


Abstract
A number of previous studies have predicted transcription factor binding sites (TFBSs) by exploiting the position of genomic landmarks like the transcriptional start site (TSS). The studies’ methods are generally too computationally intensive for genome-scale investigation, so the full potential of ‘positional regulomics’ to discover TFBSs and determine their function remains unknown. Because databases often annotate the genomic landmarks in DNA sequences, the methodical exploitation of positional regulomics has become increasingly urgent. Accordingly, we examined a set of 7914 human putative promoter regions (PPRs) with a known TSS. Our methods identified 1226 eight-letter DNA words with significant positional preferences with respect to the TSS, of which only 608 of the 1226 words matched known TFBSs. Many groups of genes whose PPRs contained a common word displayed similar expression profiles and related biological functions, however. Most interestingly, our results included 78 words, each of which clustered significantly in two or three different positions relative to the TSS. Often, the gene groups corresponding to different positional clusters of the same word corresponded to diverse functions, e.g. activation or repression in different tissues. Thus, different clusters of the same word likely reflect the phenomenon of ‘positional regulation’, i.e. a word's regulatory function can vary with its position relative to a genomic landmark, a conclusion inaccessible to methods based purely on sequence. Further integrative analysis of words co-occurring in PPRs also yielded 24 different groups of genes, likely identifying cis-regulatory modules de novo. Whereas comparative genomics requires precise sequence alignments, positional regulomics exploits genomic landmarks to provide a ‘poor man's alignment’. By exploiting the phenomenon of positional regulation, it uses position to differentiate the biological functions of subsets of TFBSs sharing a common sequence motif.
INTRODUCTION
In the postgenomic era, the identification of signals regulating transcription remains an outstanding problem (1,2). The problem has frustrated standard methods in computational sequence analysis, and experiments still provide one of the few consistently reliable sources of information about transcriptional signals (3). Even simple cis-regulatory transcription-binding sites (TFBSs) have proved notoriously difficult to identify de novo, because they usually correspond to short, degenerate motifs whose sequence information is insufficient on its own for dependable predictions. In particular, sequence analysis alone is generally unable to address the information that higher-order chromatin structure contributes to gene regulation (4).
Consider, however, a transcriptional complex anchored on a transcription start site (TSS). Each transcription factor (TF) within the complex occupies a particular position. Thus, if a TF interacts with a TFBS, the TFBS probably is constrained positionally with respect to the TSS. Moreover, as classic experiments on the lambda repressor and its operator-binding sites showed, by occupying TFBSs in different positions, a single TF can assume different biological functions (5). Rather like a receptor antagonist occupying a binding site, a TFBS corresponding to the TF might activate in one position relative to the TSS, but repress in another. Because of position, therefore, a single TFBS motif might regulate gene expression in a tissue- or temporal stage-specific manner (or both). Positional regulation of function generalizes obviously and broadly, to regulatory elements and genomic landmarks other than TFBSs and TSSs.
In the presence of positional regulation, sequence alone would be insufficient to predict TFBS function. Fortunately, many modern databases annotate their sequences. Consequently, where the traditional conference slide in computational biology once displayed an endless sea of letters, it should now display letters punctuated regularly by genomic landmarks like the TSS, exon boundaries, etc. Presently, genomic investigations are not exploiting the position of annotated landmarks as much as they might.
Positional regulomics therefore holds promise, but it requires in hand a rich source of interesting regulatory positions. With regard to TFBSs, some computational studies have examined position (6–10), but few new putative motifs emerged. In contrast, our previous work discovered 791 eight-letter DNA words displaying positional preferences with respect to the TSS (11). To summarize the work, the Database of Transcription Start Sites (DBTSS) contained many human TSSs determined from oligo-capping experiments (12–14). False positive TSSs were eliminated by precise transcript mapping, yielding a database of 4737 putative promoter regions (PPRs) containing positions −2000 to +1000 bp relative to the corresponding TSS (15). For each of the 48 eight-letter DNA words, a local maximum statistic (similar to the BLAST statistic) assessed the word's positional preferences with respect to the TSS (11). After multiplying by 48 to correct for multiple testing, the analysis yielded 791 statistically significant words (P ≤ 0.05). Of the 791 words, 388 had perfect matches in TRANSFAC database (16), an event with a P-value of 4 × 10−42. The biological function of the other 413 of the 791 words remained unidentified, but suggested the potential of positional regulomics to discover unknown sequence elements and their function.
To give an overview of the present study, with recent TSS data (17), the PPR dataset now contains 7914 sequences (see the Methods section). Within the new PPR dataset, the local maximum statistic identified words w displaying positional preferences with respect to the TSS. (To avoid unnecessary repetition of the phrase ‘with respect to the TSS’, all bp coordinates and positions below refer implicitly to the corresponding TSS, unless stated otherwise.) Each statistically significant positional preference yielded a ‘cluster’ of positions containing the corresponding word w, and each of the clusters corresponded to a group of genes (‘gene group’). Occasionally, a single word w corresponded to more than one cluster, hinting at the possibility of a TFBS under positional regulation, and rendering such words particularly interesting to us.
Two external sources of information implicated the positional clusters in the co-regulation of the corresponding gene group. First, quantitative functional relationships were determined using a semantic similarity method (18) based on the Gene Ontology (GO) annotation. The functional analysis suggested that many individual gene groups had a common biological function. Second, the microarray experiments in the GNF Atlas 2 (19) suggested that many individual gene groups identified here were co-expressed across multiple tissues. In addition to validating the biological functionality of words and helping to classify the corresponding putative TFBS, the two sources of information permitted us to formulate some novel biological hypotheses. In accord with the notion of positional regulation, our analysis sometimes linked different tissues to specific positions of a word, to our knowledge yielding the first computational evidence that a TFBS's position can influence the tissue-specificity of its regulatory functions. Furthermore, in accord with the analogy to receptor antagonists, our analysis sometimes linked different levels of activation or repression of the same gene group in different tissues to specific positions of a word. Thus, it is not an isolated phenomenon in human gene regulation, that the position of a TFBS influences its function in a regulatory module.
METHODS
The PPR database
Recently, (17) determined new TSSs with about 1.8 million 5′-end clones of full-length human cDNAs, extending the DBTSS. DBTSS yielded 30 924 TSSs for 14 628 RefSeq (20) human genes, indicating that many genes have alternative TSSs. The PPR database was constructed using every TSS within ±1000 bp of the start of an annotated RefSeq transcript for annotated genes. If several TSSs were within ±1000 bp of the same RefSeq 5′ end, the closest TSS was used. The corresponding PPRs in DBTSS were aligned to the human genome (NCBI, build 36). Each PPR that mapped unambiguously was extended to include from −2000 to +1000 bp relative to the TSS (which was at 0 bp), as in our previous study (11). The final PPR database contained 7914 sequences. An ungapped block alignment then anchored the PPRs, placing all TSSs in a single column. Supplementary Figure S1 shows systematic variations in base composition over the alignment columns, confirming that the anchored alignment generally placed the TSSs correctly.
Our previous study describes in detail the remaining procedures, applied to every one of the 48 eight-letter DNA words. For each word and for each PPR, one instance of the word was chosen uniformly at random, and the remaining instances masked. At the end of the masking procedure, each PPR contained at most one unmasked instance of the word, in a random position. The unmasked instances in each column of the block alignment were counted, and a local maximum statistic (similar to the gapless BLAST statistic) assessed whether the unmasked instances of the word were unusually clustered by columns within the block alignment (see Supplementary data—Section 1.1). The randomized masking step reduces the density of ubiquitous repetitive elements or low complexity regions (e.g. poly A, poly T), which are biologically uninteresting in the present context but which tend to be statistically significant without masking. Our study examined clusters with a significant local maximum statistic, a ‘cluster’ being simply a statistically significant set of positions within certain PPRs.
Pairwise correlation coefficient for microarray data
Given the set of n = 74 tissue-specific microarray expression values (Xi,Yi) for two genes g1 and g2, the corresponding Pearson correlation coefficient is
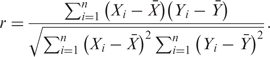 |
1 |
Pairwise correlation coefficient for significant words
Each significant word W provided a pairwise similarity corresponding to TFs in TRANSFAC (16), as follows.
We used 522 count matrices from TRANSFAC Professional 11.1, many of which represent the same or similar factors. To make the set nonredundant, we skipped all nonvertebrate matrices, and if a family of related factors shared a single matrix, the matrix appeared once, to represent the entire family. For each of the 145 nonredundant count matrices remaining, the standard log-likelihood ratio yielded a PSSM as follows: Let pn represent the background probability of nucleotide n ∈ {a, c, g, t} in the 7914 PPRs. Let cn,k represent the count of nucleotide n in column k. Then, the score for nucleotide i at column k is
| 2 |
where c = Σn∈ {a,c,g,t} cn,k is the total number of counts, which is independent of the column k (being the total number of TFBSs in TRANSFAC corresponding to the TF in question,); an is the pseudo-count, which regularizes count matrices based only on a few TFBSs; and α = Σn∈ {a,c,g,t} αn. As in previous studies (11), we took an = 1.5 × pn.
With the 145 nonredundant PSSMs in hand, we calculated match scores for each word W and PSSM M, as follows: Each PSSM was padded with eight columns of 0s on each side. As above, let sn,k denote the score for of nucleotide n in column k, where k = −8,.., − 1, 0,.., j − 1, j,.., j + 7, the columns k = 0, …, j −1 being from the original PSSM. The word W = W(0), …,W(7) receives a maximum score
| 3 |
The summed score on the right of Equation (3) can be related to the binding energy of the TF for the putative TFBSs (21). The maximum score SM,W is the best summed score that the word W receives in any offset against PSSM M.
With the maximum scores SM,W in hand, we calculated empirical P-values for each word W from our significant clusters, as follows. For each PSSM M, all eight-letter words yielded 65 536 maximum scores SM,W against the PSSM. For any word W, consider the corresponding maximum score SM,W. The empirical P-value pM,W for the sequence W against the PSSM M is the fraction of the 8-mers that have a maximum score higher than SM,W. The complement 1 − PM,W of the P-value then should increase with the binding energy for the word W and the TF generating the PSSM M. The complement 1 − PM,W is also normalized between 0 and 1.
Now, let i = 1, …,145 index the nonredundant PSSMs M; and let g = 1, …,3589 index the genes in our dataset. If the words W1, …,Ww correspond to the gene with index g, define Tg,i = maxw= 1, …,w (1 − Pi,w) (i = 1, …,145) if w > 0 and 0 otherwise.
In the table {Tg,i}, the rows represent the genes; the columns, TFs. When Equation (1) is applied to Xi = Tg1, i and Yi = Tg2, i, which correspond to the genes g1 and g2, it yields the Pearson correlation coefficients (PCCs) between rows in the table {Tg,i}. Two genes therefore receive a high PCC, if they correspond to similar words, regardless of the words’ positions. The resulting network still reflects putative TFBSs as predicted by positional preference, however.
The integration of positional, functional and co-expression data
Three networks were constructed using positional, functional and co-expression data. In the corresponding networks, an edge joined a gene pair, if the pair scored above the 95th percentile for the corresponding measure: (i) 0.566 for the Pearson correlation coefficient quantifying TF positional similarity; (ii) 0.588 for the GO semantic similarity or (iii) 0.546 for the PCC from the microarray Atlas data. The networks were analyzed using Cytoscape (22), software freely available from http://www.cytoscape.org for visualizing molecular interaction networks. The Graph Merge plug-in, also freely available from http://www.cytoscape.org, produced the intersection network (see Supplementary Figure S3) whose edges lie in all three networks. Supplementary Table 2 lists numbers of nodes and edges, and average degrees for each of the four networks. The MCODE Cytoscape plug-in (23) identified 24 densely connected sets of genes in the intersection network.
RESULTS
Many words displaying positional preferences are probably functional
After multiplying P-values by 48 to correct for multiple testing, our methods yielded 1226 eight-letter words with significant positional preferences (P ≤ 0.05). Out of the 1226 words, 71 words corresponded to two significant clusters with distinct positions and seven words corresponded to three significant clusters with distinct positions, for a total of 1311 significant clusters. (To avoid unnecessary repetition, all the ‘words’, ‘clusters’, and ‘gene groups’ mentioned below are significant at P ≤ 0.05 after multiple test corrections, unless stated otherwise.) Supplementary data file 1 contains the words and their clusters. To identify similar or overlapping words, we varied one base within each word, but few words were similar or overlapped. Only 608 of the 1226 words exactly matched subsequences of experimentally determined TFBSs in the TRANSFAC database. To discover relationships between the words and basal promoter elements like the CAAT box, SP1, CREB and TATA box (recognized by the constitutive human factors NF-Y, SP1, CREB and TBP, respectively), we again varied one base within each word. With a change of at most one base, 540 of the 1226 words exactly matched a consensus subsequence of one of the basal promoter elements. Because the regions surrounding many human genes are GC-rich, we examined the sequence composition of the words. Within the 1226 words, the frequencies of A, C, G and T were 0.159, 0.318, 0.376 and 0.146, respectively. Moreover, 123 words (≈10%) contained only G and C, but only eight words (≈0.65%) contained only A and T. Thus, the words do indeed reflect the elevated GC content around the TSS (Supplementary Figure S1).
Our previous study only found TFBSs from −200 bp to +100 bp relative to the TSS at 0 bp. Moreover, to permit a genome-scale study, our methods here ruthlessly sacrificed statistical power in favor of computational speed, so they probably found a small fraction of all functional TFBSs (which our unpublished data estimates loosely at a site-level sensitivity of about 15%). As expected, clusters upstream of the TSS were all within −200 bp relative to the TSS, indicating that our methods do not find TFBSs distant from the TSS. Clusters downstream of the TSS usually occurred within +100 bp. Cluster density peaked roughly at the TSS. Some 44 clusters were positioned more than +100 bp downstream of the TSS. A consensus GT dinucleotide appeared in 22 of the corresponding words, suggesting their role in mRNA splicing.
Many clusters correspond to gene groups with a common function
We investigated the (significant) gene groups for common functions, analyzing annotations from the GO database (24). Although several tools for analyzing GO annotations are publicly available (25–27), none was entirely suitable for our study, so we developed other methods ourselves.
Accordingly, we used semantic similarity measures to quantify the commonalities of molecular function for each pair of the 15 536 Homo sapiens gene products with GO annotations (18) (see Supplementary data—Section 1.3). The semantic measures yielded a maximum average pairwise functional similarity (APFS) within each gene group. Similarly, we calculated an APFS for 106 random gene groups, each random group chosen uniformly from the PPR dataset to match the size of the original gene group. The fraction of random groups with a larger APFS than the original group yielded an empirical p-value for the original group's APFS. Of the 1311 clusters, 502 had a significant APFS [P ≤ 0.05; false-discovery rate (FDR) = 5.3%] (see Figure 1).
Figure 1.

Empirical P-values of clusters estimated from simulation. The figure plots the count of clusters whose empirical P-value did not exceed a particular threshold against the P-value threshold. The empirical P-values were estimated from microarray data (closed triangles) and GO-derived functional similarity data (open triangles). The empirical P-values of nonconserved clusters are shown separately for the microarray data (closed circles) and GO-derived functional similarity data (open circles).
Many clusters correspond to co-expressed gene groups
If a (statistically significant) cluster represents TFBS instances with a common function, the corresponding gene group might be co-expressed. Accordingly, we analyzed expression patterns in microarray experiments from the GNF Atlas 2 (19). The microarray Atlas facilitated the generation of a cross-table, where the rows correspond to 7914 genes downstream of the PPRs and the columns to normalized expression values for 74 human tissues. Consider two clusters and the two corresponding gene groups. For each gene group, the cross-table yielded a pairwise Pearson correlation coefficient (PCC) for their expression values. The substitution of the PCC for the APFS in the procedure above yielded an empirical PCC P-value. Of the 1311 clusters, 529 had a statistically significant PCC (P ≤ 0.05; FDR = 2.6%) (Figure 1). As further validation of biological functionality, 273 words had both a significant APFS (functional) and a significant PCC (co-expression) similarity (P ≤ 0.05).
Having established that the gene groups tended to have common GO functions or co-expression, we then examined their tissue-specificity. In a particular tissue (column), the cross-table from the microarray Atlas implicitly ranks each gene (row) according to its expression. For each gene group, the Mann–Whitney rank sum statistic quantifies the expression enrichment for a particular tissue in the gene group relative to other genes. Among the 1311 × 74 = 97 014 gene group-tissue pairs, 1737 showed enriched expression (at P ≤ 0.001 without multiple test-correction, corresponding to a FDR 5.58%). Of the 1311 gene groups, 450 showed enrichment in at least one tissue, with 58 groups showing enrichment in more than 10 tissues. The vast majority of the gene groups (about 525 of the 1311 groups) showed enriched expression specifically in white blood cells (dendritic, NK, B and T cells), generally agreeing with the conclusion of a recent study on motif discovery in the human genome (28).
Recall that 273 gene groups had both common GO functions and co-expression at significant levels (P ≤ 0.05). Of the corresponding 273 words, 114 exactly matched subsequences of known human TFBSs in TRANSFAC. Additionally, experimental evidence linked 44 of the TF motifs in TRANSFAC matching positionally significant words to one or more of the tissues showing enrichment of the matched word's gene group. Table 1 lists the predicted tissue of enriched expression and the TRANSFAC TF for a randomly selected subset of these 44 words. Supplementary Table 1 in the additional files gives the complete information for all 44 words.
Table 1.
Tissue specificity of DNA words and their association with known transcription factors
| DNA Word | Factor | Enriched Tissues | P-value (expression similarity) | P-value (GO functional similarity) |
|---|---|---|---|---|
| CCGGAAGC | Sp1, c-Ets-1, Ets-1, GABP-alpha, GABP-beta, STAT1, STAT3 | PBCD4+Tcells, PBCD8+Tcells, Prostate | 1.00E-06 | 3.24E-03 |
| CGCGATGG | Egr-1 | Adrenal gland | 1.00E-06 | 1.49E-02 |
| GCCGCCAT | YY-1 | PBCD4+Tcells, PBCD8+Tcells | 1.00E-06 | 4.30E-05 |
| GCCTGCGC | NRF1, Sp1, Sp3 | Thyroid | 1.00E-06 | 8.88E-03 |
| GGCGGGGC | Sp1, Sp3, NF-Y | Amygdala, Prostate | 1.00E-06 | 7.42E-03 |
| GGTCACGT | Sp1, Sp3, ATF-1 | PLACENTA | 1.00E-06 | 1.81E-02 |
| TTCCGCGC | E2F1, Sp3 | PBCD4+Tcells, PBCD8+Tcells, Thymus | 2.20E-02 | 2.24E-02 |
The last columns give the P-values of clusters (estimated by simulation from microarray data and GO-derived functional similarity data). The rows in the table reflect the lexicographic order of the words in column 1.
Positional preference is essential to establishing the trends described above; standard sequence analysis alone is insufficient. Two additional lines of evidence support our hypothesis linking TFBSs’ locations with tissue-specific usage. First, if a single word corresponded to two or three different positional clusters, the clusters often corresponded to gene groups expressed in strikingly different tissues. For instance, the TRANSFAC binding element for AP-2alphaA, c-Ets-2, Sp1, represented by consensus CGCCGCCG, yields two significant clusters at −17 and +14 bp, respectively. While the upstream cluster showed overexpression in fetal brain, the downstream cluster showed overexpression in BM-CD71+EarlyErythroid and Thyroid. Thus, these TFs might use position-specific binding to drive differential tissue-specific activation. Other factors exhibiting a similar phenomenon include ER-alpha, T3R-beta1, Sp1, Ets (CAGGTGAG) and Sp1, Sp3, MyoD, AP-2beta (GCGGGGCC). On the other hand, some factors might use position-specific binding to cause tissue-specific repression. Such factors include p53 (GCGGCGGG), Sp1, HIF-1, GKLF, NF-Y, CTCF (GGCGGCGC) and Sp1, Sp3, MyoD, AP-2beta (GCGGGGCC) (Table 2). Second, for each of the 273 clusters described earlier, we selected a gene group of equal size as a negative control. Each gene in the control group had a PPR containing the relevant word between −200 and +100 bp, but with no other positional restriction. As expected, the Mann–Whitney rank sum test showed that unlike the actual gene groups, the control gene groups did not display any noticeable tissue specificity (see Supplementary Figure S2a and S2b).
Table 2.
TFBS words corresponding to two clusters, and thereby displaying possible positional regulation of TF tissue specificity
| DNA Word | Factor | Distance from TSS (bp) | Tissues | Activation(+)/ Repression(−) |
|---|---|---|---|---|
| CAGGTGAG | ER-alpha, T3R-beta1, Sp1, Ets | 158,a 104b | WHOLEBLOOD,a Amygdalab | + |
| CGCCCCGC | E2F-1, AP-2alphaA, NRF-1, Egr-1 | −59,a −176b | Cardiac Myocytes,a Uterus Corpusb | − |
| CGCCGCCG | AP-2alphaA, c-Ets-2, Sp1 | 14,a −17b | BM-CD71+EarlyErythroid,a Thyroid,a fetalbrainb | + |
| GCGGCGGG | p53 | 20,a −56b | TrigeminalGanglion,a Appendixb | − |
| GCGGGGCC | Sp1, Sp3, MyoD, AP-2beta | −57,a −15b | Bronchialepithelialcells,a 721_B_lymphoblasts,b SmoothMuscleb | + |
| GGCGGCGC | Sp1, HIF-1, GKLF, NF-Y, CTCF | −39,a 19b | Ovary,a AdrenalCortex,b Appendix,b OlfactoryBulbb | − |
For simplicity, only three examples have been provided for each case (activation and repression). The rows in the table reflect the lexicographic order of the words in column 1. Each word corresponds to TFs in column 2. Each word corresponds to two clusters, whose average positions relative to the TSS are in column 3. Superscripts link the entries in columns 3 and 4, to indicate the relation between the position-specific binding and the tissue-specific regulation of each TF.
The position of a TFBS can influence its function
There were 78 words (about 6.4% of all 1226 words) with two or three different significant clusters. These 78 words presented a unique opportunity to see whether the sequence of a TFBS is sufficient to determine its biological function. The 78 words generated 92 pairs of gene groups, each pair corresponding to a single eight-letter word but to two different positional clusters. If a TFBS had diverse roles (e.g. activation and repression) in different tissues, it might yield a pair of gene groups with significantly different expression patterns across the 74 tissues in the GNF Atlas 2 microarray dataset. For each of the 92 pairs, a one-sided Mann–Whitney P-value quantified the relative expression of the two gene groups in the 74 tissues. The Fisher inverse chi-squared test (29) assessed the product of the 74 one-sided Mann–Whitney P-values, and its two-sided P-value for the product indicated the overall differences in expression between the two groups (see Supplementary data––Section 1.4). After multiplying by 92 to correct for multiple testing, seven pairs were statistically significant (P ≤ 0.05). Table 3 presents results for significant pairs of gene groups (P ≤ 0.05, after multiplying by 74 to correct for multiple testing).
Table 3.
TFBS words corresponding to two clusters, whose gene groups have significantly different microarray expression patterns
| DNA Word | Factor | Distance from TSS (bp) | P-value |
|---|---|---|---|
| CCCCGCCC | c-Myc, AP-2alphaA, E2F-1, NF-AT1, MAZ | −65, −20 | 2.24E-04 |
| CCGCCGCC | YY1, Egr-1, AP-2alphaA, Sp1, Sp3 | 63, 13 | 4.18E-02 |
| CGCCCCGC | Sp1, Sp3, E2F-1, Egr-1 | −176, 41 | 4.04E-02 |
| CGCCGCTG | Unidentified | 15, 39 | 4.66E-04 |
| CGGGCGGC | DP-1, E2F:DP, Sp1, GKLF | −15, 23 | 1.16E-07 |
| GAGGCGGC | Unknown | −16, 20 | 2.85E-02 |
| GGGCGGCG | Sp1, NF-Y, GKLF | −20, 143 | 1.15E-11 |
The rows in the table reflect the lexicographic order of the words in column 1. Each word corresponds to TFs in column 2. Each word corresponds to two clusters, whose average positions relative to the TSS are in column 3. Fisher inverse chi-squared test yielded a (multiple test corrected) two-sided P-value (in column 4), which quantifies the overall differences in expression between the gene group pair in the 74 tissues.
A comparison of results from positional and sequence-based methods
For a TFBS conserved across several species, comparative genomics uses a multiple alignment across the species to narrow the TFBS search to regions of high conservation (7,30). Positional regulomics might have at least two potential advantages over comparative genomics in identifying TFBSs. First, because positional regulomics does not require accurate sequence alignments, it can find TFBSs in poorly conserved regions. Second, it does not depend on undependable details of the background DNA sequence, thereby reducing the false positive rate of its predictions.
The first potential advantage suggests the following question. Do comparative genomics and its requirement for sequence conservation obscure TFBSs that positional regulomics might find? Let a cluster be partially or less conserved if >20% of positions in it occur in nonconserved regions within the human genome, as determined by human/mouse genome alignment (hg17/mm7 assembly) of the UCSC Genome Browser. Of the 1311 clusters, 42 clusters contained 20% or more positions in nonconserved regions; of these, 12 contained 75% or more positions in nonconserved regions. Thus, sequence conservation considerations had little influence on the 1311 clusters of positions. Out of the 42 nonconserved clusters, 26 and 29 clusters appeared significant (P < 0.05) under our analysis using expression and functional similarity data, respectively (Figure 1).
To assess the second potential advantage and to compare false positives from positional and comparative genomics, consider a recent study that identified 54 702 putative human TFBSs by aligning human, mouse, rat and dog genomes (7). The present study identified 46 670 putative TFBSs, a comparable number. The spatial distribution of transposable elements (TEs) around the TSS may be an indicator of the relative false positive rates in the two studies. TEs comprise about 45% of the human genome and might contribute a substantial fraction of regulatory elements (31,32). However, a sharp decline of TEs around the TSS (33) indicates selection against their insertion in functionally important regions like core promoters where many regulatory elements are positioned. RepeatMasker <http://www.repeatmasker.org> was used to determine TE locations using the RepBase library of repeats (34). The total TE count was 24 878, including SINEs, LINEs, LTR elements, DNA elements and other unclassified elements. Overall, the masked regions represented 23% of our dataset.
Figure 2 shows distributions of positions relative to the TSS: Figure 2A, of TE-rich regions; Figure 2B, of ‘comparative TFBSs’ [predicted in (7)]; and Figure 2C, of ‘positional TFBSs’ [predicted in the present study]. TE-rich regions overlapped with 122 comparative TFBSs but with only 50 positional TFBSs (two-sided Fisher exact P = 7.8 × 10−6). Positional TFBSs had a tight distribution from about −200 to +100 bp relative to the TSS, whereas comparative TFBSs were relatively widespread, from about −500 to +500 bp. The positional TFBSs become rare as TEs become common away from the TSS. Figure 2 suggests that the positional methods are relatively insensitive to input sequence lengths, because they predict TFBSs only near their genomic anchor, namely, the TSS in the present study. In any case, Figure 2 suggests that in the cases examined, the putative positional TFBSs contain fewer false positives than the putative comparative TFBSs.
Figure 2.

Density of regulatory and repetitive DNA sequences in human core promoters. The plot displays results for 7914 human core promoters. Its X-axis runs from −1000 bp to +1000 bp, relative to the TSS for each promoter at 0 bp. The Y-axis represents the normalized count of: (A) TE-derived sequences; (B) TFBSs predicted with our positional methods and (C) TFBSs predicted with phylogenetic footprinting. In each case, the raw counts were normalized to make the area under each graph 1. The boundaries of the three curves indicate the density of predicted sequences in the different regions. Our methods tend to predict TFBSs in the [−200, +100] region of core promoters.
Positional regulomics can identify sets of co-regulating TFBSs and co-regulated genes
TFs combine to form cis-regulatory modules (CRMs), complexes controlling gene transcription. Thus, a CRM interacts with certain TFBSs and controls certain genes. The following graphical method predicts co-regulating TFBSs and co-regulated genes, without prior knowledge of the specific TFs in the CRM. By applying the techniques of systems biology to CRMs, the method enhances the dependability and interpretability of predictions.
We assembled a dataset containing all genes with complete GO and microarray GNF Atlas 2 data and corresponding to at least one significant cluster. The resulting 3589 genes constituted nodes in each of three networks (i.e. graphs), corresponding to three sources of information: (i) the positionally significant words, (ii) the GO annotation and (iii) the microarray Atlas. A Pearson correlation coefficient quantified pairwise similarity between genes, based on the significant words occurring in their promoters (see the Methods section). In the corresponding networks, an edge joined a gene pair, if the pair scored above the 95th percentile for the corresponding measure: (i) 0.566 for the Pearson correlation coefficient quantifying TF similarity; (ii) 0.588 for the GO semantic similarity or (iii) 0.546 for the PCC from the microarray Atlas data. The 95th percentile was an arbitrary choice, because considerations of computational time precluded a thorough exploration of possible thresholds.
The three sources of information validated each other's conclusions as follows. In Figure 3A, UPGMA (unweighted pair group method with arithmetic mean) clustered the genes by GO semantic similarity; in Figure 3B, by the similarity of the set of positionally significant words contained in the corresponding promoters. The organized patterns of color in Figure 3 display the correlations between the three sources of information (GO, microarray Atlas data and positional regulomics), so the sources validated each other. Integration of positional, functional and co-expression information generated an intersection network (see the Methods section). Figure 4 shows the gene expression profiles for the most densely connected set of genes, sharing common positional, functional and co-expression properties. Some other profiles appear in Supplementary Figure S4. Therefore, positional regulomics can be combined with (and validated by) other sources of information, to identify modules of TFBSs and coregulated genes.
Figure 3.

Gene profile correlation matrices. The UPGMA method clustered genes by their GO-derived functional similarity. The matrix in Figure 3A orders the genes identically on both axes by functional similarity. Each off-diagonal element in the matrix corresponds to a pair of different genes. The color of an element codes the Pearson correlation coefficient for the co-expression of the corresponding gene-pair in the microarray data. The off-diagonal blocks of consistent color indicate that functionally similar groups of genes have similar expression patterns. For comparison, the inset in the plot shows a negative control. The inset's matrix orders the genes identically on both axes, but randomly. Accordingly, the matrix lacks off-diagonal blocks of consistent color. The matrix in Figure 3B orders the genes identically on both axes, according to the similarity of the set of positionally significant words contained in the corresponding promoters (see the Methods section). The off-diagonal blocks of consistent color indicate that positional regulomics predicted groups of genes with similar expression patterns.
Figure 4.

A highly interconnected sub-network of genes from the intersection network. The top of the figure shows the genes’ microarray co-expression matrix. Its rows correspond to genes; its columns, to tissues. The bottom of the figure shows the intersection sub-network for the genes. Clearly, the sub-network of genes shares a common expression pattern.
Algorithm and Datasets
A C++ computer program implemented the algorithm identifying significant clusters of eight-letter words in anchored promoter sequences. A UNIX-compatible version of the program with user-tunable parameters is available for download at the following URL: ftp://ftp.ncbi.nlm.nih.gov/pub/marino/published/positional_regulomics/, along with the pairwise GO functional similarities for 3589 transcripts.
DISCUSSION
Historically, the lambda repressor was the first experimental system known to us to show that position (as well as sequence) influences a TFBS's function. Using the TSS as a genomic landmark, positional regulomics provides strong statistical evidence that in human transcription, the phenomenon is not isolated: if not commonly, at least not rarely, a TFBS's position as well as its sequence can influence the strength of activation or repression of a gene. Some TFs (e.g. AP-2alphaA, ER-alpha, Sp1, Sp3, p53, NRF-1) appear to bind to different positions relative to the TSS, to regulate different genes in different tissues. Moreover, a TFBS's position appears to influence biological function, not just strength of that function. These conclusions rely on data about exact words (i.e. a single sequence pattern with no alternatives), so an analysis based on sequence alone, without position, has no obvious opportunity to draw similar conclusions.
Some experimental results for specific TFBSs support our conclusions about position. The word CCGCCGCC matches the TRANSFAC motif for the YY1 factor and clusters at two different locations (+13 and +63 bp relative to the TSS) (Table 3). The cluster at +63 bp contains transcripts significantly overexpressed in T cells (PB-CD4+Tcells, PB-CD8+Tcells). In contrast, the cluster located at +13 bp contains transcripts significantly underexpressed in medulla oblongata (Medulla Oblongata). In fact, experimentally, YY1 acts as an activator or repressor, depending on its binding context within a promoter (35,36). Moreover, YY1 enhances transcription in T cells but represses it elsewhere (37). In addition to YY1, our predictions concerning the dual regulatory roles of several other TFs, notably Sp-1 (38), Sp3 (39), and AP-2alphaA (40) matched evidence from experimental literature.
Despite its interesting strengths, our study has some limitations, particularly with respect to alternative promoters. Our dataset contained PPRs corresponding to as many as 4603 genes with putative alternative promoters. In each of these genes, the alternative TSS were spaced at least 500 bp apart (17). Typically, data about functional similarity and microarray expression do not specify possible alternative start sites: the basic unit in both types of data is usually the gene. Alternative promoter usage can have tissue and sequence-context specificity, so the lack of information about alternative promoters probably restricted the precision and scope of our conclusions. If a complete catalogue of annotated promoters and alternative transcripts were available, however, a microarray could use probes with transcript-specific 5′ ends to distinguish among alternative promoters. Similarly, GO annotation could distinguish alternative promoters, if it contained the relevant additional information.
In this study, most positions in most clusters were in conserved regions relative to the mouse genome. Because the positions likely represent TFBSs with a common functionality in the human, most such TFBSs likely represent functionality common to both human and mouse. Our methods could not judge, however, the conservation of individual TFBSs in the two genomes or the TFBSs missed (41–44) by phylogenetic analysis (7,30). Variation of individual TFBSs might be one process differentiating species, but our results suggest that only relatively small subsets of TFBSs with a common function display nucleotide changes between human and mouse.
Finally, exact words yield a limited representation of TFBSs. Position-specific scoring matrices (PSSMs) are much more flexible. We are currently implementing improvements to A-GLAM (11), our Gibbs sampler program for finding TFBSs, to combine sequence information with positional information from datasets with genomic anchors, e.g. the TSS. Initial results indicate that position can contribute substantially to the accuracy of sequence motif predictions. Genomic landmarks serve as a ‘poor man's alignment’, even when precise sequence alignment is impossible. For genes that contain a common TFBS, suggesting co-regulation, our results indicate that positional regulomics can detect positional regulation and thereby unravel the mechanisms underlying diverse functionality and/or expression patterns, by exploiting the location of the TFBS. Further, the resulting models from positional regulomics systematically identify additional genes regulated in a similar manner. Thus, given the success of comparative genomics and its basis in sequence alignment, positional regulomics appears promising indeed.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This research was supported by the Intramural Research Program of the NIH, NLM, NCBI. Funding to pay the Open Access publication charges for this article was provided by NIH, NLM.
Conflict of interest statement. None declared.
REFERENCES
- 1.Banerjee N, Zhang MQ. Functional genomics as applied to mapping transcription regulatory networks. Curr. Opin. Microbiol. 2002;5:313–317. doi: 10.1016/s1369-5274(02)00322-3. [DOI] [PubMed] [Google Scholar]
- 2.Elnitski L, Jin VX, Farnham PJ, Jones SJ. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006;16:1455–1464. doi: 10.1101/gr.4140006. [DOI] [PubMed] [Google Scholar]
- 3.Rando OJ. Chromatin structure in the genomics era. Trends Genet. 2007;23:67–73. doi: 10.1016/j.tig.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev. Proteomics. 2005;2:719–729. doi: 10.1586/14789450.2.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ptashne M, Johnson AD, Pabo CO. A genetic switch in a bacterial virus. Sci. Am. 1982;247:128–130,. doi: 10.1038/scientificamerican1182-128. 132, 134–140. [DOI] [PubMed] [Google Scholar]
- 6.Kielbasa SM, Korbel JO, Beule D, Schuchhardt J, Herzel H. Combining frequency and positional information to predict transcription factor binding sites. Bioinformatics. 2001;17:1019–1026. doi: 10.1093/bioinformatics/17.11.1019. [DOI] [PubMed] [Google Scholar]
- 7.Xie XH, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C, Xuan Z, Otto S, Hover JR, McCorkle SR, Mandel G, Zhang MQ. A clustering property of highly-degenerate transcription factor binding sites in the mammalian genome. Nucleic Acids Res. 2006;34:2238–2246. doi: 10.1093/nar/gkl248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FitzGerald PC, Shlyakhtenko A, Mir AA, Vinson C. Clustering of DNA sequences in human promoters. Genome Res. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marino-Ramirez L, Jordan IK, Landsman D. Multiple independent evolutionary solutions to core histone gene regulation. Genome Biol. 2006;7:R122. doi: 10.1186/gb-2006-7-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tharakaraman K, Marino-Ramirez L, Sheetlin S, Landsman D, Spouge JL. Alignments anchored on genomic landmarks can aid in the identification of regulatory elements. Bioinformatics. 2005;21:I440–I448. doi: 10.1093/bioinformatics/bti1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl Acad. Sci. USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki Y, Yamashita R, Nakai K, Sugano S. DBTSS: DataBase of human transcriptional start sites and full-length cDNAs. Nucleic Acids Res. 2002;30:328–331. doi: 10.1093/nar/30.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- 15.Marino-Ramirez L, Spouge JL, Kanga GC, Landsman D. Statistical analysis of over-represented words in human promoter sequences. Nucleic Acids Res. 2004;32:949–958. doi: 10.1093/nar/gkh246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azuaje F, Wang H, Bodenreider O ISCB. The International Society for Computational Biology. San Diego, CA: 2005. Proceedings of the ISMB’2005 SIG meeting on Bio-ontologies, Detroit, MI; pp. 9–10. [Google Scholar]
- 19.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem. Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 22.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 26.Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 27.Hvidsten TR, Laegreid A, Komorowski J. Learning rule-based models of biological process from gene expression time profiles using gene ontology. Bioinformatics. 2003;19:1116–1123. doi: 10.1093/bioinformatics/btg047. [DOI] [PubMed] [Google Scholar]
- 28.Vardhanabhuti S, Wang J, Hannenhalli S. Position and distance specificity are important determinants of cis-regulatory motifs in addition to evolutionary conservation. Nucleic Acids Res. 2007;35:3203–3213. doi: 10.1093/nar/gkm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 30.Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- 31.Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 32.Marino-Ramirez L, Jordan IK. Transposable element derived DNaseI-hypersensitive sites in the human genome. Biol. Direct. 2006;1:20. doi: 10.1186/1745-6150-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marino-Ramirez L, Lewis KC, Landsman D, Jordan IK. Transposable elements donate lineage-specific regulatory sequences to host genomes. Cytogenet. Genome Res. 2005;110:333–341. doi: 10.1159/000084965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Seto E, Chang LS, Shenk T. Transcriptional repression by YY1, a human GLI-Kruppel-related protein, and relief of repression by adenovirus E1A protein. Cell. 1991;67:377–388. doi: 10.1016/0092-8674(91)90189-6. [DOI] [PubMed] [Google Scholar]
- 36.Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl Acad. Sci. USA. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji HB, Gupta A, Okamoto S, Blum MD, Tan L, Goldring MB, Lacy E, Roy AL, Terhorst C. T cell-specific expression of the murine CD3delta promoter. J. Biol. Chem. 2002;277:47898–47906. doi: 10.1074/jbc.M201025200. [DOI] [PubMed] [Google Scholar]
- 38.Innocente SA, Lee JM. p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett. 2005;579:1001–1007. doi: 10.1016/j.febslet.2004.12.073. [DOI] [PubMed] [Google Scholar]
- 39.Ammanamanchi S, Freeman JW, Brattain MG. Acetylated sp3 is a transcriptional activator. J. Biol. Chem. 2003;278:35775–35780. doi: 10.1074/jbc.M305961200. [DOI] [PubMed] [Google Scholar]
- 40.Rietveld LE, Koonen-Reemst AM, Sussenbach JS, Holthuizen PE. Dual role for transcription factor AP-2 in the regulation of the major fetal promoter P3 of the gene for human insulin-like growth factor II. Biochem. J. 1999;338(Pt 3):799–806. [PMC free article] [PubMed] [Google Scholar]
- 41.O’Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004;18:1859–1875. doi: 10.1210/me.2003-0044. [DOI] [PubMed] [Google Scholar]
- 42.Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, Margulies EH, Blanchette M, Siepel AC, Thomas PJ, McDowell JC, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- 43.Roh TY, Wei G, Farrell CM, Zhao K. Genome-wide prediction of conserved and nonconserved enhancers by histone acetylation patterns. Genome Res. 2007;17:74–81. doi: 10.1101/gr.5767907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alkema W, Wasserman WW. Understanding the language of gene regulation. Genome Biol. 2003;4:327. doi: 10.1186/gb-2003-4-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.