

Abstract
Vaccination for autoimmune and alloimmune diseases has long been an attractive idea. Yet, there is no suitable adjuvant to forcefully steer the immune response toward tolerance. In this study we show that dexamethasone, a potent glucocorticoid immunosuppressant, can function as a tolerogenic adjuvant when applied together with peptide immunogen. BALB/c mice with pre-established delayed-type hypersensitivity to hen OVA were immunized with an OVA-derived, MHC II-restricted peptide (OVA323–339) in the presence of dexamethasone. The treatment caused long-term desensitization in treated animals to hen OVA via a dexamethasone-dependent tolerogenic mechanism that blocks maturation of dendritic cells and expands OVA323–339-specific CD4+CD25+Foxp3+ regulatory T cells in vivo. Similar treatment of NOD mice using dexamethasone and an insulin-derived, MHC II-restricted peptide (B:9–23) prevented predisposed spontaneous diabetes. Remarkably, in both models, dexamethasone-augmented immunization induced long-term persistent, Ag-specific regulatory T cells responsive to recall Ags. These results reveal for the first time the potential usefulness of immunosuppressants as tolerogenic adjuvants.
Immunosuppressive drugs have long been used for treating allergy, autoimmune disease, and transplant rejection (1). Although effective, they only temporally dampen but do not permanently modify the immune responses toward pathogenic recall Ags and thus are not a cure. However, these drugs are generally anti-inflammatory, a condition that has been hypothesized to favor immune tolerance (2). To explore this drug-induced condition for the induction of Ag-specific tolerance, we have devised a method of “suppressed immunization” where a glucocorticoid immunosuppressant, dexamethasone (DEX),4 is administered together with a peptide immunogen during the course of active immunization. In this report we show that DEX indeed induces tolerogenic responses to active immunization that are associated with preferential expansion of Ag-specific regulatory T cells (Treg) directly in vivo.
Materials and Methods
Mice and reagents
DO11.10 TCR-transgenic BALB/c mice were bred in the University of Illinois College of Medicine at Rockford (Rockford, IL) animal facility. Other mice were from The Jackson Laboratory. All animals were maintained in pathogen-free rooms and used in accordance with the institutional guidelines for animal care. The OVA323–339 (ISQAVHAAHAEINEAGR) and B:9–23 (SHLVEALYLVCGERG) peptides were from AnaSpec. DEX, hen OVA (grade VII), and hen lysozyme were from Sigma-Aldrich. IFA was from Pierce Biotechnology. All Abs were from eBioscience. The mouse CD4+ T cell negative selection kit was from Miltenyi Biotec. CFSE was from Invitrogen.
Ag sensitization
OVA in IFA was injected subcutaneously into BALB/c mice (100 μg/mouse) twice in a 2-wk interval. The mice were also similarly sensitized for hen lysozyme. Delayed-type hypersensitivity (DTH) to either sensitizing Ag was determined by rechallenge at a footpad (10 μg/injection) and measuring the net increase in footpad thickness (swelling) at 24 h.
Suppressed immunization
Mice were injected four times (on days 1, 4, 7, and 10) with DEX in the two hind footpads (8 μg/footpad). For the day-7 injection, OVA323–339 (1 μg/footpad) was coinjected with DEX. This regimen was given twice in a 2-wk interval.
Analysis of Treg in the blood
White cells from tail blood were blocked with anti-CD16/32 mAb, immunostained for CD4 and CD25, fixed (1% paraformaldehyde) and permeabilized (0.5% Tween 20), and intracellularly immunostained for Foxp3. CD4+CD25+Foxp3+ Treg were counted relative to total CD4+ cells by flow cytometry. To determine the Ag specificity of blood Treg, white cells were labeled with CFSE (Invitrogen) and stimulated in a 96-well plate with the immunizing peptide (10 μg/ml) or an irrelevant peptide as control in the presence of syngeneic accessory cells (2.5 × 104/well) and IL-2 (800 IU/ml). Three days later, cells were stained for CD4, CD25, and Foxp3 as described above and analyzed for CFSE dilution by flow cytometry, gating on Treg.
Analysis of dendritic cells (DC) and T cells in draining lymph nodes (LN)
Total cellular content of popliteal LN was blocked with anti-CD16/32 mAb, immunostained with anti-CD11c, anti-CD83, and anti-CD86 mAbs (for DC analysis) or with anti-CD4, anti-CD25, and anti-Foxp3 mAbs (for T cell analysis), and analyzed by flow cytometry. To detect IL-10+ DC, LN cells were stained with anti-CD11c, fixed (1% paraformaldehyde), permeabilized (0.5% Tween 20), and intracellularly immunostained for IL-10.
In vivo proliferation assay
CFSE-labeled DO11.10 TCR-transgenic CD4+ T cells (2.5 ∼ 5 × 105/footpad) were coinjected with OVA323–339 (1 μg) into a footpad of BALB/c mice that had been pretreated with DEX on days −6, −3, and 0. Draining LN were recovered on day 4; total cellular content was immunostained with KJ1–26, anti-CD25, and anti-Foxp3 mAbs and analyzed by flow cytometry.
In vitro suppression assay
The assay was performed as previously described (3).
Treatment of NOD mice
Female NOD mice (6 wk of age) were injected four times (on days 1, 4, 7, and 10) with DEX in the two hind footpads (8 μg/footpad). For the day-7 injection, B:9–23 (1 μg/footpad) was coinjected with DEX. This regimen was given twice in a 2-wk interval. Glycosuria was checked weekly using Diastix strips (Bayer). Mice tested positive (≥250 mg/dl) twice consecutively were deemed diabetic.
Analysis of Treg in the spleen
Mice were rechallenged (day 0) at a footpad with the immunizing peptide emulsified in IFA (50 μg/footpad). On day 5, splenic CD4+ T cells were isolated, labeled with CFSE, and stimulated in a 96-well plate with the immunizing peptide (10 μg/ml) or an irrelevant peptide (as control) in the presence of naive syngeneic accessory cells (2.5 × 104/well) and IL-2 (800 IU/ml). On day 8, cells were stained for CD4, CD25, and Foxp3 and analyzed for CFSE dilution by flow cytometry, gating on CD4+CD25+ cells.
Statistic analysis
Two-sided Student's t test was used except for analysis of diabetic incidence, where the Kaplan-Meier method and log-rank test were applied. Differences are deemed significant if p < 0.05.
Results and Discussion
To test the idea of suppressed immunization, we first sensitized BABL/c mice for hen OVA. Using the resulting DTH to OVA as a model for recall response, we next tested suppressed immunization in these mice using an OVA-derived, MHC II-restricted peptide (OVA323–339) (4) as immunogen and DEX as adjuvant (test group). Some of the DTH-positive mice were control immunized with either or both the immunosuppressant and the peptide replaced by PBS (control groups).
Two weeks after the completion of the immunization, all groups were retested for DTH. Although the control groups remained fully sensitive to the recall Ag, the test group was markedly desensitized to OVA (Fig. 1A). The desensitization was OVA specific because the test group showed normal DTH to an irrelevant protein, hen lysozyme (data not shown). Importantly, the test group remained hyporesponsive to OVA when rechallenged 4–5 mo later (Fig. 1B), suggesting the establishment of long-term tolerance to the Ag. Consistent with such tolerance, a blood sample taken from the test group at 48 h following the Ag rechallenge showed an elevated count of CD4+CD25+Foxp3+ Treg compared with that of the non-treated control group (Fig. 1C). At least part of the increased Treg count consisted of OVA323–339-specific Treg, because they proliferated in culture in response to OVA323–339 (Fig. 1D) but not to an irrelevant peptide (data not shown). In comparison, no such Treg were detected in the blood of the non-treated control group (Fig. 1D).
FIGURE 1.
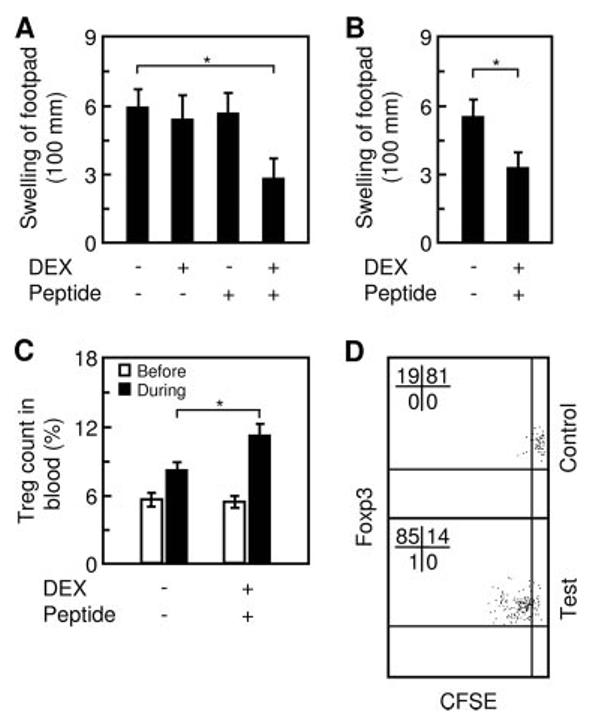
Suppressed immunization induces immune tolerance against established DTH. A, BALB/c mice with pre-established DTH were divided into four groups and each was treated with an indicated combination of DEX and OVA323–339. All groups were then retested for DTH at a footpad. B, The test and negative control groups were tested again 4 –5 mo later. C, Treg (CD4+CD25+Foxp3+) in blood samples, taken immediately before (open bar) and 48 h after (filled bar) the second DTH test, were quantified relatively to total CD4+ cells. D, The same blood taken at 48 h from non-treated mice (indicated as “Control”) or mice treated with DEX and peptide (“Test”) was further analyzed for the presence of OVA323–339-specific Treg. White cells were labeled with CFSE, stimulated in culture with OVA323–339, and analyzed for CFSE dilution (cell division) by flow cytometry, gating on CD4+CD25+Foxp3+ cells (Treg). The observed cell division was Ag dependent because no CFSE dilution was seen in the absence of OVA323–339 (data not shown). Bar, mean and SD of two or three independent experiments, each using six mice per group (n & 6); *, p < 0.05 between the indicated pair.
Treg expansion is known to be linked to the function of immature DC (5, 6) and DEX was previously reported to prevent DC maturation in vitro (7, 8), suggesting that DEX might help expand Treg by increasing immature DC. We therefore analyzed the change in the ratio of mature to immature DC in the draining LN (popliteal) following footpad injection of DEX and OVA323–339. CD83 is a maturation marker for DC (9, 10). Using CD83 in conjunction with CD86, a costimulator up-regulated in DC upon maturation, we were able to identify immature and mature DC as CD11c+CD83−CD86low and CD11c+CD83+CD86high cells, respectively. Initial experiments in BALB/c mice showed that DEX blocked DC maturation in the draining LN (data not shown). However, it was difficult to detect corresponding changes in Ag-specific Treg, as they represented only a tiny fraction of total Treg in the LN.
Consequently, to allow simultaneous observation of DC and Ag-specific Treg, we switched to DO11.10 TCR-transgenic mice, where OVA323–339-specific CD4+ T cells are abundant (4). Injecting the mice with DEX alone increased immature DC count while decreasing mature DC count, indicating that DEX blocks DC maturation in vivo (Fig. 2A). Injecting the mice with OVA323–339 alone, however, stimulated DC maturation, as was indicated by an increase in the mature DC count and a decrease in the immature DC count. Coinjection of both DEX and OVA323–339 led to an effect similar to that of DEX alone, indicating the dominance by DEX. Additional experiments showed that DEX augmented the differentiation of IL-10+ DC in the presence of OVA323–339 (Fig. 2B), suggesting that DEX not only blocks DC maturation but also alters DC function. In parallel to the immature DC count, total Treg count was also increased in a DEX-dependent manner (Fig. 2C). Importantly, DEX and OVA323–339 combined gave rise to more Treg than did DEX alone, suggesting Ag-specific expansion of Treg even in the presence of DEX.
FIGURE 2.
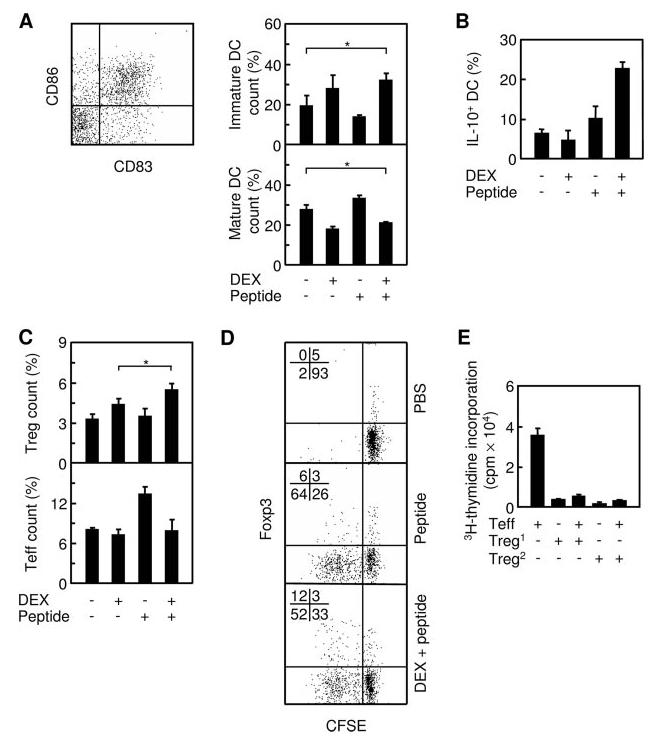
Suppressed immunization blocks DC maturation and preferentially expands Ag-specific Treg. A, DO11.10 mice were divided into four groups, each injected at a hind footpad with the indicated combination of DEX and OVA323–339. Three days later, draining LN (popliteal) were recovered and analyzed by flow cytometry, gating on CD11c+ cells (total DC). Immature (CD83−CD86low) and mature (CD83+CD86high) DC were quantified relatively to total DC. B, IL-10-producing (IL-10+) DCs were quantified relatively to total DC. C, Activated Teff (CD4+CD25+Foxp3−) and Treg (CD4+CD25+Foxp3+) in same LN were quantified relatively to total CD4+ T cells. D, CFSE-labeled DO11.10 CD4+ T cells were coinjected with OVA323–339 into BALB/c mice that had been either pretreated (indicated as “DEX + peptide”) or non-pretreated (“peptide”) with DEX. As a negative control, the T cells were also injected without OVA323–339 into non-pretreated mice (“PBS”). Four days later, total cells from draining LN were stained for KJ1–26, CD25, and Foxp3 and analyzed by flow cytometry, gating on KJ1–26+/CD25+ cells. Proliferating Treg are shown in the upper (Foxp3+) left quadrant and proliferating Teff in the lower (Foxp3−) left quadrant. E, Treg from DO11.10 mice treated with DEX and OVA323–339 were cocultured with Teff (CD4+CD25−) from naive DO11.10 mice, along with syngeneic accessory cells and OVA323–339. Proliferation was assessed by [3H]thymidine incorporation. Treg1 and Treg2 denote Treg from immunized and nonimmunized DO11.10 mice, respectively. Bar, mean and SD from 2–4 independent experiments, each using at least two mice per group (n ≥ 2); *, p < 0.05 between the indicated pair.
To determine whether this is the case, we isolated CD4+ T cells from DO11.10 mice, labeled them with CFSE to allow tracking of cell division (11), and adoptively transferred the cells into syngeneic BALB/c mice where they were stimulated with DEX and OVA323–339. Upon recovery and subsequent flow cytometric analysis of the donor cells, we found that both the Foxp3+ (Treg) and Foxp3− (effector T cell (Teff)) subsets (12) proliferated in the absence or the presence of DEX (Fig. 2D). However, the fraction of proliferating Treg in the presence of DEX rose to twice of that in the absence of DEX. At the same time, the presence of DEX slightly dampened the proliferation of Teff. Thus, the overall effect of combining DEX and Ag is preferential proliferation of Treg over Teff. Additional experiments showed that the Treg expanded in the presence of DEX are functional, as they effectively inhibited the proliferation of Teff in coculture (Fig. 2E). In aggregate, these data established the capability of DEX to expand Ag-specific Treg, which may at least partly account for the efficacy of suppressed immunization.
We further tested suppressed immunization in the NOD autoimmune diabetes model (13) using DEX and the insulin-derived, MHC II-restricted peptide Ag B:9–23 (14, 15). Although nontreated mice developed the disease with a median incidence of 19 wk, mice treated with either DEX or B:9–23 showed a delayed median incidence of 24.5 and 26 wk, respectively (Fig. 3A). In contrast, most mice treated with both DEX and B:9–23 remained disease-free during the entire period of the experiment (35 wk), indicating that suppressed immunization is effective for delaying or preventing the progression of autoimmune diabetes. Moreover, ∼40% of the animals (5 of 12 mice) in this group lived for 61 wk without diabetes and, upon rechallenge of these animals with B:9–23, Ag-specific Treg could be detected in the spleen, the primary site of memory T cells (Fig. 3B). These Treg showed greater sensitivity to restimulation with B:9–23, but not with control Ag, in comparison with control Treg (naive Treg isolated from nonimmunized, prediabetic mice). They also dominated the proliferative response to the recall Ag over splenic Teff (Foxp3−), suggesting that these Treg might be able to elicit adaptive response to recall stimulation.
FIGURE 3.
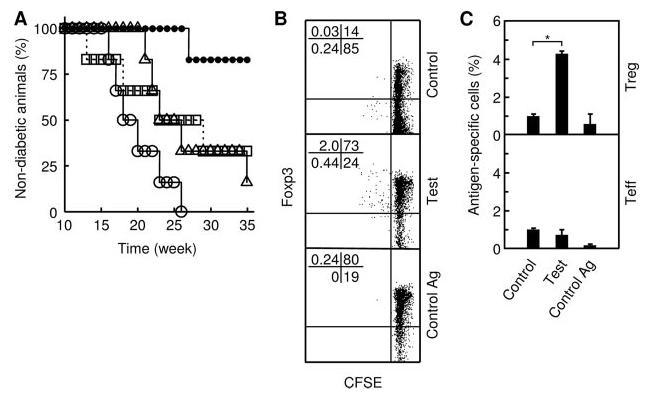
Suppressed immunization protects animals from predisposed autoimmune diseases. A, Prediabetic NOD mice (n = 6 per group) were treated with PBS (open circle), DEX alone (triangle), B:9–23 alone (square), or both DEX and B:9–23 (filled circle). Mice were checked weekly for glycosuria. Shown is one of two independent experiments with similar results. The difference between the “DEX and B:9–23” and “DEX alone” or “B:9–23 alone” groups is statistically significant (p < 0.002). B, Mice that were treated with DEX and B:9–23 and subsequently remained diabetes-free at 61 wk of age were rechallenged with B:9–23. Splenic CD4+ T cells were isolated 5 day later, labeled with CFSE, and stimulated in culture with B:9–23 (indicated as “Test”) or OVA323–339 (“Control Ag”). Control splenic T cells were obtained from nonimmunized prediabetic NOD mice that had been similarly rechallenged with B:9–23. The control cells were stimulated in culture with B:9–23 (“Control”). CFSE dilution was analyzed 3 days later by flow cytometry, gating on CD4+CD25+ cells (Treg and activated Teff). Shown are quadrant plots separating Treg (Foxp3+), Teff (Foxp3−), Ag-specific (CFSE-low), or non-antigen-specific (CFSE-high) cells. The percentage of cells in each quadrant is indicated. Shown is one of two independent experiments with similar results. C, Prediabetic NOD mice were treated with PBS (indicated as “Control”) or DEX and B:9–23 (“Test”), as described in A. At the completion of the treatment, blood samples were taken from both groups. White cells were labeled with CFSE and stimulated with B:9–23 in culture. As control for Ag specificity, an aliquot of white cells from the test mice were stimulated with OVA323–339 (“Control Ag”). Cells were analyzed by flow cytometry as described in B. Ag-specific cells were identified by CFSE dilution and quantified relatively to total CD4+CD25+ cells gated. Bar, mean and SD of two independent experiments using two or three mice per group; *, p < 0.0016 between the indicated pair.
To ascertain that suppressed immunization can preferentially prime Ag-specific Treg during the immunization phase and is therefore responsible for the generation of the “memory-like” Treg, we monitored the appearance of Ag-specific Treg and Teff in two different groups of prediabetic NOD mice that had just been treated with either DEX and B:9–23 (test) or PBS (control). At the completion of the treatment, peripheral blood was taken from the animals, and white cells were restimulated in culture with B:9–23. Compared with control group, the test group showed a significant increase in Ag-specific Treg, but not in Teff, that proliferated specifically to B:9–23 in culture (Fig. 3C), confirming that Ag-specific Treg are preferentially expanded during the immunization phase. Collectively, these results suggest that suppressed immunization may stop the progression of autoimmune diseases by preferential priming of Ag-specific Treg and provide long-term protection by generating long-term persistent Treg.
We have shown in this study that DEX can function as a tolerogenic adjuvant when applied in conjunction with a peptide immunogen. The particular effect of DEX on Treg expansion is consistent with prior findings by Chen et al. that DEX alone could alter the ratio of CD4+CD25− Treg to CD4+CD25− conventional T cells in vivo in favor of the former (16) and that DEX could also amplify IL-2-dependent expansion of functional CD4+CD25+ Foxp3+ Treg in vivo (17). The novel finding from the present study is that when combined with antigenic immunization, the use of DEX can lead to preferential induction of Treg that are Ag-specific and long-term persistent. Furthermore, based on the mechanism revealed here, one may speculate that DEX, or glucocorticoids in general, may not be unique in its ability to serve as a tolerogenic adjuvant. Other small-molecule immunosuppressive drugs, such as vitamin D3 and analogues, cyclosporine A, FK506, and rapamycin, have been shown to have similar pharmacological effects on DC maturation and function (18) and might therefore also be useful for tolerogenic immunization. Lastly, Van Overtvelt et al. recently reported their search for IL-10-inducing adjuvants and the observation that a combination of 1,25-dihydroxyvitamin D3 and dexamethasone augmented the efficacy of sublingual immunotherapy in a murine asthma model in association with the induction of CD4+CD25+Foxp3+ Treg (19). Both their work and ours support the notion of tolerogenic adjuvants.
Acknowledgments
We thank John Javaherian for providing care for the animals and Dr. M. Nawal Lutfiyya for assistance with statistics.
Footnotes
This work was supported by National Institutes of Health Grant RO1CA92243 (to A.C.) and by a grant from the Illinois Department of Public Health (to G. Z.). Y.K. was a recipient of a scholarship from the China Scholarship Council.
Abbreviations used in this paper: DEX, dexamethasone; DC, dendritic cell; DTH, delayed-type hypersensitivity; LN, lymph node; Teff, effector T cell; Treg, regulatory T cell.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Allison AC. Immunosuppressive drugs: the first 50 years and a glance forward. Immunopharmacology. 2000;47:63–83. doi: 10.1016/s0162-3109(00)00186-7. [DOI] [PubMed] [Google Scholar]
- 2.Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 3.Zheng G, Wang B, Chen A. The 4–1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J. Immunol. 2004;173:2428–2434. doi: 10.4049/jimmunol.173.4.2428. [DOI] [PubMed] [Google Scholar]
- 4.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 5.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J. Exp. Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, Socci C, Di Carlo V. Glucocorticoids affect human dendritic cell differentiation and maturation. J. Immunol. 1999;162:6473–6481. [PubMed] [Google Scholar]
- 8.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur. J. Immunol. 2000;30:1233–1242. doi: 10.1002/(SICI)1521-4141(200004)30:4<1233::AID-IMMU1233>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 9.Lechmann M, Berchtold S, Hauber J, Steinkasserer A. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol. 2002;23:273–275. doi: 10.1016/s1471-4906(02)02214-7. [DOI] [PubMed] [Google Scholar]
- 10.Prechtel AT, Steinkasserer A. CD83: an update on functions and prospects of the maturation marker of dendritic cells. Arch. Dermatol. Res. 2007;299:59–69. doi: 10.1007/s00403-007-0743-z. [DOI] [PubMed] [Google Scholar]
- 11.Wells AD, Gudmundsdottir H, Turka LA. Following the fate of individual T cells throughout activation and clonal expansion. Signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J. Clin. Invest. 1997;100:3173–3183. doi: 10.1172/JCI119873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziegler SF. FOXP3: of mice and men. Annu. Rev. Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 13.Wicker LS, Todd JA, Peterson LB. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 1995;13:179–200. doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- 14.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur. J. Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 15.Jasinski JM, Yu L, Nakayama M, Li MM, Lipes MA, Eisenbarth GS, Liu E. Transgenic insulin (B:9–23) T-cell receptor mice develop autoimmune diabetes dependent upon RAG genotype, H-2g7 homozygosity, and insulin 2 gene knockout. Diabetes. 2006;55:1978–1984. doi: 10.2337/db06-0058. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Murakami T, Oppenheim JJ, Howard OM. Differential response of murine CD4+CD25+ and CD4+CD25− T cells to dexamethasone-induced cell death. Eur. J. Immunol. 2004;34:859–869. doi: 10.1002/eji.200324506. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, Howard OM. Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3+CD4+CD25+ T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur. J. Immunol. 2006;36:2139–2149. doi: 10.1002/eji.200635873. [DOI] [PubMed] [Google Scholar]
- 18.Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 2004;4:24–34. doi: 10.1038/nri1256. [DOI] [PubMed] [Google Scholar]
- 19.Van Overtvelt L, Lombardi V, Razafindratsita A, Saint-Lu N, Horiot S, Moussu H, Mascarell L, Moingeon P. IL-10-inducing adjuvants enhance sublingual immunotherapy efficacy in a murine asthma model. Int. Arch. Allergy Immunol. 2008;145:152–162. doi: 10.1159/000108140. [DOI] [PubMed] [Google Scholar]