

Abstract
The mapping and alignment of transcripts (cDNA, expressed sequence tag or amino acid sequences) onto a genomic sequence is a fundamental step for genome annotation, including gene finding and analyses of transcriptional activity, alternative splicing and nucleotide polymorphisms. As DNA sequence data of genomes and transcripts are accumulating at an unprecedented rate, steady improvement in accuracy, speed and space requirement in the computational tools for mapping/alignment is desired. We devised a multi-phase heuristic algorithm and implemented it in the development of the stand-alone computer program Spaln (space-efficient spliced alignment). Spaln is reasonably fast and space efficient; it requires <1 Gb of memory to map and align >120 000 Unigene sequences onto the unmasked whole human genome with a conventional computer, finishing the job in <6 h. With artificially introduced noise of various levels, Spaln significantly outperforms other leading alignment programs currently available with respect to the accuracy of mapped exon–intron structures. This performance is achieved without extensive learning procedures to adjust parameter values to a particular organism. According to the handiness and accuracy, Spaln may be used for studies on a wide area of genome analyses.
INTRODUCTION
The identification of gene structure corresponding to a transcript, i.e. cDNA, EST (expressed sequence tag) or protein, is a fundamental step for genome annotation. The rapid increase in the number of sequences of both genomes and transcripts including millions of ESTs has created an acute demand for tools that efficiently map transcripts onto genomes and determine accurate gene structures, because the information thus obtained is essential for finding genes, evaluating the transcriptional activity of the genes, analyzing alternative splicing/transcription and finding polymorphic sites. So far, a large number of methods have been developed for this purpose. Although a dynamic programming (DP) algorithm called ‘spliced alignment algorithm’ may be used to solve the problem (1–8), it is too expensive to apply them to whole genomes of most eukaryotes. Hence, a majority of practical approaches employ a multi-phase strategy (9,10). In the first ‘mapping’ phase, genomic segments considered to be candidates for the target gene are sought with a rapid similarity search program, such as Blast (11), MegaBlast (12), Blat (13) or SSAHA (14). In the following ‘alignment’ phase, the precise locations of exon boundaries are identified by DP or other methods (15,16). However, this composite approach is tedious and sometimes inefficient for general users. Only a handful of programs, such as Blat, Exonerate (10), ESTmapper (17), Squall (18), MGAlign (19) and Gmap (20), can perform both mapping and alignment in a single job. However, most of these programs require large memory; e.g. the data structure adopted by ESTmapper, a write-only top-down suffix tree, requires several times as large memory as the size of the genome, ∼3 Gb for a mammalian genome. Other programs use a common strategy in the first phase, in which relatively long exact matches of length 18–33 nt are searched with a word index table. Thus, a considerably large memory of O(4k) is necessary to store the word index table, where k is the length of a word. Blat and Gmap circumvent this increase in memory by finding a hit in multiple steps with a few consecutive words of length k. However, they still require memory equivalent to the size of the genome to run swiftly or to format the genomic sequence within a reasonable period.
In this report, we show a novel algorithm that also uses a table of words but in a considerably different way from the aforementioned algorithms; instead of finding matches of long words, our algorithm first finds rough location(s) in the genomic region within which the target gene may reside by means of a statistical examination of occurrence of shorter words. The precise exon–intron structure of the target gene is then explored within that region by recursively applying seed extension and DP procedures. We implemented the algorithm to develop a computer program named Spaln (space-efficient spliced alignment) in C/C++ program language. Spaln requires only a small memory, typically <800 Mb for a mammalian genome, for the entire process. Accordingly, Spaln may run on conventional personal computers running under the Unix/Linux operating system. Spaln runs reasonably fast, about two and six times faster than the latest versions of Gmap and Blat, respectively, to map and align >120 000 Unigene sequences onto the whole human genome under our computational environment (a single 32-bit CPU, 4 Gb memory).
Besides high speed and space efficiency, accuracy may be the most important aspect of mapping/alignment programs, especially when such programs are applied to the detailed analyses of alternative splicing, which often involves shifts of splice sites by only a few nucleotides (21,22). In fact, our primary motivation for developing Spaln was to facilitate automatic classification of alternative splicing and transcriptional initiation events observed in various organisms (23,24). As EST sequences are prone to sequencing errors and the opportunity for cross-species comparison has become feasible with the increase in the number of closely related genomic sequences publicly available, it is desired that mapping/alignment tools should be tolerant to noise, such as substitutions and short indels. Therefore, we examined the performance of Spaln and other leading programs under various levels of artificially introduced noise. The results indicate that Spaln significantly outperforms other programs particularly at increased noise levels. It is also shown that a set of species-independent ‘generic’ parameters work fine for intra-species comparison, while a species-adjusted parameter set is more suitable for cross-species comparison. Hence, Spaln may be flexibly used in a wide area of genomic studies of various organisms. The web server at http://www.genome.ist.i.kyoto-u.ac.jp/~aln_user supports the facility of Spaln for several species. The source code is available for free for academic users from the same site.
MATERIALS AND METHODS
Outline
Like most existing algorithms for mapping and aligning cDNA sequences onto the genomic sequence, our algorithm consists of multiple phases. In the first phase, we look for a pair of ‘blocks’ between which the gene corresponding to the transcript may reside, where a block is a predefined segment of the genome with a fixed length. The second phase is a recursive procedure consisting of search and selection. The search procedure is similar to that used by popular homology search programs, such as Fasta (25), Blast (11) and PatternHunter (26) to find high-scoring segment pairs (HSPs). The selection procedure uses a sparse DP algorithm to rearrange the found HSPs in a co-linear order. In the third phase, the precise locations of exon boundaries are identified with the standard spliced alignment DP procedure (6), in which some modifications are made to adapt it to a query of DNA, to reduce memory requirement and to speed up computation. The details of each phase are described in the following subsections.
Partitioning genomic sequence into blocks and a greedy algorithm for calculating block scores
Let g and q be the genomic sequence and the query cDNA sequence, and N and M be their lengths, respectively. For convenience, we regard g as a single chain in which several chromosomes or contigs may be concatenated with a special delimiter. A subsequence of g is denoted by g[i,j] = gigi+1, …, gj, where gi is the i-th nucleotide of g. A similar notation is used for the subsequence of q. The mapping phase starts with the division of g into ‘blocks’ of fixed length B (Figure 1), except that the last block of a chromosome or contig may be shorter than B. The genomic sequence is also covered by non-overlapping oligomers of length k, g[1, k], g[k + 1, 2k], etc. ‘Code’ c of an oligomer is the quaternary expression of subsequence g[i, i + k − 1], where nucleotides A, C, G, and T are converted into numerals 0, 1, 2 and 3, respectively. If g[i, i + k − 1] contains one or more ambiguous nucleotides, such as N, c is assigned to the outlier, 4k, and the word is ignored in the subsequent processes. Otherwise, we express g[i, i + k − 1] = w(c), where c takes a value between 0 and 4k − 1.
Figure 1.

Schematic diagram showing blocks, words and the greedy algorithm for finding significant blocks. Arrow corresponds to a k-mer word, and its direction indicates direct or reverse strand. Circle indicates presence of the word shown on the left within the block shown on top. Big circles are used to discriminate true hits from background noise indicated by small circles. Three blocks (numbers 10–12) containing three or more hits are considered to be significant and indicated by yellow color.
We read the genomic sequence twice. At the first read, the total number of w(c), n(c), in the genome is counted. At the same time, the number of blocks that contain w(c), or more precisely, that contain the first nucleotide of w(c), is also counted. This information is used to allocate the memory for the ‘block index table’ constructed at the second read. This two-dimensional table stores the lists of blocks that contain w(c). The table is similar to a widely used look-up table, but its elements point not to the genomic positions of w(c) but to the blocks that contain at least one w(c). In practice, the table of n(c) (or s(c) described subsequently) and the block index table are constructed only once for each genome, stored in a file in binary forms and read into memory at run time.
If we assume a uniform distribution of w(c) over g, p(c) = B·n(c)/N indicates the probability of w(c) being found in a block. We estimate the average value of p(c) by 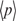 = B/Z, where Z ≤ 4k is the number of w(c) for which n(c) > 0. Then, we define the ‘word score’ s(c) of w(c) by
= B/Z, where Z ≤ 4k is the number of w(c) for which n(c) > 0. Then, we define the ‘word score’ s(c) of w(c) by
| 1 |
The second equation is intended to eliminate spurious matches derived from repetitive elements dispersed over the genome, and constant a (=10 by default) adjusts the elimination level. Accordingly, Spaln runs comfortably without previous masking of the genomic sequence. A similar idea has been used in other programs, such as SSAHA (14), Blat (13) and WindowMasker (27). The average value for s(c), 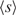 , is obtained by:
, is obtained by:
| 2 |
where the summation is taken over all the k-mers for which n(c) > 0.
When a set of words, w(c) = {w(c1), w(c2), …, w(cm)}, are given, we will define the ‘block score’, S(w(c), b), for a particular block b as
| 3 |
where δ(w(ch), b) = 1 or 0 depending on whether w(ch) is present in b or not. If 〈p〉≪1, exp[−S(w(c), b)] approximates the probability that the observed number of words is found in b by chance alone. If w(c) is extracted from a subsequence of q, q[i, j], in some way, the smaller probability [the larger value for S(w(c), b)] implies the greater likelihood that some subsequence in b is homologous to q[i, j]. To find such ‘homologous’ blocks with as few test words as possible, we use the greedy algorithm depicted in Figure 1. We choose test words on both strands from both ends of q moving toward the interior. Thus, four series of word sets are examined in parallel. Starting with q[1, k], for example, we recurrently choose the words in a depth-first manner as long as at least one block contains the series of words w(c1) = q[1, k], w(c2) = q[k + 1, 2k], …, w(cm) = q[(m − 1)k + 1, mk] simultaneously. The blocks that contain w(ch) are quickly identified with the block index table, and the block score of these blocks is incremented by s(ch). If a block score exceeds the predefined threshold discussed subsequently, the block is called ‘significant’, and the recurrence is broken. The next recurrence starts with w(cm+1) = q[2, k + 1], …, and the final recurrence of this round starts with q[k, 2k − 1]. Analogous procedures are conducted on the complementary strand and in the backward direction as well.
Gene ends corresponding to a majority of full-length cDNAs are identified by the bi-directional search described earlier. However, this approach sometimes fails for EST sequences with high error rates at the 3′-ends, chimerical cDNAs and unfinished genomic sequences. Hence, we also use a unidirectional approach in which we search consecutive blocks with positive block scores starting with a significant block. If the sum of the first and the last block scores exceeds the threshold, the pair of start and end blocks are passed to the second phase.
The four series of block search recur from the points at which the previous round of recurrence was broken, if neither bi-directional nor unidirectional search succeeds. This procedure is repeated until the forward search and the backward search meet each other. After the first significant block pair was found, the above rounds of recurrence are repeated maximally three more times to search for possibly additional significant block pairs. This procedure allows us to find plural significant blocks corresponding to paralogs in each of the four series. We examine all combinations of significant blocks found in the forward and backward series on the same strand. Block pairs that are sufficiently close to each other to accommodate a single gene in the correct direction are selected and passed to the second phase. For a mammalian genome, the maximal distance between the block pairs is set to be about 3.4 Mb, the size of the longest known human gene (28). If no block pair satisfies the criteria of block scores and mutual genomic locations, the gene corresponding to the transcript is judged to be missing in the genome.
Statistical consideration of block scores
By the analogy with local alignment scores in sequence similarity search (29,30), the maximum block scores for foreign query sequences are expected to follow an extreme-value distribution with mean and variance of the forms:
| 4 |
as a function of the number of test words m < B, where α, β and σ2 are constants. This is actually the case as shown in Figure 2a. The instability at the right extreme is probably due to sparsity of samples. Thus, we set the threshold value for a significant block with the formula:
| 5 |
where the values for coefficient α and bias β+ are treated as adjustable parameters. In the present version of Spaln, the default values for α and β+ are set to 0.4 and 3.0, respectively. It must be noted that the Equations (4) and (5) are derived from empirical observations, and their theoretical foundation remains to be established.
Figure 2.

Block score as a function of number of test words. (a) Human Unigene sequences were shuffled and used as queries against the whole human genome. The mean (open triangle) and the SD (cross symbol) are fitted to logarithmic and constant curves, respectively. Each bin corresponds to the number of rounds at which the recurrence has finished. The number of occasions in each bin is shown by filled circle in the logarithmic scale. (b) Mean block score (filled square) for real Unigene sequences mapped on the human genome. SDs are shown by error bars. A part of (a) is also shown for comparison. The number of sequences in each bin is shown by filled circle in the logarithmic scale.
The condition 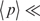 indicates that B
indicates that B
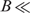 Z ⩽ 4k. On the other hand, the smaller value of B and the larger value of k require a large memory to store the block index table. To balance the requirements, we choose B and k so that
Z ⩽ 4k. On the other hand, the smaller value of B and the larger value of k require a large memory to store the block index table. To balance the requirements, we choose B and k so that  , and k > ln(N)/2ln(4). For a mammalian genome with
, and k > ln(N)/2ln(4). For a mammalian genome with 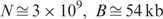 and we found k = 12 to be the most appropriate. The condition that both B and
and we found k = 12 to be the most appropriate. The condition that both B and 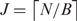 are <216 has a practical benefit in that 2-byte unsigned integers can be used to realize block indexes, thereby contributing to the reduction in memory usage.
are <216 has a practical benefit in that 2-byte unsigned integers can be used to realize block indexes, thereby contributing to the reduction in memory usage.
Construction of chained HSP lists
Matches of words of a fixed length (seeds) are searched for with a classical word index table (31,32) constructed on the genomic region sandwiched by a pair of blocks found in the first phase. Instead of a contiguous seed pattern, however, we use discrete seed patterns as suggested by Ma et al. (26). The seeds are connected to form a large matching segment if they are separated from each other by less than a fixed distance along the same diagonal. Each matching segment is evaluated with nucleotide match and mismatch scores. When the sum of the scores is larger than the given threshold, the matching segment is regarded as an HSP.
The HSPs are then chained into a co-linear order by the sparse DP algorithm (33). Although more efficient algorithms have been proposed (34), we adhere to the simple quadratic algorithm, as the number of HSPs to be considered is usually of the order of ten or smaller.
If an inter-HSP region is wide enough to suggest the presence of at least one additional exon, the above procedures are recursively repeated up to the prespecified depth (third by default) with progressively decreasing word size. We use fixed seed patterns ‘110010110111’, ‘110011101’ and ‘11101’ by default. If the presence of an additional exon is improbable, the algorithm moves to the third phase.
Spliced alignment algorithm
The third phase is the standard spliced alignment with DP, as described previously (6). However, several simplifications and extensions have been made. First, the concept of coding frame or coding potential is no longer valid because the query is a DNA rather than an amino acid sequence. Second, either ‘generic’ or ‘species-specific’ splicing signals may be chosen. The generic signal depends only on the dinucleotides at both ends of an intron. By default, Spaln accepts only consensus tetra-nucleotides, ‘GT … AG’, ‘GC … AG’ and ‘AT … AC’ as intron ends, although this limitation can be relaxed with ‘-ya’ option (Table 1). The species-specific signal is learned from known sequences around the intron–exon boundaries of a specific genome by means of a first- or second-order Markov model. Third, the distribution of intron lengths is scored and may be involved in the alignment score. Fourth, we also adopt the strategy of ‘attack by both sides’ or ‘Sandwich algorithm’ used in Exonerate (10) and Gmap (20), provided that the presence of only one intron is expected in the relevant region. The X drop-off method used in the algorithm can eliminate voluminous computation otherwise spent within the intron region. Fifth, in addition to the default semi-global alignment mode, the Smith–Waterman type local mode (35) may also be chosen with ‘-LS’ option (Table 1). Finally, Spaln implements a hybrid space-saving traceback algorithm in which Hirschberg's linear-space algorithm (36) is used to locate the ‘midpoint’ of an alignment if the expected space for storing the traceback information exceeds the predefined threshold; otherwise, an ordinary traceback algorithm (1) is applied.
Table 1.
List of programs and options used for experiment
| Name | Version | Options for alignment | Options for mapping | Reference |
|---|---|---|---|---|
| Blat | 34 | -noTrimA -fine -q=rna | -noTrimA -fine -q=rna -ooc=11.ooc | (13) |
| Exalin | 2005-05-06 | default | (15) | |
| –showalignment n | ||||
| Exonerate | 1.4.0 | –showtargetgff y -n 1 | (10) | |
| -S n –refine region -m e2g | ||||
| Gmap | 2007-09-28 | default | -f 2 -B 2 | (20) |
| GmapX | 2007-09-28 | -X | (20) | |
| MegaBlast | 2.2.14 | -D 3 -F'm R;V;D’ | (12) | |
| Sim4 | 2003-09-21 | default | (40) | |
| Spaln | 1.2.1 | -Q3 -pa | -Q7 -M | This work |
| SpalnA | 1.2.1 | -Q3 -pa -ya | This work | |
| SpalnS | 1.2.1 | -Q3 -pa -yS -yX | This work | |
| SpalnX | 1.2.1 | -Q3 -pa -yX | -Q7 -yX -M | This work |
| SpalnXL | 1.2.1 | -Q3 -pa -yX -LS | -Q7 -yX -LS -M | This work |
Blank cell indicates ‘not examined’. The ‘-X’ option of Gmap indicates that canonical intron boundaries are favoured. Conversely, the ‘-ya’ option of Spaln indicates that non-canonical intron boundaries are accepted. The ‘-LS’ (local similarity) option was used only in combination with the ‘-yX’ (cross-species) option.
Preparation of test data
Human (Build 36) and mouse (Build 35) genomic sequences and the human Unigene (Build #203) sequence were downloaded from NCBI (ftp://ftp.ncbi.nih.gov/). We used cDNA sequences and annotations provided by the Projector web site (http://www.sanger.ac.uk/Software/analysis/projector) as test samples. After removal of duplications and correction for trivial errors, the data set contains 491 homologous pairs each for human and mouse. Comparison of the results of Spaln with the annotations indicated 13 discrepancies in 9 human genes. Inspection of the alignments shown in Table S1 in the Supplementary Data indicated that all the discrepancies were due to single-nucleotide indels at some exon–intron junctions, all of which were located in 5′ or 3′ UTRs. We believe that the results of Spaln are correct because they exactly follow consensus splicing rules. An earlier version of Gmap (Gmap-2006-12-18) also suggested the same exon–intron junctions as Spaln, whereas the latest version of Gmap produced slightly different results from the earlier one.
The results for mouse genes were somewhat more complicated than those of human genes. In addition to single-base indels at splicing junctions such as those found in human genes, the Projector annotations contain several short ‘introns’ unlikely to be real ones because of their shortness, biased nucleotide compositions and lack of consensus sequences at the ends. Although Spaln discards most of these obviously false introns, it reports three relatively long inserts as introns. Thus, we regard these predictions as mistakes of Spaln. We also removed four duplicated genes from our test samples, leaving 487 mouse genes. Note that only the annotations were corrected and cDNA sequences were unmodified, and that all the 2 × 491 = 982 gene–cDNA pairs were tested when only CDS regions were used for cross-species comparison.
Artificial introduction of noise
To simulate sequencing errors and cross-species comparisons, we introduced various levels of mutations into the test cDNA sequences. First, a specific number of nucleotide positions were randomly selected except for the 10 nt from both ends that were kept intact. The selected nucleotide was randomly mutated into one of the others in 98% of the cases. In the remaining 2% of cases, insertion or deletion was chosen at equal probability, and indel length was decided according to the geometric distribution with the common ratio of 0.6. The segment to be inserted was generated by a random series of nucleotides chosen at the equal probability of 0.25.
Tested programs
The tested programs and the options used are summarized in Table 1. Megablast was used with repeat filtering with RepBase12.0.7 (http://www.girinst.org/). As the psl format of Blat does not discriminate intron from ordinary insertion, an insertion longer than 30 bp is regarded as an intron. The ‘-pa’ option of Spaln indicates that the terminal polyA or polyT sequence is not trimmed; the ‘-ya’ option indicates that intron ends may not be confined to the canonical dinucleotides; the ‘-yX’ option adjusts parameter values suitable for cross-species comparison and the ‘-yS’ option indicates the use of species-specific boundary signals and intron-length distribution information. The actual parameter values used by Spaln are listed in Table S2 in Supplementary Data. For genome mapping, ‘-M’ option is also set to locate potentially multiple genomic regions homologous to the query. When Unigene sequences were used as queries, ‘-yX -LS’ option was further set to obtain local rather than semi-global alignments. All calculations were performed on the same computer with 4 Gb memory and a 3.0 GHz Intel® Pentium® D CPU running under Linux.
RESULTS AND DISCUSSION
Sensitivity and specificity of block-based mapping
Most existing programs quickly locate the genomic position(s) corresponding to the query cDNA sequence by looking for relatively long (18–33 nt) exact matches. In contrast, Spaln first finds the block(s) in which the target gene may reside, where a block is a predefined genomic region with a fixed length (see Materials and methods section). This search relies on statistical property of occurrence of a set of short (10–12 nt) words extracted from the query sequence. If the test words are concentrated in a particular block more frequently than expected from the random distribution, the block is likely to contain at least a part of the target gene. To test the hypothesis, we observed the distribution of block scores for the entire 124 355 sequences in the ‘unique’ set of Unigene Build #203 and that of their randomized sequences, where a block score is the negative logarithm of probability of observation by chance alone.
In contrast to the logarithmic distribution of block scores as a function of the number of test words extracted from random sequences (Figure 2a), the block scores for real cDNA sequences scatter above those for random sequences (Figure 2b). Based on the former distribution, we estimate the thresholds as a function of the number of test words (see Materials and methods section), and discard a query with the maximum block score below the threshold value. Thus, of the 124 355 sequences tested, 243 failed to be mapped on the genome. This observation results in the nominal sensitivity of 99.8%. The results in comparison with those of Blat, Gmap and Megablast are summarized in Table 2. The genomic sequence was formatted before the search according to the protocol of each program. For Blat, both genomic and query sequences were converted into the.2-bit format. The CPU time shown in Table 2 does not include the computation time used in the formatting processes. Note that Blat, Gmap and Spaln involve the process for identification of exon–intron boundaries to generate gene models (records), while Megablast does not. Hence, the number of records of Megablast was roughly estimated as the number of unique combinations of the cDNA and contig identifiers. Table 2 indicates that the sensitivities of Blat, Gmap and Spaln are nearly identical to one another in spite of the quite different mapping strategies, while Megablast is slightly less sensitive than the others. In contrast, the numbers of records reported by the four programs vary extensively, leading to nearly 30-fold difference in the nominal selectivity between the extremes, Blat and Spaln. This is not surprising because Blat and Megablast are designed for general purpose to detect as many similarities as possible, whereas Spaln is specialized to detect only the cognate or orthologous genes and their close relatives on the genome.
Table 2.
Mapping of Unigene sequences onto human genome
| Program | Mappeda | Recordsb | Sensitivity (%)c | Selectivity (%)d | CPU (h) |
|---|---|---|---|---|---|
| Blat | 124 103 | 37 74 716 | 99.80 | 3.29 | 37.37 |
| Gmap | 123 373 | 162 700 | 99.21 | 75.83 | 11.00 |
| Megablast | 118 643 | 304 138 | 95.41 | 39.01 | 63.05 |
| SpalnXL | 124 112 | 142 316 | 99.80 | 87.21 | 5.58 |
aThe number of query sequences that were mapped on at least one genomic locus.
bThe total number of gene models reported.
cThe percentage of mapped queries of 124 355 cDNA sequences examined.
d100× number of mapped queries divided by the total number of records reported.
To get some insight into the quality of the mapping results, we asked how much fraction of records obtained by different methods is common to one another. In this test, each query is counted only once even if it is mapped on more than one separate genomic region. A query is considered to be commonly mapped if the genomic regions mapped by different methods overlap in part. The proportion of pair-wise, triple-wise or tetra-wise commonality among the mapped query sequences (Table S3 in Supplementary Data) indicates again that the sensitivity of Splan is very similar to those of Blat and Gmap. Meanwhile, Megablast is slightly less sensitive despite the one order of magnitude longer computation time than Splan.
Although the above observations indicate that the four programs tested are more or less similar to each other in mapping sensitivity, it is difficult to stringently assess the quality of mapping and alignment from the gross data because the true locations and structures of the genes corresponding to individual Unigene sequences are not known. For assessment purposes, we relied on a smaller, human-curated data set as described in the following subsections.
Evaluation of alignment
In this subsection, we examine the quality of alignment produced by various programs listed in the Materials and methods section. In this test, the genomic segment that encompasses the gene corresponding to the cDNA is input to the programs. The test data are 491 human and 487 mouse genes in the Projector data set (37) with some modifications in the annotations (see Materials and methods section). All cDNA sequences are virtually full-length consisting of 5′ UTR, CDS region and 3′ UTR. Even with no artificially introduced noise, only Spaln and an earlier version of Gmap (Gmap-2006-12-18) perfectly reproduced the reference human gene structures, and no program succeeded in completely reproducing the mouse gene structures; Spaln erroneously regarded three possibly polymorphic inserts as introns in addition to another small discrepancy, while other programs were even less accurate in these respects. It is noteworthy that Spaln found all the micro-exons present in the Projector data set [listed in (38)], although Spaln does not implement such special routine as that proposed by Volfovsky et al. (39).
Figure 3a and b show error rates at the gene level and the exon level, respectively, under various levels of artificially introduced noise. Each error rate is the average of six experiments, and the numerical values for the average and the SD are listed in Tables S4 and S5 in Supplementary Data. An error at the gene level implies that at least one of the exon boundaries is incorrectly assigned, and an error at the exon level implies that either or both exon boundaries are incorrect. Tables S6 and S7 in Supplementary Data show the exon-level error rates broken down into those for internal and terminal exons, respectively. Tables S4–S7 also present the results of one more setting of Gmap and two more settings of Spaln (Table 1) that are not shown in Figure 3. Although error rates slightly vary with the run time options, these results as a whole clearly indicate that Spaln is significantly more tolerant to noise than the other programs. Even at the highest noise level examined, Spaln correctly identified >99% of exons, whereas >3% error is commonly observed with the other programs. An example of variation in alignment produced by Spaln, Gmap and Exonerate around a micro-exon is presented in Figure S1 in Supplementary Data.
Figure 3.

Comparison of performance of various spliced alignment methods. Error rates at gene level (a) or exon level (b) are shown as a function of artificially introduced noise levels. The actual error rates of Sim4 and Blat are, respectively, twice and five times as large as those indicated by the bar heights. Letters X and S attached to Spaln indicate that ‘-yX’ and ‘-yX –yS’ options were applied, respectively. Each error rate is the mean of six trials, the numerical value and the SD of which are shown in Tables S4 and S5.
Although the error rates naturally increase with increasing noise level for most programs and settings, the results of Spaln with ‘-yX -yS’ options are almost invariable regardless of noise level, and are the most accurate at the highest noise level. This result is considered to be a consequence of the statistical power to recognize intron boundaries that dominates information derived from relatively weak sequence similarities. Gene structures inferred with ‘-yX -yS’ options might actually correspond to splicing variants. In general cases, however, the use of these options is not recommended for the identification of the gene structure corresponding to a given transcript.
To examine whether the above observations are extrapolated to the distance as far as human and mouse homologs, we conducted cross-species comparisons with the combinations of human genomic fragments that contain the human gene and homologous mouse transcripts and vice versa. Although such applications might be outside the scope of some tested programs, all results are shown for references. As UTR sequences are not necessarily well conserved between human and mouse, we used only the CDS regions of all the 491 gene pairs contained in the Projector data set. The results are summarized in Table 3, in which accuracies rather than error rates are presented. As expected, the general tendency was the same as that of the aforementioned observations with artificially introduced noise; Spaln, particularly with ‘-yX -yS’ options, outperformed other programs. Note, however, that our Aln program (6) that used a translated amino acid sequence as query, performed considerably better than Spaln with a CDS query, when the parameter sets were adjusted for mammalian genes. In fact, the results of Aln were even better than those of Projector (37) and GeneAlign (38) that used knowledge about the exon–intron structure of the homologous gene. To incorporate the mapping phase into the Aln algorithm, we are now constructing an extended version of Spaln that accepts either nucleotide or amino acid sequence as query. A detailed discussion of these topics will be presented in a separate report.
Table 3.
Accuracy in cross-species comparison between human and mouse cDNA and genomic sequences
| Measure | Blat | Exalin | Exonerate | Gmap | Sim4 | Spaln | SpalnS | SpalnX |
|---|---|---|---|---|---|---|---|---|
| #Answer | 266 | 981 | 982 | 937 | 978 | 982 | 982 | 982 |
| #Gene | 70 | 429 | 458 | 367 | 161 | 593 | 752 | 714 |
| %Gene | 7.13 | 43.69 | 46.64 | 37.37 | 16.40 | 60.39 | 76.58 | 72.71 |
| %Exon | 67.63 | 89.51 | 80.97 | 81.41 | 59.54 | 90.09 | 96.21 | 95.11 |
| %IntExon | 64.17 | 94.26 | 76.36 | 78.06 | 62.59 | 93.77 | 96.87 | 95.81 |
| %TermExon | 66.22 | 69.94 | 70.44 | 63.58 | 47.41 | 82.40 | 93.20 | 92.10 |
| %Junction | 79.50 | 93.97 | 85.97 | 86.03 | 73.54 | 93.27 | 97.91 | 97.26 |
| %Nucleotide | 95.30 | 98.20 | 93.22 | 92.60 | 91.18 | 97.77 | 99.38 | 99.31 |
| CPU (s) | 143.1 | 3181.3 | 221.2 | 52.5 | 33.9 | 220.8 | 190.3 | 145.5 |
A total of 982 human–mouse cross-species pairs of genomic segments and CDS sequences in Projector data set are examined. The numbers of ‘Answer’ and ‘Gene’ are the number of runs with any outputs and that with perfectly correct gene structures, respectively. The accuracies at the exon, junction and nucleotide levels are respective harmonic averages of the specificity and sensitivity defined by 200C/(T + P), where C, T and P are correctly predicted, true and predicted quantities, respectively. For internal (% IntExon) or terminal (% TermExon) exons, the sensitivity defined by 100 × C/T is presented.
Table S8 in Supplementary Data and the last row of Table 3 present the computation times spent in the experiments mentioned earlier. Each execution time includes that used to read sequence files. Except for Exalin that adopts a full-DP procedure, all programs adopt heuristics to accelerate computation. Sim4 is the fastest, but its speed does not seem to compensate for the high error rates, as demonstrated in Figure 3 and Table 3. On the other hand, the heuristics adopted by Gmap and Spaln, in particular, is quite valuable as it speeds up computation by more than one order of magnitude without loss of accuracy (or contribute to produce even better quality) compared to the full-DP procedure.
Evaluation of mapping and alignment
We used again the Projector data set and the artificial mutants to assess the total performance of Spaln involving both mapping and alignment phases. Only Blat and Gmap were used as references because they are the only programs that work with a whole genome without having to rely on external software under our computational environment. The results summarized in Figure 4 and Table S9 in Supplementary Data reveal that Spaln is significantly better than the others at reproducing the correct gene structures particularly at high noise levels. In contrast, Spaln is slightly less sensitive than Blat and Gmap in locating genomic region(s) corresponding to relatively dissimilar query sequences. Except for the highest noise level, however, Spaln correctly locates >99% of genes, and hence the slightly lower sensitivity of Spaln may not be much problematic in most practical applications.
Figure 4.

Accuracy in mapping and alignment of CDS sequences onto the human or mouse genomic sequence. Error rates at the gene level (a) and at the exon level (b) are shown as functions of artificially introduced noise levels. The actual error rates of Blat are three times as large as those indicated by the bar heights. SpalnXL indicates that ‘-yX –LS’ options were applied to Spaln at run time. Each error rate is the mean of six trials, the numerical value and the SD of which are shown in Table S9 with other information.
For some queries, several genomic regions have nearly or perfectly identical nucleotide sequences within the transcribed parts. In the default settings, Spaln reports only one gene structure corresponding to each query, and occasionally reports a paralog rather than the true correspondents. By setting the ‘-M’ option for reporting multiple loci, many of the missed loci are rescued. However, the rescue is still imperfect, which accounts for a significant portion of missed loci shown in Figure 4 and Table S9. Finding a way to thoroughly deal with paralogs is one of the issues to be resolved in the further improvement of Spaln.
SUPPLEMENTARY DATA
Supplementary Data are at available NAR Online.
ACKNOWLEDGEMENTS
I would like to thank Mr M. Yamamura and Dr H. Nagasaki for technical assistance and discussions, and Dr T. Yada and Dr N. Ichinose for valuable discussions. I also would like to thank Ms K. Sugimori and Mr M. Morita for help in Website preparation. This study was funded by KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas ‘Comparative Genomics’ (No.18017017) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Funding to pay the Open Access publication charges for this article was provided by National Institute of Advanced Industrial Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gotoh O. Optimal sequence alignment allowing for long gaps. Bull. Math. Biol. 1990;52:359–373. doi: 10.1007/BF02458577. [DOI] [PubMed] [Google Scholar]
- 2.Huang X, Zhang J. Methods for comparing a DNA sequence with a protein sequence. Comput. Appl. Biosci. 1996;12:497–506. doi: 10.1093/bioinformatics/12.6.497. [DOI] [PubMed] [Google Scholar]
- 3.Gelfand MS, Mironov AA, Pevzner PA. Gene recognition via spliced sequence alignment. Proc. Natl Acad. Sci. USA. 1996;93:9061–9066. doi: 10.1073/pnas.93.17.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- 5.Usuka J, Zhu W, Brendel V. Optimal spliced alignment of homologous cDNA to a genomic DNA template. Bioinformatics. 2000;16:203–211. doi: 10.1093/bioinformatics/16.3.203. [DOI] [PubMed] [Google Scholar]
- 6.Gotoh O. Homology-based gene structure prediction: simplified matching algorithm using a translated codon (tron) and improved accuracy by allowing for long gaps. Bioinformatics. 2000;16:190–202. doi: 10.1093/bioinformatics/16.3.190. [DOI] [PubMed] [Google Scholar]
- 7.Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulze U, Hepp B, Ong CS, Rätsch G. PALMA: mRNA to genome alignments using large margin algorithms. Bioinformatics. 2007;23:1892–1900. doi: 10.1093/bioinformatics/btm275. [DOI] [PubMed] [Google Scholar]
- 9.Wheelan SJ, Church DM, Ostell JM. Spidey: a tool for mRNA-to-genomic alignments. Genome Res. 2001;11:1952–1957. doi: 10.1101/gr.195301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 13.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang M, Gish W. Improved spliced alignment from an information theoretic approach. Bioinformatics. 2006;22:13–20. doi: 10.1093/bioinformatics/bti748. [DOI] [PubMed] [Google Scholar]
- 16.van Nimwegen E, Paul N, Sheridan R, Zavolan M. SPA: a probabilistic algorithm for spliced alignment. PLoS Genet. 2006;2:e24. doi: 10.1371/journal.pgen.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Lee W-J, Tseng C-W. 2005. 19th IEEE International Parallel and Distributed Processing Symposium (IPDPS'05) – Workshop 7; p. 196a. [Google Scholar]
- 18.Ogasawara J, Morishita S. A fast and sensitive algorithm for aligning ESTs to the human genome. J. Bioinform. Comput. Biol. 2003;1:363–386. doi: 10.1142/s0219720003000058. [DOI] [PubMed] [Google Scholar]
- 19.Ranganathan S, Lee BTK, Tan TW. MGAlign, a reduced searchspace approach to the alignment of mRNA sequences to genomic sequences. Genome Inform. 2003;14:474–475. [Google Scholar]
- 20.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 21.Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat. Genet. 2004;36:1255–1257. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- 22.Hiller M, Huse K, Szafranski K, Rosenstiel P, Schreiber S, Backofen R, Platzer M. Phylogenetically widespread alternative splicing at unusual GYNGYN donors. Genome Biol. 2006;7:R65. doi: 10.1186/gb-2006-7-7-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagasaki H, Arita M, Nishizawa T, Suwa M, Gotoh O. Automated classification of alternative splicing and transcriptional initiation and construction of visual database of classified patterns. Bioinformatics. 2006;22:1211–1216. doi: 10.1093/bioinformatics/btl067. [DOI] [PubMed] [Google Scholar]
- 24.Nagasaki H, Arita M, Nishizawa T, Suwa M, Gotoh O. Species-specific variation of alternative splicing and transcriptional initiation in six eukaryotes. Gene. 2005;364:53–62. doi: 10.1016/j.gene.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 25.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma B, Tromp J, Li M. PatternHunter: faster and more sensitive homology search. Bioinformatics. 2002;18:440–445. doi: 10.1093/bioinformatics/18.3.440. [DOI] [PubMed] [Google Scholar]
- 27.Morgulis A, Gertz EM, Schäffer AA, Agarwala R. WindowMasker: window-based masker for sequenced genomes. Bioinformatics. 2006;22:134–141. doi: 10.1093/bioinformatics/bti774. [DOI] [PubMed] [Google Scholar]
- 28.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 29.Karlin S, Altschul SF. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl Acad. Sci. USA. 1990;87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waterman MS. Introduction to Computational Biology; Maps, Sequences and Genomes. London: Chapman & Hall; 1995. [Google Scholar]
- 31.Dumas J-P, Ninio J. Efficient algorithms for folding and comparing nucleic acid sequences. Nucleic Acids Res. 1982;10:197–206. doi: 10.1093/nar/10.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilbur WJ, Lipman DJ. Rapid similarity searches of nucleic acid and protein data banks. Proc. Natl Acad. Sci. USA. 1983;80:726–730. doi: 10.1073/pnas.80.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilbur WJ, Lipman DJ. The context-dependent comparison of biological sequences. SIAM J. Appl. Math. 1984;44:557–567. [Google Scholar]
- 34.Eppstein D, Galil Z, Giancarlo R, Italiano G. Sparse dynamic programming I: Linear cost functions. J. Assoc. Comp. Mach. 1992;39:519–545. [Google Scholar]
- 35.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 36.Hirschberg DS. A linear-space algorithm for computing maximal common subsequences. Commun. ACM. 1975;18:341–343. [Google Scholar]
- 37.Meyer IM, Durbin R. Gene structure conservation aids similarity based gene prediction. Nucleic Acids Res. 2004;32:776–783. doi: 10.1093/nar/gkh211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh SJ, Lin CY, Liu NH, Chow WY, Tang CY. GeneAlign: a coding exon prediction tool based on phylogenetical comparisons. Nucleic Acids Res. 2006;34:W280–W284. doi: 10.1093/nar/gkl307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volfovsky N, Haas BJ, Salzberg SL. Computational discovery of internal micro-exons. Genome Res. 2003;13:1216–1221. doi: 10.1101/gr.677503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.