|---|---|---|---|
| Necrosis | −− | −− | ++ | +++ | Knight et al., 2001, 2002; Gujral et al., 2002; Bajt et al., 2003 |
| mGSH | +++ | −− | + | ++ | Knight et al., 2001, 2002 |
| mGSSG | (+) | −− | + | ++ | Knight et al., 2001, 2002 |
| mNT | −− | + | ++ | ++ | Cover et al., 2005 |
| MDNA | +++ | +++ | ++ | + | Cover et al., 2005 |
Mitochondrial oxidant stress and peroxynitrite formation in C3Heb/FeJ mice after 300 mg/kg acetaminophen (APAP) as published in previous papers. Symbols: −−, not detectable; (+) +, minor increases; ++ and +++ progressive higher levels/content of analyte in tissue. Abbreviations: mGSH, mitochondrial glutathione; mGSSG, mitochondrial glutathione disulfide; mNT, mitochondrial nitrotyrosine protein adducts; mDNA, intact mitochondrial DNA.
Based on these earlier findings, we isolated mitochondria from APAP-treated livers and evaluated changes in protein thiol groups of mitochondrial proteins. Using 2D IEF/SDS-PAGE in combination with BIAM thiol labeling, we generated high-resolution protein gels of liver mitochondria, which were immunoblotted onto nitrocellulose membranes and analyzed by western blotting techniques to identify APAP-dependent changes in mitochondrial protein thiol status. The BIAM approach was developed by Rhee and colleagues as a method to identify protein cysteinyl groups susceptible to oxidative modification by hydrogen peroxide and other oxidizing species (Kim et al., 2000). BIAM reacts with and covalently labels reduced, unmodified cysteines but not cysteines that have been oxidized or modified by reactive oxygen, nitrogen, or lipid species. Similarly, reaction of NAPQI with reduced cysteine residues to form protein adducts would also block BIAM labeling of thiol groups within proteins. BIAM-labeled proteins can then be visualized and quantified after gel electrophoresis and immunoblotting using streptavidin-HRP (Kim et al., 2000). Thus, protein thiols that have been oxidized or modified are identified by decreased labeling with BIAM using proteomics approaches. Figure 2A shows representative protein thiol immunoblots from liver mitochondria of untreated mice and mice exposed to APAP for 1, 3, and 6 h. Treatment with APAP resulted in no significant changes in total (i.e. global) protein thiol intensity as measured by BIAM labeling (Figure 2A and B).
Figure 2. Effect of APAP treatment on mitochondrial protein thiol content.
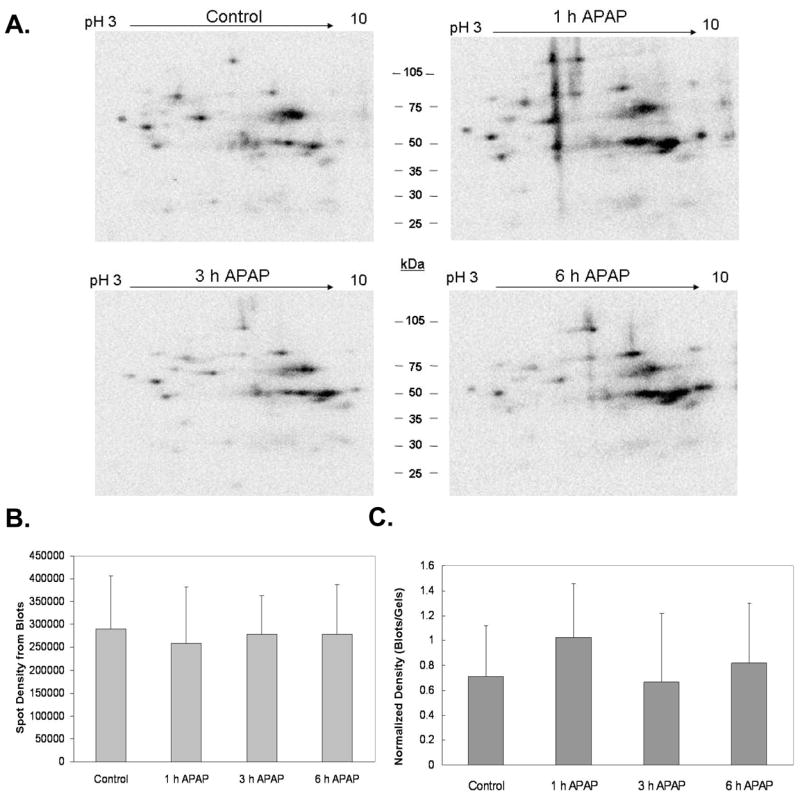
Mitochondria isolated from liver of mice treated with 300 mg/kg APAP were incubated with BIAM, a compound that labels reduced thiols. Labeled proteins were separated by 2D IEF/SDS-PAGE and gels were subjected to immunoblotting to visualize BIAM-labeled proteins. Immunoblot analysis showed no significant difference in thiol labeling intensity between control and APAP treated groups over the time period of the experiment. Panel A shows representative BIAM blots for each treatment group, panel B shows the density of the BIAM blots, and panel C shows the normalized density of total BIAM labeled protein from blots as compared to the total protein density of matched gels shown in Figure 3. Data represent mean ± SD of n = 3–5 animals per time point.
To further analyze changes in proteins and protein thiol groups, protein gels were analyzed to determine whether APAP treatment had effects on total mitochondria protein content. Figure 3A shows representative total protein gels for each treatment group. Densitometry revealed that there were no significant changes in total (i.e. global) mitochondrial protein content due to APAP exposure (Figure 3B). When the protein thiol densitometry data for each time point was normalized to its corresponding total protein gel density, there was no significant difference in total mitochondrial protein thiol content in response to APAP treatment (Figure 2C). However, it is still possible that changes in the thiol content of specific mitochondrial proteins occurred in response to APAP-induced toxicity and oxidative stress.
Figure 3. Effect of APAP treatment on mitochondrial proteins.
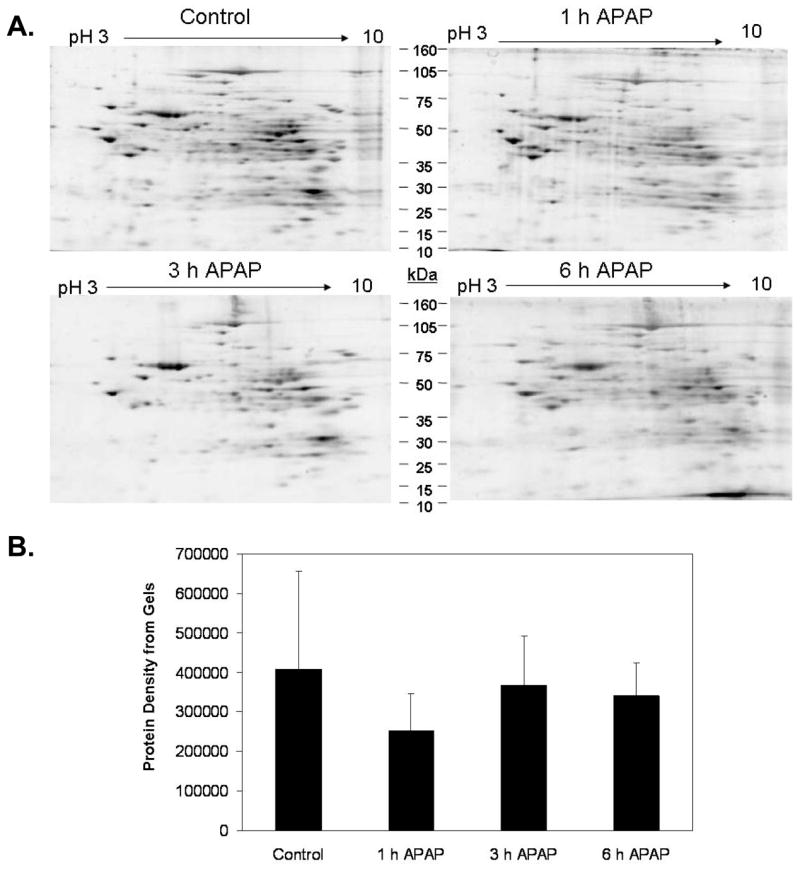
Mitochondria isolated from liver of mice treated with 300 mg/kg APAP were subjected to 2D IEF/SDS-PAGE proteomics protocol. Gels were stained with Sypro Ruby™ protein stain. Panel A shows representative gels for each treatment. Panel B, Image analysis revealed that mitochondrial protein content in APAP treated groups was no different when compared to the untreated control. Data represent mean ± SD of n = 3–5 animals per time point.
To further elucidate APAP-mediated modifications to the mitochondrial protein thiol proteome, we performed a more extensive analysis of protein thiol blots and protein gels. For this, we matched 18 protein spots immuno-reactive for the BIAM thiol label that were in common to all treatment groups and their corresponding protein on the gel (Figure 4A and B). When the density for each BIAM-labeled protein from blots (Figure 4A) was divided by the protein gel density of the same matched spot (Figure 4B), we observed alterations in the thiol status of protein spots 12 and 13 (Figure 4C). Specifically, the thiol content of protein spot 12 was significantly increased at 1 h and decreased at 6 h APAP treatment compared to control. The thiol content of protein spot 13 was also decreased at 3 h APAP treatment when compared to 1 h APAP. These 18 BIAM-labeled proteins were identified using MALDI-TOF mass spectrometry and proteins with highest confidence against the Matrix Science database are reported in Table 2. Of the 18 individual protein spots we were able to identify 15, with 12 of the spots being unique proteins. Spot 12 was identified as 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (HMG-CoA synthase), a liver specific enzyme involved in ketogenesis, and spot 13 was identified as catalase, a peroxisomal enzyme that is a common mitochondrial contaminant. As highlighted in Figure 5, there was clearly a significant loss in the thiol content of HMG-CoA synthase as a consequence of APAP treatment (Panels B and D). While there was no change in the amount of this specific protein in response to APAP treatment (Panels A and C), there was indeed a decrease in thiol content even when thiol blot intensity of this protein was normalized to the corresponding protein in the gel (Panel E). Moreover, these changes in thiol content were associated with a 40% decrease in HMG-CoA synthase activity in mitochondrial extracts from APAP-treated animals compared to controls (Panel F).
Figure 4. Master maps of BIAM-labeled proteins and matched proteins from 2D gels.

The spots circled on these master images represent BIAM-labeled protein spots (A) that were matched across all treatment groups. The corresponding proteins for these BIAM-labeled proteins on gels are shown in Panel B. Densitometry of individual BIAM-labeled protein thiols from blots and protein gels were calculated and normalized values of BIAM-labeled proteins is shown in Panel C (blot spots/gel spots). Two proteins showed significant differences in thiol labeling as a consequence of APAP treatment. Data represent mean ± SD of n = 3–5 animals per time point. *p < 0.05, compared to control; #p < 0.05, compared to 1 h APAP treatment.
Table 2.
Proteins Containing BIAM-Reactive Thiol Groups
| Spot Number | Protein identification | MOWSE | MW (kDa) |
|---|---|---|---|
| 1 | heat shock protein 70 kDa | 246 | 72.3 |
| 2 | ND | ||
| 3 | Succinyl- CoA Ligase Beta Chain and Ubiquinol- cytochrome-c reductase complex core protein I | 205 | 46.8 & 52.7 |
| 4 | Hspd1 Protein | 186 | 59.4 |
| 5 | Albumin | 96 | 69.3 |
| 6 | Enoyl Coenzyme A hydratase 1, peroxisomal | 75 | 36.1 |
| 7 | sterol carrier protein 2, liver | 88 | 59.0 |
| 8 | Sdha protein | 109 | 72.2 |
| 9 | ND | ||
| 10 | 3-hydroxy-3-rnethylglutaryl-coenzyme A synthase 2 | 80 | 56.8 |
| 11 | Catalase | 135 | 59.7 |
| 12 | 3-hydroxy-3-rnethylglutaryl-coenzyme A synthase 2 | 105 | 56.8 |
| 13 | Catalase | 85 | 59.7 |
| 14 | Catalase | 180 | 59.7 |
| 15 | Hydroxyacid oxidase 1 | 125 | 40.9 |
| 16 | ND | ||
| 17 | carnitine palmitoyltransferase 2 | 106 | 73.8 |
| 18 | acetyl-coenzyme A acyltransferase 2 (mitochondrial 3-oxoacyl-coenzyme A thiolase) | 144 | 41.8 |
Proteins listed above were matched on all immunoblots and gels from the control, 1, 3, and 6 h exposure to APAP. Thiol content in protein spot 12, HMG-CoA synthase, was shown to be significantly altered by APAP treatment (Figure 5). Spot numbers are the same as those used to identify BIAM labeled proteins spots in Figure 4A and B. ND = no designation, MOWSE is an algorithmic calculation used to assign a statistical weight to each peptide match; therefore the higher MOWSE score implies higher statistical weight of a match.
Figure 5. Decreased thiol content and activity in HMG-CoA synthase in response to APAP treatment.

Mitochondrial samples isolated from the liver of mice treated with 300 mg/kg APAP were analyzed as described in Figures 2 and 3. APAP treatment resulted in significant changes in the thiol content of HMG-CoA synthase. Panel A shows representative HMG-CoA synthase protein from 2D gels with corresponding BIAM-labeling shown in Panel B for each treatment. Panels C and D show the densitometry of HMG-CoA synthase protein and thiol content, respectively. Panel E shows the normalized blot density over protein density. There was a significant increase in thiol content at 1 h with significant decreases in thiol content of HMG-CoA synthase at both 3 and 6 h time points. Panel F shows the decrease in HMG-CoA synthase activity at 6 h time point. HMG-CoA synthase activity was determined as described in the methods sections. Data (panel A–E) represent mean ± SD of n = 3–5 animals per time point. Data (panel F) represent mean ± SE of n = 6 animals per group. *p < 0.05, compared to control; #p < 0.05, compared to 1 h APAP treatment.
DISCUSSION
The objective of the present study was to evaluate potential changes in mitochondrial protein thiol content after APAP overdose. Previous studies have shown that high doses of APAP decrease GSH levels and induce formation of reactive oxygen species and peroxynitrite in mitochondria (Knight et al. 2001; Cover et al., 2005). Because GSH is a source of reducing equivalents in the organelle, significant changes in the levels of GSH and GSSG and in reactive oxygen species formation may have significant consequences for protein thiol status and mitochondrial function (Garcia-Ruiz et al., 1995). Increased GSH oxidation and an increased GSSG/GSH ratio are also seen in other liver disease states such as alcoholic liver disease (Sastre et al., 2007). Enhanced glutathione oxidation can lead to increases in glutathionylation of mitochondrial proteins at cysteine residues (Hurd et al., 2005). For example, glutathionylation is associated with alterations in the functionality of NADH dehydrogenase (i.e. Complex I) of the respiratory chain (Beer et al., 2004). Important to this study is the previous observation that a glutathione-NAPQI adduct can oxidize thiol groups within proteins (Chen et al., 1999). Moreover, it should be pointed out that other post-translational modifications that may occur in response to APAP-mediated increases in reactive species include S-nitrosation reactions and oxidation of thiols to higher oxidation states such as SOH, SO2H, and SO3H (Cooper et al., 2002). Venkatraman and colleagues have also shown increased protein thiol modifications in mitochondria related to chronic alcohol induced oxidative/nitrosative stress (Venkatraman et al., 2004b). Based on this, we predicted that post-translational modifications of mitochondrial protein thiols may occur in an experimental mouse model of APAP hepatotoxicity.
To determine whether APAP treatment altered mitochondrial protein thiol content, experiments were performed using 2D proteomics in combination with the thiol-specific label, biotinylated iodoacetamide (BIAM). As mentioned previously, protein thiols that have been oxidized or modified are identified by decreased labeling with the thiol label BIAM (Kim et al., 2000). It should be pointed out that while the BIAM technique can be used to detect proteins with modified thiol groups, it does not provide information regarding the type of thiol modification. Using this approach, we observed that APAP treatment did not induce a significant loss in global thiol content in mitochondrial proteins (Figure 2). However, this finding does not mean that individual proteins may not have experienced alterations in cysteine residues as a consequence of APAP treatment.
To determine whether individual proteins contained modified thiols in response to APAP exposure, protein gels and BIAM blots were overlaid and aligned so that each BIAM labeled protein could be matched to the corresponding protein on its duplicate stained protein gel. This analysis allowed us to demonstrate that two proteins had statistically significant decreases in thiol content as a consequence of APAP treatment. One protein identified as containing decreased thiol content 6 h post APAP treatment was HMG-CoA synthase, which is the first and rate-limiting step in ketogenesis. HMG-CoA synthase catalyzes the conversion of acetoacetyl-CoA and acetyl-CoA to HMG-CoA and CoA (Zschocke et al., 2002; Hegardt, 1999). Interestingly, HMG-CoA synthase contains one active site cysteine residue (Miziorko and Behnke, 1985) and mutation of this active site cysteine has been shown to eliminate HMG-CoA synthase activity (Rokosz et al., 1994). Because HMG-CoA synthase protein level was unchanged in the current study following APAP exposure, it is proposed that the APAP-dependent decrease in HMG-CoA synthase activity (Figure 5F) is due to oxidative modification of the cysteine residue in the active site of the enzyme. We also observed that this protein showed an increase in thiol content when normalized for total HMG-CoA synthase protein at the 1 h time point (Figure 5). We propose that the increase in thiol content of the protein may be the result of an oxidant-mediated conformational change in the protein that renders the additional eight cysteine groups within the protein more reactive by either increasing their accessibility to the BIAM label or decreasing their pKa values by moving cysteines closer to charged amino acids. Indeed, several studies have shown that oxidant exposure can induce conformational changes within proteins exposing residues that are normally buried within the protein and that these structural changes can alter protein functionality (Georgiou 2002; Barford 2004). Moreover, it is highly probable that this population of highly damaged proteins in the early phase of APAP toxicity may have been cleared by the proteasome (Dunlop et al., 2002) such that by 3 and 6 h only those mildly modified forms of the protein containing only modification to the active cysteine are present. This finding is supported by studies demonstrating that the yeast homolog of HMG-CoA synthase is sensitive to oxidative modification upon exposure to hydrogen peroxide (Weber et al., 2004). In this study, oxidative modification of the HMG-CoA synthase homolog was determined via an acidic shift in the isoelectric point of the enzyme upon exposure to hydrogen peroxide (Weber et al., 2004). Similarly, we also detected an acidic shift of a small amount of the HMG-CoA synthase protein in gels (Figure 4) suggesting oxidative changes to the protein in response to APAP exposure. However, this change can not be correlated to thiol modifications as this more acidic variant of the protein showed no change in thiol content.
Taken together, these results showing oxidative modification and inactivation of HMG-CoA synthase demonstrate that APAP overdose may negatively affect an important energy conservation pathway in liver. Previously, it has been shown that aging-related defects in ketogenesis are directly related to decreased HMG-CoA synthase activity, as thiolase and lyase activities remain unaffected (Hegardt, 1999). Moreover, chronic alcohol consumption increases the formation of a 4-hydroxynonenal adduct with HMG-CoA synthase in rat liver (Patel et al., 2007). These results further support the hypothesis that HMG-CoA synthase may be a specific target of oxidative modification in diseases and/or conditions caused by oxidative/nitrosative stress. Future studies are necessary to identify the type of cysteine modification responsible for the loss of thiols and inactivation of HMG-CoA synthase following APAP exposure.
We also observed decreased thiol content in catalase in response to APAP treatment. In liver, catalase is located in the peroxisomes and functions to detoxify hydrogen peroxide (Rhee et al., 2005). It should be mentioned that catalase activity is detected in mitochondrial preparations as peroxisomes typically contaminate the mitochondrial fraction during the isolation procedure. However, emerging evidence indicates that catalase may be constitutively expressed in rodent liver mitochondria (Salvi et al, 2007). Catalase is a key antioxidant enzyme in the liver and could be of importance in detoxifying hydrogen peroxide generated during APAP toxicity. Although no cysteines are located in the active site of catalase, there is a highly conserved Cys 438 on the surface of the protein near the H2O2 entry channel (Sevinc et al., 1995), thus a modification of this cysteine might inhibit access to the heme center of the catalase enzyme. In E. coli, it was shown that the modification of Cys 438 to serine caused a 30% reduction in the specific activity (Sevinc et al., 1995). Consistent with our observation are the findings that APAP overdose caused a 30% reduction in catalase enzyme activity under similar conditions in mice (Lores Arnaiz et al., 1995). Whether a small reduction in catalase activity has any pathophysiological relevance remains unclear. The fact that massive catalase induction did not protect against APAP hepatotoxicity would argue against a critical role of this peroxisomal enzyme in APAP-induced liver injury (Chen et al., 2002).
In conclusion, we have shown that during the development of APAP-induced liver injury there are modest changes to specific mitochondrial proteins and their thiol content. Using high resolution proteomics techniques, we were able to determine that while there were no global changes to the mitochondrial thiol proteome in response to APAP, there was a significant change in the thiol content of HMG-CoA synthase, the key regulatory protein of ketogenesis, which was associated by a significant decrease in enzyme activity. There are also changes in thiol content of catalase, a regulator of hydrogen peroxide detoxification in peroxisomes. These data support the idea that early in the process of APAP toxicity there are changes in the thiol status of several proteins, which may directly or indirectly contribute to modifications in mitochondrial function. However, the overall changes in mitochondrial protein thiols appear relatively minor compared to the substantial oxidative/nitrosative stress in this cell organelle. Therefore, other protein modifications, e.g. nitration of tyrosine residues, may be more important for APAP hepatotoxicity.
Acknowledgments
This investigation was supported in part by National Institutes of Health Grants R01 AA12916 and DK070195 (HJ), P20 RR016475 and P20 RR 021940 (HJ), and R01 AA15172 and R21 DK073775 (SMB). The authors thank Margitta Lebosfky, University of Kansas, for expert technical assistance and Dr. Sudheer K. Mantena, University of Alabama at Birmingham, for thoughtful discussions and comments regarding this manuscript.
Footnotes
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey SM, Landar A, Darley-Usmar V. Mitochondrial proteomics in free radical research. Free Radic Biol Med. 2005;38:175–188. doi: 10.1016/j.freeradbiomed.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exper Therap. 2003;307:67–73. doi: 10.1124/jpet.103.052506. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94:217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol. 2004;14:679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1–2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chen C, Hennig GE, Whiteley HE, Manautou JE. Protection against acetaminophen hepatotoxicity by clofibrate pretreatment: role of catalase induction. J Biochem Mol Toxicol. 2002;16:227–234. doi: 10.1002/jbt.10043. [DOI] [PubMed] [Google Scholar]
- Chen W, Shocker JP, Tonge R, Hunter A, Gartner C, Nelson SD. Protein and nonprotein cysteinyl thiol modifications by N-acetyl-p-benzoquinone imine via a novel ipso adduct. Biochemistry. 1999;38:8159–8166. doi: 10.1021/bi990125k. [DOI] [PubMed] [Google Scholar]
- Clinkenbeard KD, Reed WD, Mooney RA, Lane MD. Intracellular localization of the 3-hydroxy-3-methylglutaryl coenzyme A cycle enzymes in liver. J Biol Chem. 1974;250:3108–3116. [PubMed] [Google Scholar]
- Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- Cooper CE, Patel RP, Brookes PS, Darley-Usmar VM. Nanotransducers in cellular redox signaling: modifications of thiols by reactive oxygen and nitrogen species. Trends Biochem Sci. 2002;27:489–492. doi: 10.1016/s0968-0004(02)02191-6. [DOI] [PubMed] [Google Scholar]
- Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exper Therap. 2005;315:879–87. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- Dietze EC, Schafer A, Omichinski JG, Nelson SD. Inactivation of glyceraldehydes-3-phosphate dehydrogenase by a reactive metabolite of acetaminophen and mass spectral characterization of an arylated active site peptide. Chem Res Toxicol. 1997;10:1097–1103. doi: 10.1021/tx970090u. [DOI] [PubMed] [Google Scholar]
- Dunlop RA, Rodgers KJ, Dean RT. Recent developments in the intracellular degradation of oxidized proteins. Free Radic Biol Med. 2002;33:894–906. doi: 10.1016/s0891-5849(02)00958-9. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Morales A, Colell A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC. Feeding S-adenosyl-L-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology. 1995;21:207–213. doi: 10.1002/hep.1840210133. [DOI] [PubMed] [Google Scholar]
- Geogiou G. How to flip the (redox) switch. Cell. 2002;111:607–610. doi: 10.1016/s0092-8674(02)01165-0. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis. Toxicol Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J. 1999;338:569–582. [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- Kim J, Yoon HW, Kwon K, Lee S, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- Kim TS, Jeong DW, Yun BY, Kim IY. Dysfunction of rat liver mitochondria by selenite: induction of mitochondrial permeability transition through thiol-oxidation. Biochem Biophys Res Commun. 2002;294:1130–1137. doi: 10.1016/S0006-291X(02)00612-5. [DOI] [PubMed] [Google Scholar]
- Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen-induced liver injury: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrotic and apoptotic cell death to cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- Landar A, Oh JY, Giles NM, Isom A, Kirk M, Barnes S, Darley-Usmar VM. A sensitive method for the quantitative measurement of protein thiol modification in response to oxidative stress. Free Rad Biol Med. 2006;40:459–468. doi: 10.1016/j.freeradbiomed.2005.08.046. [DOI] [PubMed] [Google Scholar]
- Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol Appl Pharmacol. 1999;156:179–186. doi: 10.1006/taap.1999.8635. [DOI] [PubMed] [Google Scholar]
- Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- Lores Arnaiz S, Llesuy S, Cutrin JC, Boveris A. Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med. 1995;19:303–310. doi: 10.1016/0891-5849(95)00023-q. [DOI] [PubMed] [Google Scholar]
- Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;93:378–387. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187:211–217. [PubMed] [Google Scholar]
- Miziorko HM, Behnke CE. Active-site-directed inhibition of 3-hydroxy-3-metylglutaryl coenzyme A synthase by 3-chloropropionyl coenzyme A. Biochem. 1985;24:3174–3179. doi: 10.1021/bi00334a015. [DOI] [PubMed] [Google Scholar]
- Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Dypbukt JM, Rossi AD, Manzo L, Orrenius S. Thiol modification and cell signalling in chemical toxicity. Toxicol Lett. 1992;64–65:563–567. doi: 10.1016/0378-4274(92)90232-9. [DOI] [PubMed] [Google Scholar]
- Patel VB, Spencer CH, Young TA, Lively MO, Cunningham CC. Effects of 4-hydroxynonenal on mitochondrial 3-hydroxy-3-methylglutaryl (HMG-CoA) synthase. Free Radic Biol Med. 2007;43:1499–1507. doi: 10.1016/j.freeradbiomed.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumford NR, Roberts DW, Benson RW, Hinson JA. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol. 1990;40:573–579. doi: 10.1016/0006-2952(90)90558-3. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Benet LZ, Burlingame AL. Identification of hepatic protein targets of the reactive metabolites of the non-hepatotoxic regioisomer of acetaminophen, 3′-hydroxyacetanilide, in the mouse in vivo using two-dimensional gel electrophoresis and mass spectrometry. Adv Exp Med Biol. 2001;500:663–673. doi: 10.1007/978-1-4615-0667-6_99. [DOI] [PubMed] [Google Scholar]
- Ramsay RR, Rashed MS, Nelson SD. In vitro effects of acetaminophen metabolites and analogs on the respiration of mouse liver mitochondria. Arch Biochem Biophys. 1989;273:449–457. doi: 10.1016/0003-9861(89)90504-3. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Yang K-S, Kang SW, Woo HA, Chang T-S. Controlled elimination of intracellular H2O2: Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- Rokosz LL, Boulton DA, Butkiewicz EA, Sanyal G, Cueto MA, Lachance PA, Hermes JD. Human cytoplasmic 3-hydroxy-3-methylglutaryl coenzyme A synthase; expression, purification, and characterization of recombinant wild-type and Cys129 mutant enzymes. Arch Biochem Biophys. 1994;312:1–13. doi: 10.1006/abbi.1994.1273. [DOI] [PubMed] [Google Scholar]
- Salvi M, Battaglia V, Brunati AM, La Rocca N, Tibaldi E, Pietrangeli P, Marcocci L, Mondovi B, Rossi CA, Toninello A. Catalase takes part in rat liver mitochondria oxidative stress defense. J Biol Chem. 2007;282:24407–24415. doi: 10.1074/jbc.M701589200. [DOI] [PubMed] [Google Scholar]
- Sastre J, Serviddio G, Pereda J, Minana JB, Arduini A, Vendemiale G, Poli G, Pallardo FV, Vina J. Mitochondrial function in liver disease. Front Biosci. 2007;12:1200–1209. doi: 10.2741/2138. [DOI] [PubMed] [Google Scholar]
- Sevinc MS, Ens W, Loewen PC. The cysteines of catalase HPII of Escherichia coli, including Cys438 which is blocked, do not have a catalytic role. Eur J Biochem. 1995;230:127–132. [PubMed] [Google Scholar]
- Shiva S, Crawford JH, Ramachandran A, Ceaser EK, Hillson T, Brookes PS, Patel RP, Darley-Usmar VM. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem J. 2004;379:359–366. doi: 10.1042/BJ20031758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CV, Mitchell JR. Acetaminophen hepatotoxicity in vivo is not accompanied by oxidant stress. Biochem Biophys Res Commun. 1985;133:329–336. doi: 10.1016/0006-291x(85)91879-0. [DOI] [PubMed] [Google Scholar]
- Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- Tirmenstein MA, Nelson SD. Acetaminophen-induced oxidation of protein thiols. Contribution of impaired thiol-metabolizing enzymes and the breakdown of adenine nucleotides. J Biol Chem. 1990;265:3059–3065. [PubMed] [Google Scholar]
- Valera A, Pelegrin M, Asins G, Fillat C, Sabater J, Pujol A, Hegardt FG, Bosch F. Overexpression of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in transgenic mice causes hepatic hyperketogenesis. J Biol Chem. 1994;269:3117–3123. [PubMed] [Google Scholar]
- Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem. 2004a;279:22092–22101. doi: 10.1074/jbc.M402245200. [DOI] [PubMed] [Google Scholar]
- Venkatraman A, Landar A, Davis AJ, Ulasova E, Page G, Murphy MP, Darley-Usmar V, Bailey SM. Oxidative modification of hepatic mitochondria protein thiols: effect of chronic alcohol consumption. Am J Physiol Gastrointest Liver Physiol. 2004b;286:G521–G527. doi: 10.1152/ajpgi.00399.2003. [DOI] [PubMed] [Google Scholar]
- Weber H, Engelmann S, Becher D, Hecker M. Oxidative stress triggers thiol oxidation in the glyceraldehyde-3-phosphate dehydrogenase o. Staphylococcus aureus Mol Microbiol. 2004;52:133–140. doi: 10.1111/j.1365-2958.2004.03971.x. [DOI] [PubMed] [Google Scholar]
- Zschocke J, Penzien JM, Bielen R, Casals N, Aledo R, Pie J, Hoffmann GF, Hegardt FG, Mayatepek E. The diagnosis of mitochondrial HMG-CoA synthase deficiency. J Pediatrics. 2002;140:778–780. doi: 10.1067/mpd.2002.123854. [DOI] [PubMed] [Google Scholar]