


Abstract
Clustered damages—two or more closely opposed abasic sites, oxidized bases or strand breaks—are induced in DNA by ionizing radiation and by some radiomimetic drugs. They are potentially mutagenic or lethal. High complexity, multilesion clusters (three or more lesions) are hypothesized as repair-resistant and responsible for the greater biological damage induced by high linear energy transfer radiation (e.g. charged particles) than by low linear energy transfer X- or γ-rays. We tested this hypothesis by assessing human abasic endonuclease Ape1 activity on two- and multiple-lesion abasic clusters. We constructed cluster-containing oligonucleotides using a central variable cassette with abasic site(s) at specific locations, and 5′ and 3′ terminal segments tagged with visually distinctive fluorophores. The results indicate that in two- or multiple-lesion clusters, the spatial arrangement of uni-sided positive [in which the opposing strand lesion(s) is 3′ to the base opposite the reference lesion)] or negative polarity [opposing strand lesion(s) 5′ to the base opposite the reference lesion] abasic clusters is key in determining Ape1 cleavage efficiency. However, no bipolar clusters (minimally three-lesions) were good Ape1 substrates. The data suggest an underlying molecular mechanism for the higher levels of biological damage associated with agents producing complex clusters: the induction of highly repair-resistant bipolar clusters.
INTRODUCTION
Clustered DNA damages—two or more abasic sites, oxidized bases or strand breaks on opposing strands within one or two helical turns—are potentially highly mutagenic and lethal DNA alterations. Clusters are produced by ionizing radiation, by some radiomimetic drugs, and at very low frequencies by ultraviolet radiation and during aerobic metabolism of cells (1–6). Clusters are induced by X-rays in DNA in solution and in cells, in the ratio of approximately one double-strand break (DSB): one oxidized purine cluster: 0.9 oxidized pyrimidine cluster to 0.75 abasic clusters (1,7). However, charged particles—from protons to iron ions—produce a higher ratio of DSBs to non-DSB clusters than X- or γ-rays (8).
Studies of synthetic oligonucleotides containing simple two-lesion clusters of specific composition, polarity and spacing showed that many clusters can be converted to DSBs by purified DNA repair enzymes or cell extracts (9–13). Indeed, mammalian cells exposed to supralethal radiation doses generate de novo DSBs during postirradiation incubation (14–16). However, repair-proficient cells exposed to low radiation doses do not generate detectable levels of de novo DSBs (13). Even repair-deficient cells produce only ∼10% of the level of clusters in repair-generated DSBs (17). These results suggest that while lethally irradiated cells may generate DSBs in abortive cluster processing, cells with low levels of damage use other coping mechanisms and avoid production of potentially lethal DSBs.
Both high-energy photons, such as X-rays, and energetic ions can ionize the atoms and molecules through which they pass. They also generate energetic electrons that travel a short distance while also ionizing molecules that they encounter. The density of the ionized and hence chemically reactive species is higher for high-energy particles (HZE). Thus high linear energy transfer (LET) radiation may produce highly complex DNA damages containing more altered sites than low LET radiation such as γ- or X-rays (18,19). This is consistent with the results showing a higher ratio of DSB to non-DSB clusters in particle-irradiated DNA in solution (8) and in cells (Das and Sutherland, in preparation), since clusters consisting of many lesions would have a higher probability of containing two closely opposed single-strand breaks (SSBs) than clusters containing only a few lesions. Such high complexity clusters would be measured as DSBs no matter how many additional oxidized bases or abasic sites they contained (8). Further, among HZE ions, more DNA damage (8) and more biological damage [cell killing and transformation in vitro (20,21) and cancer induction in animals (22)] are inflicted (per particle) by HZE particles with higher LETs (8) with a maximum in the range of ∼200 keV/μm (23). This higher biological damage is generally assumed to stem from slower or less accurate repair of the HZE-induced complex clusters containing more lesions than the simpler clusters induced by X-rays.
Most investigations of repair of clustered damages in synthetic oligonucleotides used radioactively labeled oligonucleotides. This entails frequent preparation of radiolabeled oligonucleotides, possible scission of the oligonucleotide by radioactive decay, problems of radioactive contamination of the work area and expenses of disposal of radioactive material. Further, purchasing or synthesizing the many different lesion-containing oligonucleotides required for this study would be prohibitively expensive. To solve these problems, we used one 51mer oligonucleotide (strand A) containing one lesion, and assembled the complementary B strand from a central 21-bp cassette containing one or more lesions in specific sites, plus two invariant 5′and 3′ terminal 15-bp oligonucleotides. Either or both of the strands were labeled with visually distinctive fluorophores to aid in identification of the cleavage products.
Using these fluorophore-labeled constructs containing defined abasic clusters of known polarity and spacing, we investigated the properties of clusters that determine their cleavage by the human abasic endonuclease Ape1. The data presented herein suggest a molecular mechanism for the higher biological damage induced by high LET radiation relative to that produced by low LET radiation such as X-rays.
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA). For specific experiments, either or both of the strands were labeled with the fluorophores 6-FAM (6-carboxyfluorescein) and/or tetramethylrhodamine (TAMRA). Cluster-containing double-stranded oligonucleotides were constructed from A1, a full-length 51mer containing a single uracil, and three complementary oligonucleotides: Ba and Bc (two invariant terminal oligonucleotides) and Bb, one central, variable cassette containing one or more uracils at specific locations (Figure 1). Strand B (51 bp) was assembled from Bb (21 bp), ligated to the 3′ Ba and the 5′ Bc terminal oligonucleotides (15-bp each) as follows: equimolar quantities of oligonucleotides A, Ba, Bb and Bc were denatured for 5 min at 80°C, annealed while slowly being cooled to 20°C (at 0.01°C/s), held for 15 min at 20°C, then cooled to 4°C at 0.01°C/s. The components of B were then ligated using T4 DNA ligase according to the manufacturer's recommendations (New England Biolabs, Ipswich, MA, USA). Finally, DNA duplexes were equilibrated to a final concentration of 5 pmol/μl with 10 mM Tris, pH 7.5.
Figure 1.

True color fluorescence oligonucleotide assay. (I) Scheme for construction of dual-color fluorescently labeled oligonucleotides. The 51mer A strand contains a single uracil, whereas the opposing strand is synthesized from a central cassette (Bb, 21 bp) containing one of a number of lesion configurations, and two flanking sequences, Ba and Bc, each 15 bp. In the example shown, A contains one uracil residue, and is labeled at its 5′ end with 6-FAM; Ba is 3′ end-labeled with TAMRA, and the central Bb cassette contains one uracil residue. The components are annealed, ligated and treated with uracil DNA glycosylase to convert the uracil moieties to abasic sites. The action of Ape1 on the construct is then assessed. (II) True color denaturing gel (adjacent segments of the same gel, separated for clarity) with fluorescence of intact and Ape1-cleaved oligonucleotides. Constructs and pairs of gel lanes showing substrates (Lanes 1, 3 and 5) and products (Lanes 2, 4 and 6). Lanes 1 and 2: 51mer A1•B−5, where A1 is 5′-labeled with 6-FAM, and B-5 is 3″ TAMRA-labeled. Lane 1 intact substrate plus free, unligated TAMRA-labeled Ba); Lane 2, products of Ape1 cleavage of A1•B−5: 3′ end of B- TAMRA, 5′ end of A-FAM) plus unligated Ba. Lanes 3 and 4: A1•B−5 containing unlabelled A1 and dually labeled B-5 (3′ TAMRA and 5′ 6-FAM). Lane 3, intact substrate, a small quantity of the partial ligation product BaBb, plus unligated TAMRA-labeled Ba and 6-FAM-labeled Bc. Lane 4, Ape cleavage products: 3′ end of B, 5′ end of B plus Ba and Bc as in Lane 3. Lanes 5 and 6, Substrate and products as in Lanes 3 and 4, but Bc was 5′-labeled with JOE (6-carboxy-4′, 5′-dichloro-2′, 7′-dimethoxyfluorescein, light green) and 3′- labeled with TAMRA.
For B-strand cleavage (Figures 4–6), B was 3′ labeled with TAMRA and 5′FAM, except for examination of cleavage at the most 3′ site, B was 3′ TAMRA labeled and strand A was 5′ FAM labeled. For all constructs, both dual-label B and single-label B/single A label experiments were carried out in at least triplicate. For double-strand cleavage, A strands were 5′ FAM labeled. For one experiment, Bc was labeled with 6-carboxy-4′, 5′-dichloro-2′, 7′-dimethoxyfluorescein (JOE). Table 1 shows the sequences of the oligonucleotides and locations of the uracil residues in two-lesion and three-lesion clustered damages. Ligated constructs were checked for completeness of ligation, and preparations with <90% ligation were not used in the Ape1 cleavage experiments. For some experiments, both strands A and B were purchased as 51mer.
Figure 4.

Strand A and strand B cleavage determined on denaturing polyacrylamide gels. (I–III) negative polarity clusters: two-abasic site cluster A1•B−5, and three-abasic site clusters A1•B−5−7 and A1•B−5−9. (IV–VI) positive polarity clusters: two-abasic site cluster A1•B+1 and three-abasic site clusters A1•B+1+4 and A1•B+1+6. Strand A1, (filled triangles); 5′ end of B, (open circles); 3′ end of B, (open squares).
Figure 5.

Cleavage of strands A and B containing bipolar clustered abasic sites by 250 pg Ape1, determined on denaturing polyacrylamide gels: (I) A1•B+1−1; (II) A1•B+1−5; (III) A1•B+4−1: Solid bars, strand A; open bars, 5′ end of strand B; diagonal bars, 3′ end of strand B. Error bars, SEM.
Figure 6.

Double-strand cleavage of oligonucleotides containing negative, positive or bipolar abasic clusters, determined on nondenaturing polyacrylamide gels. (I) Oligonucleotides containing negative polarity two-lesion cluster A1•B−5 (solid bar), or three-lesion clusters A1•B−5−7 (horizontal-stripe bar) or A1•B−5−9 (diagonal-stripe bar), cleavage by 25 pg Ape1. (II) Oligonucleotides containing positive polarity two-lesion cluster A1•B+1 (solid bar), or three-lesion clusters A1•B+1+4 (open bars) and A1•B+1+6 (diagonal-stripe bar). Insert, experimental data for cleavage by 250 pg Ape1 of A1•B+1 (filled triangles), A1•B+1+4 (open circles) and A1•B+1+6 (open squares). (III) Oligonucleotides containing bipolar clusters A1•B+1−1 (crosshatch bars), A1•B+1−5 (square-grid bars) and A1•B+4−1 (open bars). Insert, experimental data for cleavage by 250 pg Ape1 of A1•B+1−1 (filled diamonds), A1•B+1−5 (open circles) and A1•B+4−1 (open squares).
Table 1.
Uracil-containing oligonucleotide duplexes
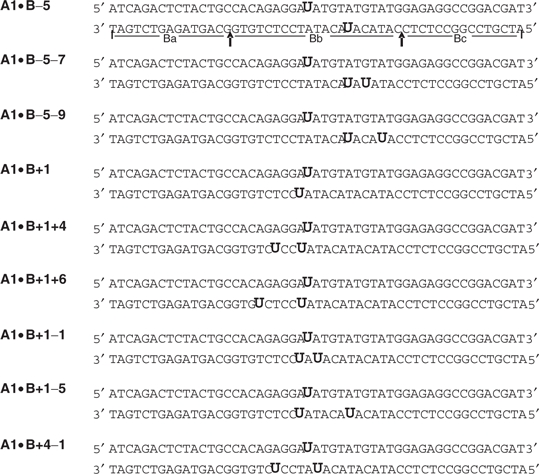 |
Sequence of duplex oligonucleotides containing abasic clusters. The upper strand A1 is invariant and contains a single Uracil (U). The B strands were constructed from the invariant 15-mers Ba and Bc (delineated by the arrows under the A1•B−5 construct) ligated to the central variable Bb cassette, containing one or more Us in specific position(s). The central Bb cassettes were the same length and position in the other constructs.
To convert uracils into abasic sites, 5 pmol duplex oligonucleotide in 5 μl buffer (50 mM HEPES pH 7.5, 50 mM KCL, 10 mM MgCl2, 1 mM DTT, 50 μg/ml BSA) was incubated with 1 U uracil-DNA glycosylase (New England Biolabs) for 30 min at 37°C, and then placed on ice. The completeness of conversion of uracil residues to abasic sites was verified by cleavage with 0.5 M NaOH or by adding 100 pg hApe1 in Ape1 buffer (100 mM HEPES pH 7.5, 50 mM KCL, 10 mM MgCl2, 1 mM DTT, 50 μg/ml BSA) to a total volume of 10 μl and incubating for 15 min at 37°C. Reactions were stopped by adding 10 μl deionized formamide and 2.5 μl loading buffer [30% glycerol, bromophenol blue in 1× TBE (100 mM Tris, 100 mM boric acid, 2 mM EDTA, pH 8.3)] and heating to 80°C for 5 min. Then 2 μl stop mix (0.1 M SDS, 30% glycerol, containing bromophenol blue in TBE buffer) was added, and the samples were incubated for 10 min at 65°C. Samples were electrophoresed on 15% denaturing polyacrylamide gels.
Enzyme reactions
The human abasic endonuclease, hApe1, was purified and characterized essentially as previously described (24). Enzyme (for readily cleaved constructs, 25 pg, shown in main panels of Figures 2, 3 and 5; for Ape1-resistant clusters, 250 pg, shown in inserts of these figures, and cleavage levels for 25 pg calculated and plotted for comparison) and 2.5 pmol of each construct were incubated at 37°C for 20 min, or as stated. Samples were electrophoresed on either 20% denaturing polyacrylamide gels for measuring single-strand cleavage, or 15% nondenaturing polyacrylamide gels for quantifying double strand cleavage.
Figure 2.

Quantitation of enzyme cleavage in dually labeled duplex oligonucleotides. (I) Linearity of TAMRA fluorescence with the quantity of dually labeled A1•B, (filled circles). (Inserts, linearity of 6-FAM fluorescence with increasing oligonucleotide quantity, linearity of ethidium bromide fluorescence with DNA mass in the same gel stained with 1 μg/ml ethidium bromide, both, filled triangles) Error bars, SD; where not shown, are smaller than the symbol showing the data point. (II) Sequence of central portion of the 51mer A1•B+1, showing abasic sites as X. Two quantitative charged coupled device-based camera images showing the time course of Ape1 action on the bistranded cluster were obtained using fluorophore-specific filters: upper image, strand A is 5′ labeled with 6-FAM. lower image, strand B is 3′ labeled with TAMRA (trace of the incompletely ligated BaBb). Strand A is poorly cleaved by Ape1, whereas the B-strand abasic site is slowly cleaved. Quantitative data from this gel (and companion gels) are shown in Figure 4 (IV).
Figure 3.

Cleavage of the A-strand abasic site in two-lesion and three-lesion abasic clusters. (I) Negative polarity clusters: A1•B−5, (filled triangles); A1•B−5−7, (open squares); A1•B−5−9, (open circles). (II) Positive polarity clusters: A1•B+1, (filled triangles); A1•B+1+4, (open squares); A1•B+1+6, (open circles). (III) Bipolar clusters: A1•B+1−1, (open triangles); A1•B+4−1, (open squares); A1•B+1−5, (open circles). In panels A and B, the solid triangles represent two-lesion simple abasic clusters; the open symbols show data for the corresponding three-lesion complex abasic clusters. Points, averages; error bars, SD; where not shown, smaller than the data symbol.
Visualizing DNA in polyacrylamide gels
After electrophoresis, two kinds of electronic images of fluorescence of DNA-bound fluorophores were obtained. For quantitative analysis, gels were illuminated by an epi-illuminator emitting UV radiation centered at 340 nm. Images were obtained by a Princeton Instruments VersArray CCD camera (Model 7380-0001; Roper Scientific, Trenton, NJ, USA) equipped with a filter that had a cut-on at 510 nm, cut-off at 530 nm, and peak transmission >65% (Omega Optical, Brattleboro, VT, USA). Quantitative electronic images were obtained (25–27), stored (28), and the FAM-fluorescence associated with each DNA species calculated using an area analysis program.
For detection of the differently colored fluorescence of the 6-FAM (green) and TAMRA (red) emissions, a true color image was obtained. Exciting radiation was from an UltraViolet Products (San Gabriel, CA, USA) Chromatovue 0-638 transilluminator (maximum emission, ∼300 nm). Images were recorded using a Canon EOS Digital Rebel XT camera using Twain imaging software.
RESULTS
To determine the roles of specific cluster properties on Ape1 cleavage, we required a substantial battery of oligonucleotides containing clusters with different numbers of lesions in specific orientations and polarities. Table 1 shows the configurations of the complete set of oligonucleotides. Rather than synthesize each strand individually, we used a constant A1 strand containing one uracil and constructed strand B from two constant terminal oligonucleotides and a central variable section. Figure 1-I shows the scheme for producing the dually labeled 51-bp duplex containing a complex bistranded clustered damage. Strand B consists of the two terminal oligonucleotides, Ba and Bc, where either or both can be labeled with a fluorophore. The 21mer central cassette Bb can contain one or more uracil residues. The components are annealed, ligated and the uracil residues converted to abasic sites by uracil-DNA glycosylase (UDG). We found that UDG effectively converted all uracil sites in the clusters shown in Table I to abasic sites (data not shown).
Figure 1-II shows a true color gel with intact 51mer A1•B−5 (Lanes 1, 3 and 5) and their Ape1 cleavage products (Lanes 2, 4 and 6). Figure 1-II shows the configuration of these oligonucleotides. In Lanes 1 and 2, strand A is 5′ labeled with 6-FAM, and strand B is 3′ labeled with TAMRA. In Lanes 3 and 4, strand A is unlabeled and strand B is labeled at its 3′ end with TAMRA and at its 5′ end with 6-FAM. Lane 5 and 6 contain unlabeled A1, and B dually labeled: 3′ with TAMRA and 5′ with JOE (6-carboxy-4′, 5′-dichloro-2′, 7′-dimethoxyfluorescein, light green). The image shows that fully, partially and unligated fragments, as well as cleavage products, can be readily identified from their fluorophore tags and migrations.
For quantitative analysis of cluster cleavage, we used a charged coupled device-based camera, in which external filters can be used to image selectively the emission of the fluorophore. We determined the direct proportionality between TAMRA and 6-FAM fluorescence and DNA amount (verified by quantifying the fluorescence of ethidium bromide staining—proportional to DNA mass—of the same bands). The fluorescence of JOE and TAMRA overlapped significantly, and therefore JOE was not used in dual-label experiments with TAMRA. Panel I of Figure 2 shows that the fluorescence—whether detected by filters that detect preferentially TAMRA or 6-FAM—of the dually labeled A•B−5 was linear with increasing DNA quantity. These data show that the TAMRA and 6-FAM fluorescence of these constructs can be used to quantify the relative proportions of the substrate and cleavage products. Representative gels for quantifying Ape1 cleavage are shown in Figure 2-II.
Ape1 cleavage of abasic sites in partial and full duplex oligonucleotides
Ape1 activity was first determined using double-stranded A1•B, containing only a single abasic site. Fifty picograms Ape 1 cleaved ∼80% of 2.5 pmol A1•B in 20 min under standard conditions. We then tested whether Ape1 action on a cluster-containing 51mer assembled from component oligomers requires ligation of those components. The cluster-containing substrate we used as a comparison standard was authentic A1•B−5, obtained as 51mer from the supplier: Ape1 cleaved 79.3 ± 4% of the substrate, comparable to the normal A1•B duplex. Notably, the enzyme had similar activities on the complete, assembled duplex (A1•BaBbBc; Figure 1), whether it was ligated (76.6 ± 5% cleaved) or not (72.7 ± 1%), suggesting that the neighboring SSBs in this context had no significant effect on Ape1 incision capacity.
We also evaluated Ape1 activity on the A-strand abasic site of three sub-components: Ape1 had no activity on single-stranded A1, as found previously by Wilson et al. (10), although we note that secondary structure can influence incision activity of Ape1 on single-stranded AP-containing oligonucleotides (29). It cleaved ∼40% of A1•Bb (a partial duplex with a 21-bp central duplex and two 15-bp single-stranded ends) and a similar level of A1•BaBb (partial duplex with a 3′ overhang of 15 bp). These results show that Ape1 can cleave a substantial range of cluster configurations. They further show that even gapped regions outside the immediate cluster region are not severe impediments to Ape1 cleavage at a central abasic site.
Cluster complexity and Ape1 cleavage
To test whether the number of lesions in a cluster determined its susceptibility to Ape1 action, we first measured Ape1 cleavage of a two-lesion cluster shown previously to be readily cleaved on both strands by this enzyme (7). Figure 3-I shows a diagram of three negative polarity clusters [B-strand abasic site(s) located 5′ to the base opposite the central A-strand abasic site]: the two-lesion A1•B−5 construct, and two three-lesion clusters formed by the addition of a second abasic site, producing A1•B−5−7 and A1•B−5−9 (Table 1). The A1•B−5−7 construct contains an additional abasic site on the B strand 2 nt from the −5 site, whereas the additional abasic site in the A1•B−5−9 duplex is 4 nt from the −5 site. The graph in Figure 3-I shows the quantitative data for cleavage of these constructs by 25 pg Ape1. The rates of cleavage of the A1 abasic site in the simpler two-lesion cluster and more complex three-lesion cluster are quite similar, with the abasic site in the two-lesion cluster actually being cleaved slightly more slowly than in the three-lesion cluster.
In contrast, the positive polarity abasic clusters [B-strand abasic site(s) located 3′ to base opposite the central abasic site on strand A] are all resistant to Ape1 cleavage (Panel II, Figure 3). The insert shows the data for cleavage of three such cluster configurations by 250 pg Ape1 (the two-lesion A1•B+1 and the three-lesion clusters A1•B+1+4 and A1•B+1+6; Table 1). The two-lesion cluster and the three-lesion cluster A1•B+1+4 were both cleaved slowly, but the A1•B+1+6 construct was resistant to cleavage.
Since these data indicated a strong polarity effect on susceptibility of abasic sites in clusters to cleavage, we investigated the cleavage of bipolar clusters, i.e. those whose B strand contains one abasic site situated in the positive direction and one in the negative direction from the abasic site on A1 (see the diagram in Figure 3-III). The graph in Figure 3-III shows that treatment with high concentrations of Ape1 (i.e. 250 pg) was required to produce cleavage of the very closely spaced three-lesion cluster A1•B+1−1 and of the A1•B+4−1 cluster. No cleavage of the A1•B+1−5 cluster could be detected.
Cleavage of A or B strands in specific clustered damages
Panel I of Figure 3 shows that the A1-abasic site in A1•B−5 is readily cleaved by Ape1, indicating that Ape1 action would produce a SSB. However, considerable previous data showed that many two-lesion bistranded clusters could be converted to DSBs (9,11–13). To determine whether Ape1 action on the A1•B−5 cluster produces a DSB, we assessed the cleavage of the abasic sites on strand B as well as strand A. Figure 4-I shows that in the two-lesion cluster A1•B−5, the abasic sites on the A and the B strands are cleaved readily. Figure 4-II shows that the addition to the cluster in A1•B−5 of another lesion can have a striking effect on specific strand cleavage, depending on the configuration of the multiple lesions. In the A1•B−5−9 cluster, the A strand and both B strand abasic sites are cleaved (Figure 4-III). However, in the more closely spaced cluster A1•B−5−7, both abasic sites on the B strand are cleaved only slowly (Figure 4-II), yet the A-strand abasic site is cleaved rapidly. We also examined Ape1 cleavage of the same B-strand lesion configurations, but with no lesions on the A strand, i.e. a unistranded multiply damaged site or tandem cluster. The comparable unistranded tandem clusters on strand B to the bistranded A1•B−5−9 were cleaved about twice as well as the bistranded cluster. Further, the comparable tandem cluster to A1•B−5−7 was cleaved about three times as well as the bistranded A1•B-5-7.
Examination of A-strand abasic site cleavage in positive polarity clusters (Figure 3-II; and for a representative gel Figure 2-II) showed the resistance of the abasic site to Ape1 action. The presence of the B-strand abasic site appears to inhibit A-strand cleavage. However, this did not preclude the possibility that the abasic sites in the B strand were susceptible to Ape1 cleavage, and thus a SSB could be generated. Panel IV of Figure 4 shows that at high Ape1 levels, the B-strand abasic site of A1•B+1 is cleaved, generating the 5′ and 3′ ends of B as shown in the insert. Thus this cluster configuration leads to the slow production of a SSB (or gap). Likewise, although the A-strand abasic sites in A1•B+1+4 (Figure 4-V) and in A1•B+1+6 (Figure 4-VI) are poorly cleaved, high levels of Ape1 cleaved both B-strand abasic sites in both constructs, thus generating SSBs. Ape1 cleavage of comparable unistranded tandem clusters, as seen in strand B of the two substrates above, was 4–10 times higher than that of the bistranded positive polarity clusters (data not shown).
Likewise, Figure 5-I shows that in bipolar clusters, high Ape1 levels produce moderate A-strand abasic site cleavage that is accompanied by substantial cleavage of both B-strand abasic sites. In A1•B+4−1, cleavage of B-strand abasic sites clusters are substantial at high Ape1 levels (Figure 5-II), whereas in A1•B+1−5, production of the 3′-labeled end is substantially slower than of the 5′ labeled end product (Figure 5-III). In A1•B+1−5, the virtual absence of A-strand cleavage precludes the generation of DSBs. In A1•B+4−1, a low level of DSBs could be generated slowly (Figure 5-III). Similarly to the positive and negative polarity clusters, the comparable tandem unistranded clusters were cleaved 3–8 times better than the corresponding bistranded bipolar cluster.
High levels of SSBs in both A and B strands probably reflect the generation of DSBs. However, in constructs with only partial cleavage of one or both strands, the outcome cannot be determined by examination of individual strand cleavage using denaturing gels. To test whether SSBs or DSBs were generated by Ape1 action, we treated the cluster-containing oligonucleotide duplexes with Ape1 and dispersed the resulting products on nondenaturing polyacrylamide gels for detection of double-strand cleavage products. Figure 6-I shows that the negative polarity clusters (A1•B−5, A1•B−5−7 and A1•B−5−9) are all cleaved by Ape1 to produce DSBs. The three-abasic site complex clusters were converted to DSBs with approximately the same kinetics as the two-lesion abasic clusters.
In contrast, two of the positive abasic site clusters were quite resistant to conversion to DSBs (Figure 6-II). Even in reactions employing 250 pg Ape1 (shown in the insert), both the two-lesion cluster A1•B+1 and the three-lesion cluster A1•B+1+4 were resistant to DSB induction. DSBs were slowly induced at the three-lesion cluster A1•B+1+6.
Low levels of Ape1 produce little cleavage of bipolar clusters. The outcome of treatment of a bipolar cluster with high levels of Ape1 depends strongly on the configuration of the cluster (Figure 6-III). None were rapidly cleaved to DSBs at Ape1 levels comparable to those readily generating DSBs in the negative polarity clusters. However, at high Ape1 levels (250 pg), generation of DSBs in both the A1•B+1-1 and A1•B+4−1 constructs was detectable. The A1•B+1−5 cluster was resistant to DSB cleavage.
DISCUSSION
Abasic clusters are potentially lethal and mutagenic damages that persist in cells long after most DSBs have been rejoined (13). The biological consequences of specific clusters depend largely on the interactions between individual clusters and the repair enzymes that process them. The properties—for example, the number of constituent lesions and their configuration—of the specific clusters are thus a major determinant of their biological impact. The higher biological damage inflicted by high LET radiation has been assumed to stem from complex, multilesion clusters that are more difficult to repair than the presumably simpler clusters induced by X- or γ-rays. To evaluate this hypothesis, we have investigated the properties of multilesion abasic clusters that affect Ape1 cleavage, and specifically the role of increased numbers of constituent lesions in a cluster.
Ape1 activity on positive and negative polarity clusters
We asked if Ape1 activity on multilesion clusters can be predicted from its action toward isolated abasic sites or component two-lesion clusters. Wilson et al. (10) showed that for isolated abasic sites, Ape1 requires double-helical structure for at least 4 bp on the 5′ side of the abasic site, but only 3 bp on the 3′ side. In addition, a mispaired nucleotide arrangement immediately 5′ to the AP lesion had a more severe negative impact on Ape1 incision efficiency relative to a 3′ mispair. This latter quality was evident with the two-lesion clusters: the negative polarity −3 and −1 abasic clusters (in which the 5′ side of the target abasic sites is free of lesions) were well cleaved, whereas Ape1 exhibited poor activity toward positive polarity clusters of the same spacing (30,31). David-Cordonnier et al. (31) also showed that positive polarity two-site abasic clusters with more widely spaced constituent lesions are more readily cleaved than closely spaced clusters.
The same Ape1 requirements are also important in cleavage of multilesion clusters. Panel I of Figure 3 clearly shows good Ape1 activity on the A-strand abasic sites in the negative polarity three-lesion clusters. In such clusters, the 5′ side contains no opposing lesions, and the inter-lesion gap is sufficiently large to exceed the 3′ 3-nt spacing requirement. Figure 3-II shows that Ape1 has poor activity on positive polarity constructs with multiple abasic sites on the opposing strand and 5′ to the A-strand abasic site. Activity on the two B-strand abasic sites of the three-member clusters also reflects this property: in A1•B−5−7 (and similarly in A1•B−5−9), the close spacing of the two abasic sites results in decreased cleavage.
Ape1 and bipolar clusters
In bipolar clusters, the A-strand abasic site is opposed on the B-strand by (at least) two abasic sites: one abasic site located 5′ and one located 3′ to the position opposite the A-strand lesion. All the three-member bipolar abasic clusters were poor substrates for Ape1, requiring high enzyme levels for detectable cleavage (Figure 3-III). In each of the bipolar clusters, both the A-strand abasic site and the +1 and +4 B-strand sites have opposing abasic sites within the critical distance for intact double strandedness required for efficient Ape1 activity. Thus, the requirement of 250 pg of Ape1 for productive cleavage at these sites is consistent with the aforementioned prerequisites. Ape1 also cleaved poorly at the −1 and −5 B-strand sites of the bipolar clusters, which are not opposed by other lesions within the 5′ 4-bp Ape1 requirement. However, the −1 strand sites are within the smaller 3′ 3-bp critical distance. In contrast, in two-membered bistranded abasic clusters, −1 sites are well cleaved by mammalian cell extracts (32).
The structures of two-member positive and negative polarity abasic clusters have been determined by NMR by Lin and de los Santos (33), revealing significant differences in the local helical configuration surrounding the damaged region. However, the structures of DNAs containing multilesion abasic clusters have not been characterized. It seems likely that structural alterations by these complex damages could perturb the local helical conformation so as to preclude good Ape1 activity in bipolar clusters whose configuration otherwise meets Ape1 spacing requirements.
Cluster complexity and radiation
Multiple mechanisms can explain the poor repair and thus higher biological impact of high LET radiation-induced damages. One possibility is that high LET radiation produces multiple-lesion clusters, and thus the two-lesion, repairable clusters of one polarity would be replaced by three-lesion, poorly repairable clusters of the same polarity. This possibility is not supported by the current data, since positive polarity clusters are poor substrates for Ape1, whether they contain two or more lesions. Moreover, negative polarity clusters, whether composed of two or more lesions, are equally good Ape1 substrates.
Instead the current data suggest a different mechanism for the decreased DNA repair and increased biological damage produced by high LET radiation. Low LET radiation induces isolated lesions (strand breaks, oxidized bases and abasic sites) and two-lesion clusters, but probably few multilesion clusters. Assuming random distribution of lesions within a cluster, the two-lesion clusters—whether induced directly by radiation or resulting from partial processing of oxidized base clusters—will consist of approximately equal levels of positive polarity and negative polarity clusters. Since positive polarity abasic clusters are poor substrates for Ape1 and negative polarity clusters are good Ape1 substrates, about 50% of the abasic clusters would be well repaired and 50% poorly repaired.
Higher LET radiation makes fewer two-lesion clusters, but predominantly three- or higher-member clusters of closely spaced lesions. There is an upper limit to the number of lesions in an oxidized base- or abasic cluster, since SSBs can also be induced as a member lesion within a cluster. As the number of lesions increases, the probability that the cluster will contain two closely opposed SSBs also increases. Such a cluster would be counted as a DSB, even if additional oxidized bases or abasic sites were nearby. With even a small increase in the average number of lesions in the high LET radiation-induced clusters, bipolar clusters are formed at an approximate ratio of 2 bipolar clusters: 1 positive polarity cluster: 1 negative cluster. Since both the bipolar clusters and the positive polarity clusters are poorly repaired by Ape1 (while negative polarity clusters constitute the one class complex damages that are repaired well), this now results in ∼75% of the abasic clusters being poor Ape1 substrates and only ∼25% being good substrates.
DSB production from minimal and complex clusters in vitro and in cells
Ape1 cleavage of both DNA strands at two- and three-lesion cluster sites is strongly dependent on the polarity and spacing of the constituent lesions relative to the spatial requirements of the enzyme. The same requirements hold for clusters containing SSB(s). We studied ligated and unligated constructs (A1•BaBb1Bc, where Bb1 contains a single abasic site) that provide closely related two-lesion and four-lesion clusters, respectively. In addition to the abasic sites, unligated A1•B−5 contains two nicks (SSBs), one immediately 3′ of nucleotide +9 and one 5′ of nucleotide −11, as well as the core bistranded abasic sites. The A-strand abasic site was readily cleaved in these two constructs since Ape1's spatial requirements are still met in these configurations. David-Cordonnier et al. (34) found that some SSB-containing clusters were incised poorly by Ape1; however, these poorly cleaved constructs contained an SSB within three bases of the abasic site. The greater inter-base spacing of the four-lesion construct we studied permitted Ape1 cleavage of these complex substrates. Such data indicate that the immediate neighborhood of the abasic site is critical in determining reparability.
Previous work on minimal clusters showed that many two-lesion clusters could be readily converted to DSBs by purified enzymes or by cell extracts (9,11–13). Further, mammalian cells exposed to high radiation doses also produced de novo DSBs during postirradiation incubation (14–16). At lower radiation doses, repair-competent human cells do not produce repair-generated DSBs (13). One possible explanation for the lack of observable de novo DSBs at low radiation doses might be that any DSB products resulting from abortive cluster repair were ligated so rapidly that they were difficult to detect. However, Gulston et al. (17) showed that in xrs-5, a nonhomologous end-joining mutant CHO line, a low level of DSBs—corresponding to only about 10% of the levels of clusters—accumulated during postirradiation incubation. Thus cells appear to have strategies for cluster repair other than DSB generation, or at least alternative mechanisms for their efficient resolution. Figures 3 and 5 suggest two possible ‘repair’ modes.
First, Ape1 induces a SSB at some lesion sites within a cluster. This strand break induction could represent an early step in normal base excision repair (BER), in which the polarity or proximity of other abasic sites within the cluster prevent simultaneous scission of the opposing strand and DSB generation. In Escherichia coli—depending on the cluster configuration and cellular genotype—some clusters are processed sequentially while others are converted to DSBs (35–37). Strong evidence indicates that component sites in two-membered bistranded clusters are repaired sequentially by extracts of Ku80-deficient Chinese hamster ovary cells (32). Eot-Houllier et al. (38,39) studied multilesion clusters containing oxidized bases and small gaps, and also found avoidance of DSB induction by both purified repair enzymes and cell extracts.
Second, some clusters are highly resistant to processing by Ape1, thus resulting in production of no or few SSBs or DSBs from these configurations. Examples include the positive clusters shown in Figure 4 and the bipolar clusters shown in Figure 5. These highly repair-refractory clusters could escape all the cellular repair mechanisms, including BER and DSB rejoining, and persist until DNA replication. An error-prone polymerase could then replicate cellular DNA on the damaged template, possibly producing mutations and ‘diluting’ the cluster into isolated lesions segregated to the two double-stranded daughter DNA products (13). This suggests that cellular responses to complex radiation-induced damages may be carried out by multiple processing mechanisms: BER to deal with component lesions of a cluster, nonhomologous end joining (and perhaps homologous recombination) for processing any de novo DSBs produced in attempted cluster repair, and conventional repair to deal with the single damaged sites that arise following error-prone replication of the original damaged template. The more complex nature of high LET-induced DNA damage would suggest that such lesions are more susceptible to mutagenic coping mechanisms or to inducing cell death outcomes. A complete resolution of the coordination and execution of cellular responses to DNA damage clusters awaits further investigation.
ACKNOWLEDGEMENTS
We thank Dr John Sutherland, John Trunk and Denise Monteleone (BNL) for use of ImageSystem; Dr John Dunn (BNL) for advice on the constructs, and Keith Thompson (BNL) for statistical analysis of the data. This work was funded by Low Radiation Dose Program of the Office of Biological and Environmental Research of the US Department of Energy and the Human Research Program of the Exploration Systems Mission Directorate of the National Aeronautics and Space Administration [joint agency funding, DEAC 02-98CH10886 (BO-086) to B.M.S.]; National Space Biomedical Institute (IIH00404 to A. Gewirtz, University of Pennsylvania and B.M.S.); Human Research Program of the Exploration Systems Mission Directorate of the National Aeronautics and Space Administration, (T-467X to B.M.S.); National Institutes of Health (R01 CA86897 to B.M.S.) Brookhaven National Laboratory Pollution Prevention Program (to B.M.S. and B.P.), and the Intramural Research Program of the NIH, NIA (D.M.W.III). Funding to pay the Open Access publication charges for this article was provided by the Low Dose Radiation Program, US Department of Energy.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sutherland BM, Bennett PV, Sidorkina O, Laval J. DNA damage clusters Induced by ionizing radiation in isolated DNA and in human cells. Proc. Natl Acad. Sci. USA. 2000;97:103–108. doi: 10.1073/pnas.97.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Povirk LF. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 3.Song JM, Milligan JR, Sutherland BM. Bistranded oxidized purine damage clusters: induced in DNA by long-wavelength ultraviolet (290-400 nm) radiation? Biochemistry. 2002;41:8683–8688. doi: 10.1021/bi020137x. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PV, Cintron NS, Gros L, Laval J, Sutherland BM. Are endogenous clustered DNA damages induced in human cells? Free Radic. Biol. Med. 2004;37:488–499. doi: 10.1016/j.freeradbiomed.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Bennett PV, Cuomo NL, Paul S, Tafrov ST, Sutherland BM. Endogenous DNA damage clusters in human skin, 3-D model, and cultured skin cells. Free Radic. Biol. Med. 2005;39:832–839. doi: 10.1016/j.freeradbiomed.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Regulus P, Duroux B, Bayle PA, Favier A, Cadet J, Ravanat JL. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc. Natl Acad. Sci. USA. 2007;104:14032–14037. doi: 10.1073/pnas.0706044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georgakilas AG, Bennett PV, Sutherland BM. High efficiency detection of bistranded abasic clusters in γ-irradiated DNA by putrescine. Nucleic Acids Res. 2002;30:2800–2808. doi: 10.1093/nar/gkf393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hada M, Sutherland BM. Spectrum of Complex DNA Damages Depends on the Incident Radiation. Radiat. Res. 2006;165:223–230. doi: 10.1667/rr3498.1. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhry MA, Weinfeld M. The action of Escherichia coli Endonuclease III on multiply damaged sites in DNA. J. Mol. Biol. 1995;249:914–922. doi: 10.1006/jmbi.1995.0348. [DOI] [PubMed] [Google Scholar]
- 10.Wilson DM, III, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 1995;270:16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 11.Harrison L, Hatahet Z, Purmal AA, Wallace SS. Multiply damaged sites in DNA: interactions with Escherichia coli endonucleases III and VIII. Nucleic Acids Res. 1998;26:932–941. doi: 10.1093/nar/26.4.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunniffe S, O’Neill P. The complexity of radiation-induced DNA damage as revealed by exposure to cell extracts. Radiat. Res. 1999;152:421–427. [PubMed] [Google Scholar]
- 13.Georgakilas AG, Bennett PV, Wilson DM, III, Sutherland BM. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 2004;32:5609–5620. doi: 10.1093/nar/gkh871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dugle D, Gillespie C, Chapman JD. DNA strand breaks, repair and survival in X-irradiated mammalian cells. Proc Natl. Acad. Sci. USA. 1976;73:809–812. doi: 10.1073/pnas.73.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahnstrom G, Bryant PE. DNA double-strand breaks generated by the repair of X-ray damage in Chinese hamster cells. Int. J. Radiat. Biol. 1982;41:671–676. doi: 10.1080/09553008214550761. [DOI] [PubMed] [Google Scholar]
- 16.Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 2004;3:1323–1334. doi: 10.1016/j.dnarep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Gulston M, de Lara C, Jenner T, Davis E, O’Neill P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004;32:1602–1609. doi: 10.1093/nar/gkh306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Radiat. Biol. 1994;65:7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 19.Nikjoo H, O’Neill P, Wilson WE, Goodhead DT. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001;156:577–583. doi: 10.1667/0033-7587(2001)156[0577:cafdts]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Yang TC, Mei M, George KA, Craise LM. DNA damage and repair in oncogenic transformation by heavy ion radiation. Adv. Space Res. 1996;18:149–158. doi: 10.1016/0273-1177(95)00801-k. [DOI] [PubMed] [Google Scholar]
- 21.Yang H, Anzenberg V, Held KD. Effects of heavy ions and energetic protons on normal human fibroblasts. Radiat. Biol. Radioecol. 2007;47:302–305. [PubMed] [Google Scholar]
- 22.Fry RJM, Powers-Risius P, Alpen EL, Ainsworth EJ. High-LET radiation carcinogenesis. Radiat. Res. 1985;104:S188. [PubMed] [Google Scholar]
- 23.Kranert T, Schneider E, Kiefer J. Mutation induction in V79 Chinese hamster cells by very heavy ions. Int. J. Radiat. Biol. 1990;58:975–987. doi: 10.1080/09553009014552291. [DOI] [PubMed] [Google Scholar]
- 24.Erzberger JP, Barsky D, Scharer OD, Colvin ME, Wilson DM., III Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 1998;26:2771–2778. doi: 10.1093/nar/26.11.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutherland JC, Lin B, Monteleone DC, Mugavero J, Sutherland BM, Trunk J. Electronic imaging system for direct and rapid quantitation of fluorescence from electrophoretic gels: application to ethidium bromide-stained DNA. Anal. Biochem. 1987;163:446–457. doi: 10.1016/0003-2697(87)90247-8. [DOI] [PubMed] [Google Scholar]
- 26.Sutherland JC, Sutherland BM, Emrick A, Monteleone DC, Ribeiro EA, Trunk J, Son M, Serwer P, Poddar SK, Maniloff J. Quantitative electronic imaging of gel fluorescence with charged coupled device cameras: applications in molecular biology. Biotechniques. 1991;10:492–497. [PubMed] [Google Scholar]
- 27.Sutherland JC, Monteleone DC, Trunk JG, Bennett PV, Sutherland BM. Quantifying DNA damage by gel electrophoresis, electronic imaging and number-average length analysis. Electrophoresis. 2001;22:843–854. doi: 10.1002/1522-2683()22:5<843::AID-ELPS843>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Sutherland JC, Monteleone DC, Sutherland BM. Computer network for data acquisition, storage and analysis. J. Photochem. Photobiol. B. 1997;40:14–22. doi: 10.1016/s1011-1344(97)00030-4. [DOI] [PubMed] [Google Scholar]
- 29.Fan J, Matsumoto Y, Wilson DM., III Nucleotide sequence and DNA secondary structure, as well as replication protein A, modulate the single-stranded abasic endonuclease activity of APE1. J. Biol. Chem. 2006;281:3889–3898. doi: 10.1074/jbc.M511004200. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhry MA, Weinfeld M. Reactivity of human apurinic/apyrimidinic endonuclease and Escherichia coli exonuclease III with bistranded abasic sites in DNA. J. Biol. Chem. 1997;272:15650–15655. doi: 10.1074/jbc.272.25.15650. [DOI] [PubMed] [Google Scholar]
- 31.David-Cordonnier M-H, Cunniffe S, Hickson I, O’Neill P. VIIth International Workshop on Radiation Damage to DNA. France: Orleans- Nouan le Fuzelier; 2001. [Google Scholar]
- 32.Lomax ME, Cunniffe S, O’Neill P. Efficiency of repair of an abasic site within DNA clustered damage sites by mammalian cell nuclear extracts. Biochemistry. 2004;43:11017–11026. doi: 10.1021/bi049560r. [DOI] [PubMed] [Google Scholar]
- 33.Lin Z, de los Santos C. NMR characterization of clustered bistrand abasic site lesions: effect of orientation on their solution structure. J. Mol. Biol. 2001;308:341–352. doi: 10.1006/jmbi.2001.4587. [DOI] [PubMed] [Google Scholar]
- 34.David-Cordonnier MH, Cunniffe SM, Hickson ID, O’Neill P. Efficiency of incision of an AP site within clustered DNA damage by the major human AP endonuclease. Biochemistry. 2002;41:634–642. doi: 10.1021/bi011682l. [DOI] [PubMed] [Google Scholar]
- 35.Malyarchuk S, Youngblood R, Landry AM, Quillin E, Harrison L. The mutation frequency of 8-oxo-7,8-dihydroguanine (8-oxodG) situated in a multiply damaged site: comparison of a single and two closely opposed 8-oxodG in Escherichia coli. DNA Repair (Amst.) 2003;2:695–705. doi: 10.1016/s1568-7864(03)00040-5. [DOI] [PubMed] [Google Scholar]
- 36.D'Souza DI, Harrison L. Repair of clustered uracil DNA damages in Escherichia coli. Nucleic Acids Res. 2003;31:4573–4581. doi: 10.1093/nar/gkg493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison L, Brame KL, Geltz LE, Landry AM. Closely opposed apurinic/apyrimidinic sites are converted to double strand breaks in Escherichia coli even in the absence of exonuclease III, endonuclease IV, nucleotide excision repair and AP lyase cleavage. DNA Repair (Amst.) 2006;5:324–335. doi: 10.1016/j.dnarep.2005.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eot-Houllier G, Eon-Marchais S, Gasparutto D, Sage E. Processing of a complex multiply damaged DNA site by human cell extracts and purified repair proteins. Nucleic Acids Res. 2005;33:260–271. doi: 10.1093/nar/gki165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eot-Houllier G, Gonera M, Gasparutto D, Giustranti C, Sage E. Interplay between DNA N-glycosylases/AP lyases at multiply damaged sites and biological consequences. Nucleic Acids Res. 2007;35:3355–3366. doi: 10.1093/nar/gkm190. [DOI] [PMC free article] [PubMed] [Google Scholar]