

Abstract
Barrett’s esophagus, a columnar metaplasia of the lower esophagus epithelium related to gastroesophageal reflux disease, is the strongest known risk factor for the development of esophageal adenocarcinoma (EAC). Understanding the signal transduction events involved in esophageal epithelium carcinogenesis may provide insights into the origins of EAC and may suggest new therapies. To elucidate the molecular pathways of bile acid–induced tumorigenesis, the newly identified inflammation-associated signaling pathway involving IκB kinases β (IKKβ), tuberous sclerosis complex 1 (TSC1), and mammalian target of rapamycin (mTOR) downstream effector S6 kinase (S6K1) was confirmed to be activated in immortalized Barrett’s CPC-A and CPC-C cells and esophageal cancer SEG-1 and BE3 cells. Phosphorylation of TSC1 and S6K1 was induced in response to bile acid stimulation. Treatment of these cells with the mTOR inhibitor rapamycin or the IKKβ inhibitor Bay 11-7082 suppressed bile acid–induced cell proliferation and anchorage-independent growth. We next used an orthotopic rat model to evaluate the role of bile acid in the progression of Barrett’s esophagus to EAC. Of interest, we found high expression of phosphorylated IKKβ (pIKKβ) and phosphorylated S6K1 (pS6K1) in tumor tissues and the Barrett’s epithelium compared with normal epithelium. Furthermore, immunostaining of clinical EAC tissue specimens revealed that pIKKβ expression was strongly correlated with pS6K1 level. Together, these results show that bile acid can deregulate TSC1/mTOR through IKKβ signaling, which may play a critical role in EAC progression. In addition, Bay 11-7082 and rapamycin may potentially be chemopreventive drugs against Barrett’s esophagus–associated EAC.
Introduction
Approximately 14,550 people were diagnosed with esophageal cancer in the United States in 2006; half of whom had esophageal adenocarcinoma (EAC; ref. 1). In fact, its incidence has risen faster than that of any other major cancer, including lung cancer, breast cancer, and melanoma in the United States since 1975. The prognosis of EAC is extremely poor, with a 5-year survival rate of <20%, even among patients who undergo combined modality therapy (2, 3). The causes underlying the dramatic increase in EAC incidence are poorly understood. A history of gastroesophageal reflux disease (GERD), which consists of the reflux of gastric contents into the lower esophagus, has been closely associated with EAC development (4, 5). Chronic GERD leads to the development of Barrett’s esophagus, a pathologic transformation of the normal squamous esophagus epithelium into metaplastic intestinal-like epithelium. Barrett’s esophagus is a true premalignant condition with an estimated 43-fold increase in the relative risk of developing EAC compared with the general population (6, 7). People with Barrett’s esophagus diagnosed with endoscopy are more likely to develop adenocarcinoma of the esophagus than the normal population (8–10). Bile acids are one of the major harmful components of refluxed gastric material, and intraesophageal bile salt concentrations are higher in patients with Barrett’s esophagus than in those with uncomplicated GERD (11, 12). Bile acids are promising candidates as esophageal carcinogens, because repeated exposure to bile acid, particularly exposure to the unconjugated bile acids deoxycholic acid and chenodeoxycholic acid (CDCA), can enhance Barrett’s esophagus transformation to EAC (13, 14). As bile salts can also stimulate the signaling effects of antiapoptotic pathways, such as c-myc, cyclo-oxygenase-2, and nuclear factor-κB (NF-κB), it is likely that continuous bile damage enhances dysregulated proliferation, ultimately contributing to genomic instability and progression to EAC (15–18). However, the molecular signaling switch for Barrett’s esophagus progression from dysplasia to invasive carcinoma is still unknown, especially in the refluxed bile acid–related cancer development and progression.
It is recognized that chronic inflammation is functionally important for the developmental process of several types of neoplasia, including lung and liver cancers (19, 20). Similarly, the multistep progression from Barrett’s esophagus metaplasia to invasive EAC evolves in a chronic inflammatory microenvironment (21–24). In Barrett’s esophagus, damaged esophageal cells and infiltrating inflammatory cells, including T lymphocytes and neutrophils, release inflammatory cytokines and chemokines, such as tumor necrosis factor α (TNFα), interleukin-8 (IL-8), and IL-10 (25–27). Published evidence suggest that the TNFα/IκB kinase β (IKKβ) signaling pathway plays a role in inflammation-mediated tumorigenesis. TNFα may be a proinflammatory cytokine involved in human cancers. When cells are exposed to TNFα stimuli, IKKβ is activated, which triggers IκBα degradation and enhances NF-κB translocation to the nucleus, ultimately activating target genes (28, 29). NF-κB has been found to be up-regulated in Barrett’s epithelium (BE), and the nuclear translocation of NF-κB in EAC is associated with poor prognosis and treatment response in patients (30).
Tuberous sclerosis complex 1 (TSC1), a well-known tumor suppressor involved in the development of tuberous sclerosis, negatively regulates the mammalian target of rapamycin (mTOR) pathway by inhibiting Ras homologue enriched brain (31, 32). Deregulation of the TSC/mTOR signaling pathway has been implicated in the development of cancer, and the activation of ribosomal protein S6 kinase (S6K1) by mTOR was found to lead to increased cell proliferation and angiogenesis (33).
Although the evidence suggests that inflammation is associated with human tumors, the underlying mechanism of tumorigenesis is not yet fully understood. In a recent study, we found that TNFα enhanced tumor angiogenesis through IKKβ-mediated deregulation of the TSC/mTOR pathway in breast cancer (34, 35). In the present study, we found that the bile acid deregulated the TSC/mTOR pathway through IKKβ signaling. We also found that treatment of Barrett’s esophagus cells and EAC cells with the IKKβ inhibitor Bay 11-7082 or the mTOR inhibitor rapamycin inhibited cell proliferation and in vitro transformation measured by the anchorage-independent growth assay. In a rat animal model, the expressions of phosphorylated IKKβ (pIKKβ) and phosphorylated S6K1 (pS6K1) were higher in Barrett’s esophagus and EAC compared with normal esophageal epithelium. We next evaluated normal esophageal epithelium, Barrett’s esophagus lesions, and tumor samples from patients with Barrett’s esophagus–associated EAC and found a concomitant sustained expression of both pIKKβ and pS6K1 in Barrett’s esophagus high-grade dysplasia and EAC. Together, our results indicate that bile acid may activate the mTOR pathway via IKKβ signaling, leading to Barrett’s esophagus and EAC.
Materials and Methods
Cell lines
We used immortalized human CPC-A and CPC-C cells which were derived from Barrett’s esophagus premalignant epithelium (CPC-A from hyperplasia and CPC-C from high-grade dysplasia) and were transfected with the human telomerase catalytic subunit to extend cell life span (kind gifts from Dr. P. Rabinovitch, University of Washington; ref. 36). These cells were maintained at 37°C in a 5% CO2 incubator with MCDB153 (Sigma) plus 10% fetal bovine serum (FBS), 20 ng/mL epidermal growth factor, 20 mg/mL adenine, 140 μg/mL bovine pituitary extract (Invitrogen), 5 μg/mL insulin-transferrin-selenium (Sigma), 4 mmol/L L-glutamine, and 0.4 μg/mL hydrocortisone. Human esophageal cell lines SKGT4, SEG-1, and BE3, which were derived from Barrett’s esophagus–associated EACs of the distal esophagus (kind gifts from Dr. D. Beer, University of Michigan), were maintained at 37°C in a 5% CO2 incubator with DMEM/F12 plus 10% FBS.
Antibodies and reagents
The antibodies used in this study were anti-TSC1 (Zymed), anti-pS6K1 (T389; Cell Signaling Technology), anti-S6K1 (Cell Signaling Technology), anti-pIKKβ (S181; Cell Signaling Technology), anti–phosphorylated AKT (S473; Cell Signaling Technology), anti-AKT (Cell Signaling Technology), anti-actin (Sigma), and anti-IKKβ (Cell Signaling Technology). Antibodies against the phosphorylation sites of TSC1 S511 were produced using the synthetic phosphorylated peptides SPFYRDpSLPGSQ as antigens and purified on a phosphopeptide column at Bethyl Laboratories, Inc. CDCA was obtained from Sigma Chemical Co. and dissolved in ethanol. Bay 11-7082 and rapamycin were purchased from Calbiochem. Recombinant human TNFα was obtained from Roche.
Chenodeoxycholate, taurochenodeoxycholate, Bay 11-7082, and rapamycin treatment
CDCA and taurochenodeoxycholate (TCDA), the major components of bile acid, were used to mimic the physiologic refluxed bile acid in Barrett’s esophagus and EAC. In brief, EAC SEG-1 and BE3 cells and Barrett’s esophagus CPC-A and CPC-C cells were grown in the complete medium and exposed to 100 μmol/L, 300 μmol/L CDCA, or 200 μmol/L TCDA for 2 h with or without Bay 11-7082 (40 μmol/L, 45 min pretreatment) or rapamycin (50 or 100 nmol/L, 3 h pretreatment). Cell proliferation/viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazo-lium bromide (MTT) assay, and the anchorage-independent growth was measured by soft agar assay. Western blotting was performed to determine the expression of members of the IKKβ/TSC1/mTOR pathway (e.g., IKKβ, TSC1, and S6K1).
Small interfering RNAs
SEG-1 cells were transfected with IKKβ SMARTpool small interfering RNAs (siRNA; Upstate Biotechnology) or control SMARTpool siRNAs (Upstate Biotechnology) by Lipofectamine with plus reagent. After 72 h of siRNA transfection, the cells were treated with 300 μmol/L CDCA for 2 h and lysed for Western blotting.
MTT and anchorage-independent growth assays
Cell growth was measured using the MTT assay (37). In brief, 5,000 cells were seeded in 96-well plates and incubated in culture medium overnight. We then treated cells with 100 μmol/L CDCA, 300 μmol/L CDCA for 2 h, then washed, and cultured with media containing10% FBS for 24 h. For acid treatment, cells were incubated for 24 h with serum-free medium that was adjusted with 0.1 mol/L HCl to pH 4.0, as monitored by pH meter. An equivalent volume of PBS was added to serum-free, normal pH medium to control the difference in osmolarity. At the end of the exposure period, 20 μL of MTT was added to the cell culture and the cells were incubated for 2 h. Subsequently, 100 μL of DMSO (Sigma) was added to each well and the plate was incubated for 5 min to dissolve the formazan crystals. Light absorbance was measured at 570 nm using a multiwell scanner (Labsystems). To determine the number of colonies under anchorage-independent growth condition, we cultured EAC cells in soft agar medium after SEG-1 and BE3 EAC cells were treated with 300 μmol/L CDCA for 16 h. In brief, aliquots of 2 × 104 cells were suspended in 1.5 mL of standard medium containing 0.3% agar at 43°C and layered on 1 mL of solidified 0.5% agar in the culture medium in 35-mm culture dishes. Dishes were incubated at 37°C in 5% CO2 for 3 wk (38). Cells were fed with the culture medium, and viable colonies were stained with p-iodo-nitrotetrazolium violet (Sigma). Each experiment was repeated thrice.
Rat model of Barrett’s esophagus and adenocarcinoma
Six-week-old male Sprague-Dawley rats were purchased from Harlan and housed at two per cage under standard laboratory conditions. Rats were given commercial rat chow (Auto 5010 test diet; Purina Mills) before and up to 4 wk after surgical intervention. The rats were allowed to acclimate for 2 wk before surgery. Rat chow was withheld 24 h before surgical intervention, and liquids were stopped on the morning of surgery. The rats were anesthetized by isoflurane (99.9%, 0.5 mL/L volume), followed by an i.m. injection of xylazine hydrochloride (12 mg/kg) and ketamine (75 mg/kg) as maintenance anesthesia. Enteroesophageal reflux was induced by Levrat’s esophagojejunostomy technique, as previously described (39).
After the reflux induction of esophageal cancer, the rats were euthanized by CO2 narcosis. A midline incision was made from the laryngopharynx to the lower abdomen. The site of anastomosis was identified by the location of the polypropylene sutures, and the esophageal tissue was removed from the larynx to 2 mm above the anastomosis site. A longitudinal slice of inflamed and tumorous esophageal tissue (<1-mm wide) was removed and stored at −70°C. The remaining esophageal tissue was fixed in 10% buffered formalin for 24 h and then transferred to 80% ethanol. The tissue was longitudinally divided into slices, with each slice representing the entire length and depth of the esophagus.
Protein expression
The expression of IKKβ, pIKKβ (S181), TSC1, pTSC1 (S511), S6K1, and pS6K1 (T389) was detected by Western blot analysis. The method of Western blot was described previously (40). In brief, Barrett’s esophagus and EAC cells were plated in 10-cm dishes until they reached confluence. They were then harvested, washed in ice-cold PBS, and resuspended in whole-cell extraction solution [0.02 mol/L HEPES (pH 7.9), 0.005% NP40, 0.15% glycerol, 0.3 mol/L NaCl, 0.001 mol/L EDTA, and 0.01 mol/L NaF] with protease inhibitors. After 30-min incubation on ice, lysates were collected by centrifugation at 12,000 rpm for 10 min at 4°C. Protein concentrations were estimated using the Bradford method (Bio-Rad protein assay). Fifty micrograms of total protein were resolved by 6% to 10% SDS PAGE, transferred to polyvinylidene difluoride membranes (NEN Life Science), and probed with a specific antibody of the target protein. After binding with horseradish peroxidase coupled with secondary antibodies (Amersham Pharmacia), the immune complex was visualized with an enhanced chemiluminescence detection system (Amersham Pharmacia) by exposure to X-ray films (Kodak).
Detection of pIKKβ and pS6K1 protein expression by immunohis-tochemical staining in human EAC
Immunohistochemical staining for pIKKβ and pS6K1 protein expression was performed on adjacent 4-μm formalin-fixed paraffin-embedded tissue sections. One hundred twenty-three EAC, assembled in a tissue microarray, were examined. Adjacent BE and normal esophageal epithelia were available in 20 of the 123 EAC cases and included in all 20 cases of BE metaplasia and 8 cases of BE high-grade dysplasia. Tissue specimens were obtained retrospectively from patients undergoing, as primary treatment, complete esophageal surgical resection at M. D. Anderson between January 1986 and December 1997. The Institutional Review Board approved this project. All specimens were reviewed by an experienced gastrointestinal pathologist (T.T. Wu). The tissue microarray contained BE premalignant (CPC-A, CPC-C) and EAC (SKGT-5, SKGT-5, BE-3, SEG-1, TT) cell lines as internal controls. Immunohistochemistry was performed as previously described using pIKKβ (S181) or pS6K1 (T389) antibodies (41). Visualization was performed with aminoethylcarbazole chromogen. Only cytoplasmic immunoreactivity in cancer cells was considered positive for scoring purposes. Protein staining was evaluated on a semiquantitative scale combining staining intensity and percentage of positive cells in the cancer fields. Staining intensity was evaluated as follows: no/weak staining, negative; moderate or strong staining, positive. Percentage of positive cells was assessed on a four-point semiquantitative scale as follows: no positive cells, >0 to 10% positive cells, >10% to 25% positive cells, >25% to 50% positive cells, and >50% positive cells. Normal, BE, and cancer samples at ≥10% of the scored cells were considered positive. Two investigators (J.G.I. and D.F.L.), independently, in a blind fashion, analyzed protein expression. Fisher’s exact test and Spearman rank correlation tests were used for statistical analysis; P < 0.05 was considered statistically significant.
Results
IKKβ/TSC1/mTOR pathway was activated in certain EAC cell lines but not in immortalized Barrett’s esophagus cell lines
To determine whether the IKKβ/TSC1/mTOR pathway may be involved in inflammation-associated Barrett’s esophagus and EAC, immortalized Barrett’s esophagus cell lines CPC-A and CPC-C and EAC cell lines SKGT-4, SEG-1, and BE3 were evaluated for the expression of pIKKβ (S181), pTSC1 (S511), and mTOR downstream pS6K1 (T389). CPC-A and CPC-C cells had a lower expression level of pIKKβ (S181) and expressed concurrently low levels of pTSC1 (S511) and pS6K1 (T389). The SKGT4 cancer cell line also had low expression of pIKKβ (S181), pTSC1 (S511), and pS6K1 (T389). Expression of pIKKβ (S181) was correlated with higher level of pTSC1 (S511) and pS6K1 (T389) in SEG-1 and BE3 EAC cancer cell lines. These phenomena were observed in two of three cancer cell lines, suggesting that the TSC1/mTOR pathway via IKKβ signaling may be activated in certain inflammation-associated EAC (Fig. 1A).
Figure 1.

IKKβ/TSC1/mTOR pathway was activated in Barrett’s esophagus–associated EAC cancer cells. A, increased expression of pIKKβ (S181) was correlated with higher level of pTSC1 (S511) and pS6K1 (T389) in SEG-1 and BE3 EAC cancer cell lines. Immortalized Barrett’s esophagus cell lines CPC-A and CPC-C had lower expression level of pIKKβ (S181) and expressed concurrently low levels of pTSC1 (S511) and pS6K1 (T389). The SKGT4 EAC cancer cell line also had lower expression of pIKKβ (S181), pTSC1 (S511), and pS6K1 (T389). B, the phosphorylation of pTSC1 (S511) and pS6K1 (T389) increased two to three times in SEG-1 cancer cells treated with 300 μmol/L CDCA (P < 0.001) and low-pH medium (P < 0.001) compared with those in untreated cells. C, conjugated bile acid TCDA increased the expressions of pTSC1 (S511) and pS6K1 (T389), and the IKKβ inhibitor Bay 11-7082 blocked the TCDA stimulation effect in SEG-1 EAC cancer cells. D, cell proliferation was increased in SEG-1 and BE3 cancer cells compared with SKGT-4 cells, which had lower expression of the IKKβ/TSC1/mTOR signaling pathway.
Bile acid induced activation of IKKβ/TSC1/mTOR pathway, leading to enhanced EAC cell proliferation
Because bile acid and a low pH environment have been thought to be important risk factors in the development of Barrett’s esophagus and EAC, we determined whether CDCA or a low pH environment might activate the IKKβ/TSC1/mTOR pathway in EAC cell lines and thus contribute to cell survival in the presence of bile acid. The relative phosphorylated TSC1 (S511) levels were increased 2.6-fold and concurrently pS6K1 (T389) levels were 2.8-fold higher under CDCA 300 μmol/L stimulation in SEG-1 EAC cancer cells (Fig. 1B) and BE3 (data not shown) cells compared with those in CDCA untreated cells. CDCA is unconjungated bile acid, and to further prove that conjungated bile acid had the same stimulation effect in EAC cancer cells, we used the TCDA and treated the SEG-1 cancer cells. Interestingly, the TCDA induced the phosphorylation of TSC1 and S6K1 in SEG-1 cancer cells, and these inductions were suppressed by the IKKβ inhibitor Bay 11-7082 (Fig. 1C). Accordingly, the cell growth rate was increased in both SEG-1 and BE3 cancer cells; however, the CDCA and acidic stimulation did not have an effect on the growth rate of SKGT4 cancer cells, in which the IKKβ/TSC1/mTOR pathway was not activated (Fig. 1D). Together, these results suggest that CDCA and acidic environment may stimulate the mTOR pathway and enhance cell growth in certain EAC cells.
Inhibition of IKKβ and mTOR diminished bile acid–induced cell growth in EAC cells
To further evaluate whether a causal relationship exists between the induction of the IKKβ/TSC1/mTOR pathway and an increased cell growth rate in response to bile acid, we used the IKKβ inhibitor and IKKβ siRNAs to block the IKKβ kinase activity. The activation of pTSC1 (S511) and pS6K1 (T389) by bile acid was suppressed by the IKKβ inhibitor Bay 11-7082 (40 μmol/L, 45 min pretreatment) and IKKβ siRNAs. CDCA did not activate AKT in SEG-1 and BE3 cancer cells (Fig. 2A and B). To ensure that the observed increase in S6K1 phosphorylation was caused by mTOR activity, we treated cells with rapamycin, a well-known mTOR inhibitor. CDCA exposure increased the level of pS6K1 (T389) in SEG-1 and BE3 cancer cells, and rapamycin (50 or 100 nmol/L, 3 h pretreatment) blocked activation of the mTOR pathway. Rapamycin inhibited both basal and CDCA-induced S6K1 phosphorylation (Fig. 2C). Consistently, both the IKKβ and mTOR inhibitors (Bay 11-7082 and rapamycin), as well as IKKβ siRNAs, suppressed the growth rates of the SEG-1 and BE3 cells (Fig. 2D). These results suggest that activation of the TSC1/mTOR pathway via IKKβ signaling may be required for the proliferation of EAC cells in the presence of bile salts.
Figure 2.

Bile acid stimulated the IKKβ/TSC1/mTOR pathway and increased cell proliferation, which was blocked by IKKβ and mTOR inhibitors. A, CDCA induced pTSC1 (S511) and pS6K1 (T389) in the SEG-1 and BE3 EAC cancer cell lines. The increased expression of pTSC1 (S511) and pS6K1 (T389) by CDCA were blocked by the IKKβ inhibitor Bay 11-7082 (40 μmol/L, 45 min pretreatment). CDCA did not activate Akt in SEG-1 and BE3 cancer cells. B, siRNA-mediated knockdown of IKKβ inhibited CDCA-induced pS6K1 (T389) in SEG-1 EAC cancer cells. C, CDCA exposure increased the expression of pS6K1 (T389) in SEG-1 EAC cancer cells, and rapamycin (50 nmol/L, 3 h pretreatment) blocked activation of the mTOR pathway. D, CDCA exposure enhanced SEG-1 and BE3 EAC cancer cell proliferation (P < 0.05). Knockdown of IKKβ by IKKβ siRNA inhibited CDCA stimulation effect (P < 0.001), and CDCA-induced cell proliferation was also blocked by the IKKβ inhibitor Bay 11-7082 and mTOR inhibitor rapamycin (P < 0.001).
Bile acid and TNFα activated IKKβ/TSC1/mTOR pathway in immortalized Barrett’s esophagus CPC-A and CPC-C cells
Bile acid and cytokine TNFα, which exist in the inflammatory microenvironment, are major factors in the development of Barrett’s esophagus. We hypothesized that CDCA and TNFα would activate the IKKβ/TSC1/mTOR pathway in nontransformed Barrett’s esophagus cell lines, resulting in enhanced cell proliferation. To test this hypothesis, we treated the Barrett’s esophagus cell lines CPC-A and CPC-C with CDCA or TNFα. We observed that TNFα increased phosphorylation of TSC1 and S6K1 in both CPC-A and CPC-C cells. The IKKβ/TSC1/mTOR pathway can also be activated by bile acid CDCA in CPC-C cell line. Both CDCA-induced and TNFα-induced phosphorylation of TSC1 and S6K1 were blocked by the IKKβ inhibitor Bay 11-7082 (Fig. 3A). Furthermore, the IKKβ inhibitor Bay 11-7082 inhibited the TNFα-induced cell proliferation of CPC-A and CPC-C cells. The Bay 11-7082 also inhibited the CDCA-induced cell proliferation of CPC-C (Fig. 3B). These results show that bile acid and the inflammatory cytokine TNFα, both important risk factors for the progression of Barrett’s esophagus, induce activation of the IKKβ/TSC1/mTOR pathway and enhance the growth of epithelium cells in Barrett’s esophagus.
Figure 3.

Bile acid and TNFα activated the IKKβ/TSC1/mTOR pathway in Barrett’s esophagus CPC-A and CPC-C cells. A, inflammatory cytokine TNFα induced expression of pTSC1 (S511) and pS6K1 (T389) in Barrett’s esophagus CPC-A and CPC-C cells (lane 2), CDCA induced expression of pTSC1 (S511) and pS6K1 (T389) in CPC-C cells (lane 4). The stimulation effect of TNFα or CDCA was blocked by IKKβ inhibitor Bay 11-7082 (lanes 3 and 5). Cells were serum-starved overnight and then treated with 20 ng/mL TNFα for 30 min or 40 μmol/L Bay 11-7082 for 45 min. B, TNFα induced CPC-A and CPC-C cell proliferation (P < 0.05) and CDCA induced CPC-C cell proliferation (P < 0.05). Both TNFα-induced and CDCA-induced cell proliferation was blocked by IKKβ inhibitor Bay 11-7082 (P < 0.001).
Bile acid–induced esophageal anchorage-independent growth was suppressed by Bay 11-7082 and rapamycin
To evaluate the effect of bile acid on the transforming activity of EAC cells, we determined the anchorage-independent growth activity of SEG-1 and BE3 EAC cells, which is a transforming phenotype frequently associated with tumorigenic cancer cells. The activity level was increased in SEG-1 and BE3 EAC cancer cells after CDCA stimulation compared with that in control untreated cells (P < 0.05). This CDCA-enhanced SEG-1 and BE3 cancer cell anchorage-independent growth was substantially blocked by treatment with Bay 11-7082 and rapamycin (P < 0.001; Fig. 4). These observations indicate that bile acid can increase the anchorage-independent growth activity of the esophageal cancer cells, and this stimulation effect may be due to activation of the IKKβ/TSC1/mTOR pathway.
Figure 4.
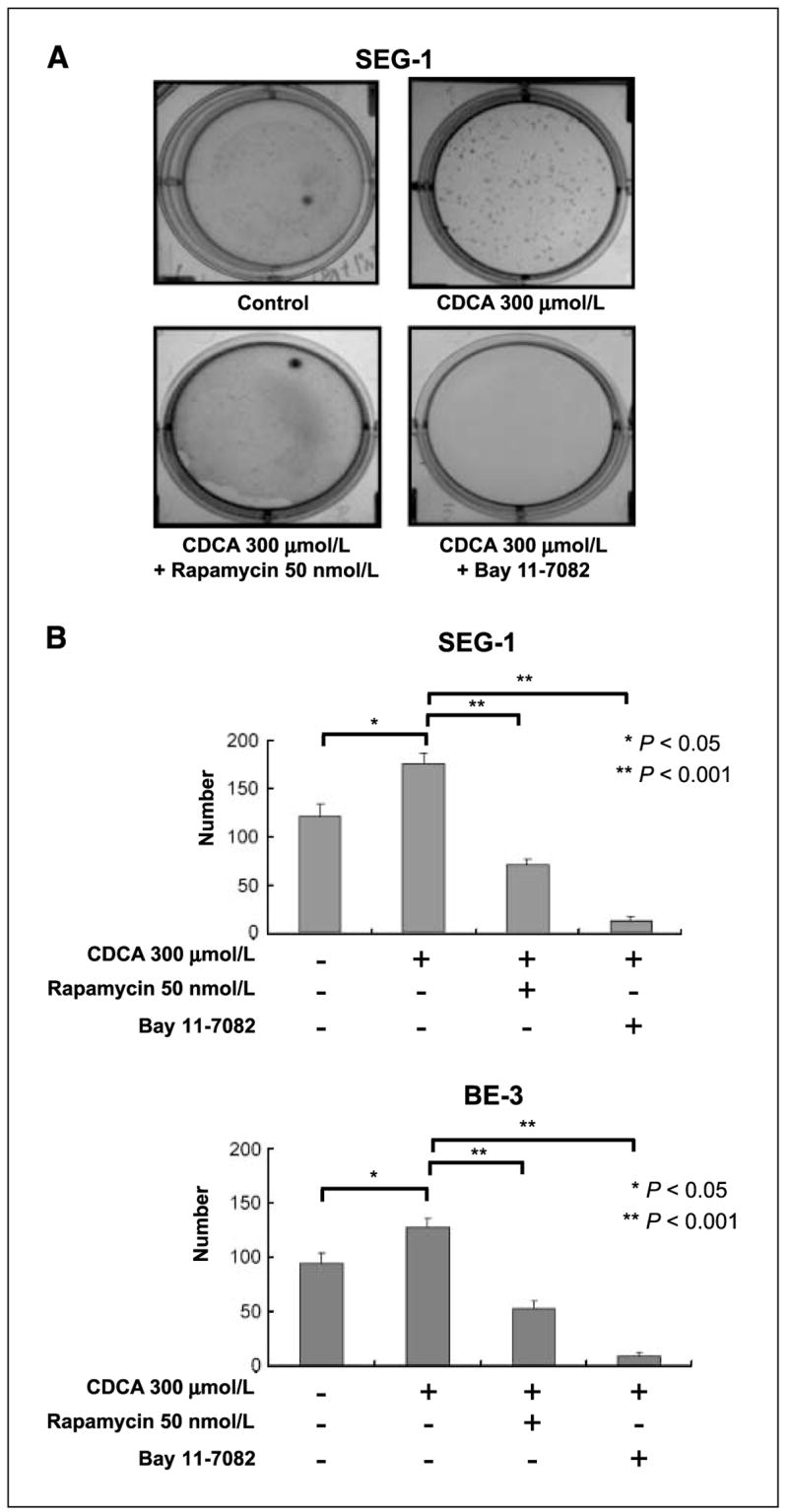
Bay 11-7082 and rapamycin suppressed bile acid–induced esophageal tumor anchorage-independent growth. A, CDCA enhanced the anchorage-independent growth abilities of SEG-1 EAC cancer cells in soft agar. B, quantitative result of A. CDCA-induced SEG-1 and BE3 EAC cancer cells’ soft agar growth ability (P < 0.05). The enhanced anchorage-independent growth could be inhibited by Bay 11-7082 and rapamycin (P < 0.001).
IKKβ signaling led to activation of the TSC1/mTOR pathway in an orthotropic animal model
To further understand the contribution of the TSC1/mTOR pathway via IKKβ signaling in the progression of Barrett’s esophagus to EAC, we used an orthotropic animal model (39). Because intestinal bile acid reflux plays an important role in the development of Barrett’s esophagus and EAC, we exposed the esophagus of the rat to intestinal bile acid by surgical anastomosis of the esophagus and intestines. After several months, esophageal inflammation and tumor developed, mimicking Barrett’s esophagus and EAC in humans. Of interest, the relative expression density of pIKKβ (S181) was 1.5 times higher in inflammatory BE and 3.4 times higher in EAC compared with normal epithelium (NE). The relative expression densities of pS6K1 (T389) was 1.6 times higher in BE and 2.8 times higher in EAC compared with NE (Fig. 5A). Immunohistochemical staining revealed the expression of pIKKβ (S181) and pS6K1 (T389) in three of five rats with enteric bile acid reflux-related BE and EAC. The adjacent normal esophagus squamous epithelia (NE) stained pIKKβ (S181) and pS6K1 (T389) negative (Fig. 5B).
Figure 5.

Activation of the IKKβ/TSC1/mTOR pathway was observed in an orthotropic animal model. A, in rats, the relative expression density of pIKKβ (S181) was 1.5 times higher in inflammatory BE and 3.4 times higher in EAC. The relative expression density of pS6K1 (T389) was 1.6 times higher in BE and 2.8 times higher in EAC compared with normal esophagus squamous epithelia (NE). B, immunohistochemical staining revealed the expression of pIKKβ (S181) and pS6K1 (T389) in BE and EAC. pIKKβ (S181) and pS6K1 (T389) staining in the adjacent NE was negative.
Thus, the animal model supported the role of the IKKβ/mTOR pathway that was identified in cell line studies.
Immunohistochemical staining revealed a correlation between pIKKβ and pS6K1 in Barrett’s esophagus and EAC human tissue samples
To further determine the importance of the TSC1/mTOR pathway via IKKβ signaling in Barrett’s esophagus–associated EAC, we evaluated a tissue microarray of 123 EAC tissue specimens. Expression of both proteins was found in most EAC cases: 73 out 98 scorable cases (74.5%) for pIKKβ and 57 of the 96 scorable cases (59.3%) for pS6K1 were positive. As shown in Fig. 6B, the degree of positivity was heterogenous between cases. In 90 cases, concomitant scoring of both proteins was possible and revealed a strong correlation between positive pIKKβ pS6K1 expression [of the 90 specimens, 45 (50%) showed both pIKKβ and pS6K1 positive, 15 (16.6%) showed both protein negative, 21 (23.3%) harbored only pIKKβ positive, and 9 (10%) had only pS6K1 positive; P = 0.016; Fig. 6]. We also examined normal esophageal mucosa and BE lesions (20 metaplasia and 8 high-grade dysplasia) adjacent to resected EAC in 20 of the 90 cases for which both protein were scorable. We found that, whereas normal esophageal squamous epithelium did not show positivity for neither proteins, 3 (15%) BE metaplasia and 5 (62.5%) BE high-grade dysplasia showed positivity for both proteins. Interestingly, in all BE-positive cases, the paired EAC showed also positivity for pIKKβ and pS6K1. These results suggest that activation of IKKβ/mTOR pathway plays a role in EAC development.
Figure 6.

pIKKβ (S181) and pS6K1 (T389) protein expression in 123 EAC human specimens. A, examples of immunohistochemical visualization of both proteins. B, degree of positivity for pIKKβ (S181) and pS6K1 (T389) in EAC tissues. C, correlation between pIKKβ (S181) and pS6K1 (T389) positive expression.
Discussion
Although the close relationship between Barrett’s esophagus and EAC has been well established, the pathogenic mechanism responsible for the transformation of Barrett’s esophagus to EAC is still far from understood, especially in view of the contribution of reflux substance, bile acid and gastric acid to cancer development and progression (42). Therefore, we undertook an investigation of the biological effects of bile acid and acidic condition on the Barrett’s esophagus and EAC cells. We found that both bile acid (CDCA) and a low pH environment, the major content of gastro-esophageal reflux, triggered the activation of IKKβ, leading to phosphorylation of TSC1 and S6K1 and activation of the mTOR pathway. The IKKβ/TSC1/mTOR pathway may be a molecular switch, allowing Barrett’s esophagus to progress to cancer. This is likely to be clinically relevant to pathogenesis as we found a statistically significant correlation between the phosphorylation of IKKβ and phosphorylation of S6K1 in Barrett’s esophagus–associated EAC.
Chronic exposure to bile acid and gastric acid can cause an inflammation process in the lower esophageal epithelium, resulting in Barrett’s esophagus, which may subsequently evolve into EAC (23, 43). Bile acids are one of the major components of the gastro-esophageal refluxate and cause esophageal epithelium damage in Barrett’s esophagus, which is highly associated with EAC (12, 44). However, the molecular mechanism by which bile acid exposure is involved in the esophageal epithelium inflammatory process and promotes malignant transformation in Barrett’s esophagus is poorly understood. The results shown in this study from cell lines support the notion that CDCA may activate IKKβ/mTOR signaling in certain types of EAC cancer cells, such as SEG-1 and BE3 cells, thereby increasing cell proliferation rate and transforming phenotype associated with tumorigenic potential. In Barrett’s esophagus cell lines CPC-A and CPC-C, the inflammatory cytokine TNFα and CDCA also resulted in up-regulation of the IKKβ/TSC1/mTOR pathway and in turn enhanced proliferation of Barrett’s related cells; some of the surviving nontransformed Barrett’s related cells may eventually develop into EAC. We validated our in vitro data using an in vivo bile salt reflux rat model, which promoted Barrett’s esophagus and EAC. We showed expression of both pIKKβ and pS6K1 in bile acid–induced Barrett’s esophagus epithelial cells and EAC. Additionally, we further showed expression of both proteins in human tissues derived from Barrett’s esophagus and EAC. These results suggest that deregulation of mTOR signaling in Barrett’s esophagus cells may promote a stepwise progression from inflammatory epithelium to cancer development. It is worthwhile to emphasize that both the animal model and human tumor tissues further supported the molecular mechanism, namely IKKβ/mTOR pathway that was established from the cell line studies.
The mTOR signaling has been established in the formation of a number of cancers, including lung cancer (45), breast cancer (46), renal cell carcinoma (47), non–Hodgkin’s lymphoma (48), and malignant melanoma (49), and activation of mTOR by IKKβ has just recently been reported in breast cancer (34). The current study further links mTOR activation through the bile acid stimulation of the IKKβ/TSC1 signaling in Barrett’s esophagus–associated EAC. EAC is currently the cancer with the fastest rate of incidence in the United States (2). Currently available treatments for EAC patients, including chemotherapy and radiotherapy, are ineffective, resulting in a 5-year survival rate of <20% (2, 3). We investigated the mechanism by which bile acid stimulates the expression of IKKβ, a downstream molecule of the TNFα signaling pathway, and further activates the TSC1/mTOR pathway. Activation of mTOR stimulates translation of ribosomal proteins via the activation of S6K1, which may be involved in the neoplastic process. Rapamycin can bind to FKBP12 and the complex blocks the mTOR activity very selectively. Bay 11-7082 has also been reported to effectively inhibit IKKβ activity (50). Treatment of the esophageal cancer cells with rapamycin and Bay 11-7082 can effectively block the CDCA-induced cell proliferation and cell anchorage-independent growth ability. These findings suggest that incorporating targeted inhibition of the IKKβ/TSC1/mTOR pathway into conventional chemoradiotherapy regimens might improve the EAC treatment outcome.
In conclusion, the results from this study clearly show that bile acid can activate mTOR activity, as measured by S6K1 through IKKβ signaling, in Barrett’s esophagus and EAC. Activation of IKKβ/mTOR pathway enhances cell growth and may initiate the development of the EAC. Furthermore, blocking IKKβ or mTOR signaling with Bay 11-7082 or rapamycin can interrupt the EAC growth and transformation. The results further suggest that Bay 11-7082 and rapamycin may be useful as chemopreventive drugs in the treatment of Barrett’s esophagus–associated EAC.
Acknowledgments
Grant support: PO1 CA099031, Kadoorie Charitable Foundations (M.-C. Hung), University of Texas M. D. Anderson Multidisciplinary Esophageal Research Grant, Riverkreek Foundation, Dallas, Cantu, Smith, and Park Families (J.A. Ajani), Cancer Center Support grant CA16672, a predoctoral fellowship from the U.S. Army Breast Cancer Research Program, grant W81XWH-05-1-0252, T.C. Hsu Endowed Memorial Scholarship, Andrew Sowell-Wade Huggins Scholarship, Presidents’ Research Scholarship from University of Texas Graduate School of Biomedical Sciences at Houston (D.-F. Lee), Andrew Sowell-Wade Huggins Scholarship (H.-P. Kuo), a predoctoral fellowship from the U.S. Army Breast Cancer Research Program, grant W81XWH-06-1-0709 (C.-K. Chou), and Taiwan Merit Scholarship from National Science Council of Taiwan (H.-L. Sun).
We thank Drs. P. Rabinovitch and D. Beer for providing reagents and Dr. S. Miller for editing.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–30. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142–6. doi: 10.1093/jnci/dji024. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB, Mason AC, Petersen N, Key CR. Epidemiological differences between adenocarcinoma of the oesophagus and adenocarcinoma of the gastric cardia in the USA. Gut. 2002;50:368–72. doi: 10.1136/gut.50.3.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pace F, Tonini M, Pallotta S, Molteni P, Porro GB. Systematic review: maintenance treatment of gastro-oesophageal reflux disease with proton pump inhibitors taken ‘on-demand’. Aliment Pharmacol Ther. 2007;26:195–204. doi: 10.1111/j.1365-2036.2007.03381.x. [DOI] [PubMed] [Google Scholar]
- 5.Spechler SJ. Clinical practice. Barrett’s esophagus. N Engl J Med. 2002;346:836–42. doi: 10.1056/NEJMcp012118. [DOI] [PubMed] [Google Scholar]
- 6.Spechler SJ, Goyal RK. The columnar-lined esophagus, intestinal metaplasia, and Norman Barrett. Gastroenterology. 1996;110:614–21. doi: 10.1053/gast.1996.v110.agast960614. [DOI] [PubMed] [Google Scholar]
- 7.Playford RJ. New British Society of Gastroenterology (BSG) guidelines for the diagnosis and management of Barrett’s oesophagus. Gut. 2006;55:442. doi: 10.1136/gut.2005.083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solaymani-Dodaran M, Logan RF, West J, Card T, Coupland C. Risk of oesophageal cancer in Barrett’s oesophagus and gastro-oesophageal reflux. Gut. 2004;53:1070–4. doi: 10.1136/gut.2003.028076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhillon PK, Farrow DC, Vaughan TL, et al. Family history of cancer and risk of esophageal and gastric cancers in the United States. Int J Cancer. 2001;93:148–52. doi: 10.1002/ijc.1294. [DOI] [PubMed] [Google Scholar]
- 10.Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology. 2005;129:1825–31. doi: 10.1053/j.gastro.2005.08.053. [DOI] [PubMed] [Google Scholar]
- 11.Dvorak K, Payne CM, Chavarria M, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett’s oesophagus. Gut. 2007;56:763–71. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nehra D, Howell P, Williams CP, Pye JK, Beynon J. Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity. Gut. 1999;44:598–602. doi: 10.1136/gut.44.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F, Altorki NK, Wu YC, Soslow RA, Subbaramaiah K, Dannenberg AJ. Duodenal reflux induces cyclooxygenase-2 in the esophageal mucosa of rats: evidence for involvement of bile acids. Gastroenterology. 2001;121:1391–9. doi: 10.1053/gast.2001.29781. [DOI] [PubMed] [Google Scholar]
- 14.Atherfold PA, Jankowski JA. Molecular biology of Barrett’s cancer. Best Pract Res Clin Gastroenterol. 2006;20:813–27. doi: 10.1016/j.bpg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Jenkins GJ, Harries K, Doak SH, et al. The bile acid deoxycholic acid (DCA) at neutral pH activates NF-κB and induces IL-8 expression in oesophageal cells in vitro. Carcinogenesis. 2004;25:317–23. doi: 10.1093/carcin/bgh032. [DOI] [PubMed] [Google Scholar]
- 16.Buskens CJ, Van Rees BP, Sivula A, et al. Prognostic significance of elevated cyclooxygenase 2 expression in patients with adenocarcinoma of the esophagus. Gastroenterology. 2002;122:1800–7. doi: 10.1053/gast.2002.33580. [DOI] [PubMed] [Google Scholar]
- 17.Cheong E, Ivory K, Doleman J, Parker ML, Rhodes M, Johnson IT. Synthetic and naturally occurring COX-2 inhibitors suppress proliferation in a human oesophageal adenocarcinoma cell line (OE33) by inducing apoptosis and cell cycle arrest. Carcinogenesis. 2004;25:1945–52. doi: 10.1093/carcin/bgh184. [DOI] [PubMed] [Google Scholar]
- 18.Tselepis C, Morris CD, Wakelin D, et al. Upregulation of the oncogene c-myc in Barrett’s adenocarcinoma: induction of c-myc by acidified bile acid in vitro. Gut. 2003;52:174–80. doi: 10.1136/gut.52.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith CJ, Perfetti TA, King JA. Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal Toxicol. 2006;18:667–77. doi: 10.1080/08958370600742821. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki K, Murata M, Yoshida K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 21.Hamilton SR, Smith RR. The relationship between columnar epithelial dysplasia and invasive adenocarcinoma arising in Barrett’s esophagus. Am J Clin Pathol. 1987;87:301–12. doi: 10.1093/ajcp/87.3.301. [DOI] [PubMed] [Google Scholar]
- 22.Montgomery E, Goldblum JR, Greenson JK, et al. Dysplasia as a predictive marker for invasive carcinoma in Barrett esophagus: a follow-up study based on 138 cases from a diagnostic variability study. Hum Pathol. 2001;32:379–88. doi: 10.1053/hupa.2001.23511. [DOI] [PubMed] [Google Scholar]
- 23.Jankowski JA, Wright NA, Meltzer SJ, et al. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol. 1999;154:965–73. doi: 10.1016/S0002-9440(10)65346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jankowski J, Sharma P. Review article: approaches to Barrett’s oesophagus treatment-the role of proton pump inhibitors and other interventions. Aliment Pharmacol Ther. 2004;19(Suppl 1):54–9. doi: 10.1111/j.0953-0673.2004.01839.x. [DOI] [PubMed] [Google Scholar]
- 25.Tselepis C, Perry I, Dawson C, et al. Tumour necrosis factor-α in Barrett’s oesophagus: a potential novel mechanism of action. Oncogene. 2002;21:6071–81. doi: 10.1038/sj.onc.1205731. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald RC, Abdalla S, Onwuegbusi BA, et al. Inflammatory gradient in Barrett’s oesophagus: implications for disease complications. Gut. 2002;51:316–22. doi: 10.1136/gut.51.3.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gough MD, Ackroyd R, Majeed AW, Bird NC. Prediction of malignant potential in reflux disease: are cytokine polymorphisms important? Am J Gastroenterol. 2005;100:1012–8. doi: 10.1111/j.1572-0241.2005.40904.x. [DOI] [PubMed] [Google Scholar]
- 28.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[κ]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 29.Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 30.Izzo JG, Malhotra U, Wu TT, et al. Association of activated transcription factor nuclear factor κB with chemoradiation resistance and poor outcome in esophageal carcinoma. J Clin Oncol. 2006;24:748–54. doi: 10.1200/JCO.2005.03.8810. [DOI] [PubMed] [Google Scholar]
- 31.Kwiatkowski DJ, Zhang H, Bandura JL, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–34. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 32.Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–6. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segrelles C, Ruiz S, Santos M, Martinez-Palacio J, Lara MF, Paramio JM. Akt mediates an angiogenic switch in transformed keratinocytes. Carcinogenesis. 2004;25:1137–47. doi: 10.1093/carcin/bgh132. [DOI] [PubMed] [Google Scholar]
- 34.Lee DF, Kuo HP, Chen CT, et al. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 35.Lee DF, Hung MC. All Roads Lead to mTOR: integrating inflammation and tumor angiogenesis. Cell Cycle. 2007;6:3011–4. doi: 10.4161/cc.6.24.5085. [DOI] [PubMed] [Google Scholar]
- 36.Palanca-Wessels MC, Klingelhutz A, Reid BJ, et al. Extended lifespan of Barrett’s esophagus epithelium transduced with the human telomerase catalytic subunit: a useful in vitro model. Carcinogenesis. 2003;24:1183–90. doi: 10.1093/carcin/bgg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueno NT, Yu D, Hung MC. Chemosensitization of HER-2/neu-overexpressing human breast cancer cells to paclitaxel (Taxol) by adenovirus type 5 E1A. Oncogene. 1997;15:953–60. doi: 10.1038/sj.onc.1201250. [DOI] [PubMed] [Google Scholar]
- 38.Yan DH, Chang LS, Hung MC. Repressed expression of the HER-2/c-erbB-2 proto-oncogene by the adenovirus E1a gene products. Oncogene. 1991;6:343–5. [PubMed] [Google Scholar]
- 39.Buttar NS, Wang KK, Leontovich O, et al. Chemo-prevention of esophageal adenocarcinoma by COX-2 inhibitors in an animal model of Barrett’s esophagus. Gastroenterology. 2002;122:1101–12. doi: 10.1053/gast.2002.32371. [DOI] [PubMed] [Google Scholar]
- 40.Chang JY, Xia W, Shao R, et al. The tumor suppression activity of E1A in HER-2/neu-overexpressing breast cancer. Oncogene. 1997;14:561–8. doi: 10.1038/sj.onc.1200861. [DOI] [PubMed] [Google Scholar]
- 41.Yu D, Hung MC. Role of erbB2 in breast cancer chemosensitivity. Bioessays. 2000;22:673–80. doi: 10.1002/1521-1878(200007)22:7<673::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 42.Maley CC, Rustgi AK. Barrett’s esophagus and its progression to adenocarcinoma. J Natl Compr Canc Netw. 2006;4:367–74. doi: 10.6004/jnccn.2006.0031. [DOI] [PubMed] [Google Scholar]
- 43.Buttar NS, Wang KK. Mechanisms of disease: carcinogenesis in Barrett’s esophagus. Nat Clin Pract Gastroenterol Hepatol. 2004;1:106–12. doi: 10.1038/ncpgasthep0057. [DOI] [PubMed] [Google Scholar]
- 44.Nishijima K, Miwa K, Miyashita T, et al. Impact of the biliary diversion procedure on carcinogenesis in Barrett’s esophagus surgically induced by duodenoesophageal reflux in rats. Ann Surg. 2004;240:57–67. doi: 10.1097/01.sla.0000130850.31178.8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinov M, Fischer B, Arcaro A. Targeting mTOR signaling in lung cancer. Crit Rev Oncol Hematol. 2007;63:172–82. doi: 10.1016/j.critrevonc.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Chan S, Scheulen ME, Johnston S, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–22. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 47.Garcia JA, Rini BI. Recent progress in the management of advanced renal cell carcinoma. CA Cancer J Clin. 2007;57:112–25. doi: 10.3322/canjclin.57.2.112. [DOI] [PubMed] [Google Scholar]
- 48.Costa LJ. Aspects of mTOR biology and the use of mTOR inhibitors in non-Hodgkin’s lymphoma. Cancer Treat Rev. 2007;33:78–84. doi: 10.1016/j.ctrv.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Margolin K, Longmate J, Baratta T, et al. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–8. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 50.Pierce JW, Schoenleber R, Jesmok G, et al. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]