

Abstract
Axonal injury is a hallmark of traumatic brain injury (TBI) and is associated with a poor clinical outcome. Following central nervous system injury, axons regenerate poorly, in part due to the presence of molecules associated with myelin that inhibit axonal outgrowth, including myelin-associated glycoprotein (MAG). The involvement of MAG in neurobehavioral deficits and tissue loss following experimental TBI remains unexplored and was evaluated in the current study using an MAG-specific monoclonal antibody (mAb). Anesthetized rats (n = 102) were subjected to either lateral fluid percussion brain injury (n = 59) or sham injury (n = 43). In surviving animals, beginning at 1 h post-injury, 8.64 μg anti-MAG mAb (n = 33 injured, n = 21 sham) or control IgG (n = 26 injured, n = 22 sham) was infused intracerebroventricularly for 72 h. One group of these rats (n = 14 sham, n = 11 injured) was killed at 72 h post-injury for verification of drug diffusion and MAG immunohistochemistry. All other animals were evaluated up to 8 weeks post-injury using tests for neurologic motor, sensory and cognitive function. Hemispheric tissue loss was also evaluated at 8 weeks post-injury. At 72 h post-injury, increased immunoreactivity for MAG was seen in the ipsilateral cortex, thalamus and hippocampus of brain-injured animals, and anti-MAG mAb was detectable in the hippocampus, fimbria and ventricles. Brain-injured animals receiving anti-MAG mAb showed significantly improved recovery of sensorimotor function at 6 and 8 weeks (P < 0.01) post-injury when compared with brain-injured IgG-treated animals. Additionally, at 8 weeks post-injury, the anti-MAG mAb-treated brain-injured animals demonstrated significantly improved cognitive function and reduced hemispheric tissue loss (P < 0.05) when compared with their brain-injured controls. These results indicate that MAG may contribute to the pathophysiology of experimental TBI and treatment strategies that target MAG may be suitable for further evaluation.
Keywords: adhesive/sticky paper test, head injury, hemispheric tissue loss, myelin-associated glycoprotein, osmotic minipumps, rat
Introduction
Each year, traumatic brain injury (TBI) affects 2 000 000−3 000 000 people in the USA (NIH Consensus Development Panel, 1999) and more than 1 000 000 people in the European Union require hospitalization as a result of TBI (International Brain Injury Association, 2005). Currently no standardized pharmacologic therapy with proven clinical benefit is available for the treatment of TBI, despite the promising evaluation of several therapeutic approaches to experimental TBI using clinically relevant experimental models (Bullock et al., 1999; Laurer & McIntosh, 2001; Maas, 2002). Most experimental studies, to date, have focused on the evaluation of acute neuroprotection in the immediate post-injury period (for review see Royo et al., 2003), rather than an assessment of novel strategies to enhance the inherent regenerative and repair capacity of the central nervous system.
Axonal injury is a common finding following experimental and clinical TBI (Gentleman et al., 1995; Povlishock et al., 1999; Graham et al., 2000; Adams et al., 2001). The regeneration of axons post-injury may be limited due to numerous factors including insufficient trophic support, the formation of a physical barrier such as the glial scar, insufficient induction and activation of appropriate growth-promoting proteins and/or the presence of a hostile axonal microenvironment including molecules inhibitory to axonal outgrowth (for review see Fawcett & Asher, 1999; Silver & Stryker, 2001; Filbin, 2003; Lee et al., 2003; Sandvig et al., 2004; Schwab, 2004). Both in vitro and in vivo evidence suggests that inhibitors of axonal growth present in myelin, such as Nogo-A, oligodendrocyte-myelin glyco-protein and myelin-associated glycoprotein (MAG), may prevent axonal outgrowth in models of nervous system injury such as cerebral ischemia, traumatic spinal cord injury and peripheral nerve injury (Caroni et al., 1988; Caroni & Schwab, 1988; McKerracher et al., 1994; Mukhopadhyay et al., 1994; Prinjha et al., 2000; Kottis et al., 2002; Wang et al., 2002b). These results suggest that myelin-associated inhibitors of axonal outgrowth may also possibly contribute to the poor functional recovery seen following TBI. Recently, we have evaluated the effects of Nogo-A inhibition using the Nogo-A-neutralizing monoclonal antibodies (mAbs) 11C7 and 7B12 following experimental TBI using the lateral fluid percussion (FP) injury model and observed improved cognitive recovery at 4 weeks post-injury compared with brain-injured rats treated with vehicle (Lenzlinger et al., 2005; Marklund et al., 2004). These findings support those in other models of central nervous system injury (Papadopoulos et al., 2002; Emerick et al., 2003; Li & Strittmatter, 2003) where antibody treatment against Nogo and its receptor has resulted in improved functional recovery compared with knockouts (Marklund et al., 2005).
Myelin-associated glycoprotein, a 100-kDa transmembrane protein (Quarles et al., 1973), plays a role in both signal transduction and cell adhesion (Paivalainen et al., 2003). MAG has been localized to cell surfaces of oligodendrocytes and Schwann cells via immunohistochemistry (Bartsch et al., 1989; Trapp et al., 1989) and interacts with neuronal receptors. The inhibition of axonal outgrowth by MAG, Nogo-66 and oligodendrocyte-myelin glycoprotein is mediated by binding to either the NOGO-66 (NgR1) or the NgR2 receptors and activation of downstream mediators (Wang et al., 2002a; Venkatesh et al., 2005). However, Nogo-A, oligodendrocyte-myelin glycoprotein and MAG may use additional neuronal receptors, indicating the potential for differing functional activities (Irving et al., 2005; Vinson et al., 2003). The inhibitory effects of MAG in vitro have been restricted to adult neurons (McKerracher et al., 1994; Mukhopadhyay et al., 1994) and during development, MAG is believed to play a beneficial role in both the process of myelination and the maintenance of myelin (Fruttiger et al., 1995; Schachner & Bartsch, 2000; Marcus et al., 2002). In vitro neutralization of a soluble form of MAG (dMAG) resulted in an increase in neurite outgrowth (Tang et al., 2001) and inactivation of MAG in vitro immediately post-optic nerve crush injury has been shown to improve regeneration of the optic nerve tract (Wong et al., 2003).
Myelin-associated glycoprotein also plays a role in the maintenance and stabilization of myelin. The cytoplasmic tail interacts with signaling molecules and cytoskeletal components and the absence of MAG results in a progressive ‘dying-back oligodendropathy’ (Weiss et al., 2000). Therefore, under non-disease conditions, the interaction between MAG and neuronal receptors triggers a signaling cascade within oligodendrocytes resulting in maintenance of structure and integrity, possibly via Fyn kinase activation. A pro-survival response in oligodendrocytes is also triggered using an anti-MAG mAb to mimic neuronal receptors (Irving et al., 2005).
Recently, the properties of an anti-MAG mAb have been investigated. This antibody binds specifically to MAG, blocks interaction with neuronal MAG receptors and prevents MAG-mediated inhibition of neurite outgrowth (Irving et al., 2005). Furthermore, the antibody prevents glutamate-mediated toxicity of oligodendrocytes in culture. In vivo, the antibody significantly improves motor function and reduces lesion volume up to 1 week following middle cerebral artery occlusion-induced focal ischemia in rat (Irving et al., 2005).
Although these studies suggest that inhibition of MAG results in neuroprotection, the inhibition of MAG as a treatment strategy has not been examined, to date, in TBI. In the present study, we evaluated the effects of MAG inhibition on neurobehavioral deficits and histological damage using neurological motor, sensory and cognitive function, and hemispheric tissue loss up to 8 weeks post-injury following administration of monoclonal mouse anti-MAG antibody into the ipsilateral ventricle of rats from 1 to 72 h following TBI.
Materials and methods
All procedures were approved by the Institutional Animal Care and Use Committee at the The University of Pennsylvania and were performed in accordance with standards published by the National Research Council (1996).
Surgery and fluid percussion brain injury
Adult male Sprague-Dawley rats (350−400 g, n = 102) were anesthetized (sodium pentobarbital, 65 mg/kg, i.p.) and placed into a stereotaxic frame. A subcutaneous injection of bupivicaine (0.25%; 0.2−0.3 ml) was administered at the incision site. At approximately 5 min following injection, a midline scalp incision was made and the underlying periosteum dissected. The scalp and left temporal muscle were reflected, exposing the skull and a left parietal 5-mm craniectomy was performed midway between lambda and bregma, and 4 mm lateral to the midline over the left parietal cortex. A modified Luer-Lok cap was cemented over the craniectomy and filled with saline. At 90 min after administration of anesthesia, lateral FP brain injury of moderate (2.9−3.1 atm) severity (n = 59) was induced as originally described by McIntosh et al. (1989). Sham (control) animals (n = 43) received anesthesia and all surgical procedures without FP brain injury. The Luer-Lok fitting was then removed and the incision sutured. Animals were placed on heating pads from the initiation of anesthesia until 60 min post-pump implantation in order to maintain normothermia.
Pump implantation and intracerebroventricular drug administration
At 1 h post-injury, surviving animals were randomized to receive an intracerebroventricular injection of either 0.12 mg/mL inhibitory anti-MAG mAb (72 μL; a kind gift from Glaxo Smith Kline, antibody originally from Chemicon, Hampshire, UK, with additional preparation as per Irving et al., 2005) or control antibody (equal volume and concentration of mouse IgG) (Irving et al., 2005), injected over 72 h at a rate of 1 μL/h. Separate groups of brain-injured or sham-injured animals received an identical volume of control Ab (IgG). The total dose of anti-MAG mAb administered per animal was 8.64 μg.
For antibody delivery, osmotic mini-pumps (1003D, ALZET, Cupertino, CA, USA) were attached to a cannula for intracerebroventricular infusion (Brain Infusion Kit I, ALZET) and implanted according to the manufacturer's instructions. The mini-pumps were filled under sterile conditions, attached to a 35-mm-long polyethylene catheter and infusion cannula, and primed overnight at 37 °C in 0.9% sodium chloride. The animals were reanesthetized (isoflurane, 1.5− 3%) and placed into a stereotaxic frame. To accommodate the mini-pumps, the existing scalp incision was reopened and a subcutaneous pocket was created between the scapulae by blunt dissection. The infusion cannula was stereotactically inserted at 0.8 mm posterior to bregma (AP 0.8), 1.3 mm lateral (left) to the midline (ML 1.3) and a depth of 3.8 mm from the surface of the cortex (DV −3.80) into the left lateral ventricle. Using sterile technique, the pump assembly was inserted into the subcutaneous pocket and the cannula was inserted at the above coordinates and secured to the bone with tissue adhesive (Vetbond, 3M, St Paul, MN, USA) over which the scalp was sutured. On day 3 post-injury, all animals were reanesthetized (isoflurane, 1.5− 3%), the polyethylene infusion catheter was tied off and the pump was removed.
Study A. Evaluation of drug penetration and myelin-associated glycoprotein expression post-injury
To examine the penetration of anti-MAG mAb into brain tissue and immunohistochemistry for MAG in the early post-injury phase, a subgroup of brain-injured animals was randomly assigned to receive either the inhibitory anti-MAG mAb (n = 6) or control IgG mAb (n = 5). Sham-injured controls similarly received either anti-MAG mAb (n = 8) or control IgG (n = 6). At 72 h post-injury, animals were overanesthetized with sodium pentobarbital (75 mg/kg) and transcardially perfused with heparinized saline followed by 4% paraformaldehyde. The brains were removed and post-fixed overnight at 4 °C in paraformaldehyde, and were then transferred into 30% sucrose solution for 3−4 days, snap frozen in 2-methylbutane at −20 °C, and stored at −80 °C. Brains were cut on a freezing microtome into 40-μm free-floating sections.
Detection of anti-myelin-associated glycoprotein monoclonal antibody or control antibody
Following blocking for 1 h with 3% normal horse serum, donkey anti-mouse IgG biotin (1 : 1000; Jackson ImmunoResearch, West Grove, PA, USA) was applied to every 12th section from Bregma −1.3 to −7.3. The initial order was determined in a random fashion. Following an overnight incubation at 4 °C, the avidin-biotin peroxidase method (Vector Laboratories, Burlingame, CA, USA) was used for visualization of the drug or control antibody. Internal controls included use of non-antibody-treated tissue sections and omission of secondary antibody from the protocol.
Expression of myelin-associated glycoprotein post-injury
Following blocking for 1 h with 3% normal horse serum, goat anti-MAG (1 : 2000; R and D Systems, Abingdon, UK) was applied to every 12th section from Bregma −1.3 to −7.3. The initial section chosen was adjacent to that chosen for drug diffusion. Following an overnight incubation at 4 °C, sections were washed and incubated in biotinylated donkey anti-goat IgG (Jackson ImmunoResearch) at a concentration of 1 : 1000. Following the 1-h secondary antibody incubation period, the avidin-biotin-peroxidase method was used for visualization of MAG within the brain sections. Internal controls included deletion of the primary antibody from the protocol.
Study B. Evaluation of neurobehavioral function and tissue loss
To examine the long-term neurobehavioral effects of anti-MAG mAb following TBI, brain-injured animals were randomized to receive either the inhibitory anti-MAG mAb (n = 25) or control antibody (n = 20, vide supra) intracerebroventricularly and sham-injured animals were also randomized to receive control IgG (n = 14) or anti-MAG mAb (n = 15). The total dose administered (8.64 μg) was identical to Study A. Following surgery or injury, neurological motor function was evaluated for 2 months in surviving animals in sham-injured (control-treated n = 13 and anti-MAG mAb-treated n = 13) and brain-injured (control-treated n = 17 and anti-MAG mAb-treated n = 18) rats using a battery of functional tests which have been previously shown to be sensitive for discriminating injury severity (Dixon et al., 1987; McIntosh et al., 1989; Mattiasson et al., 2000). Experienced observers, blinded to the injury and treatment status of each animal, performed behavioral assessments.
Neuromotor function
Gross neurologic motor function of animals was assessed at 48 h and 1, 2, 4, 6 and 8 weeks after injury using the composite neuroscore, a previously described battery of tests (McIntosh et al., 1989). The seven tests used included left and right forelimb contraflexion response during suspension by the tail, left and right hindlimb flexion while hindlimbs are lifted up and back by the tail, the ability to resist right and left lateral pulsion, and the ability to stand on an inclined plane. Scores for each test were given on an integer scale from 4 (uninjured) to 0 (severely impaired) and were combined, yielding a maximum composite neuroscore of 28.
The rotating pole test has been shown to be effective in the evaluation of neurological motor dysfunction following lateral FP brain injury (Mattiasson et al., 2000). On each of 2 days prior to injury, each animal was trained to traverse the full length of the pole. On the first training day, animals were trained on a non-rotating pole and on the second training day animals were trained on a rotating pole. At 1, 2, 4, 6 and 8 weeks post-injury, three trials per animal were performed with the pole rotating at 5 r.p.m. in the left rotational (counter-clockwise) direction. Animals were scored using the following scale: 4, rat traverses the pole with less than one footslip; 3, rat traverses the pole with two to three footslips or jumps; 2, rat traverses the pole with four or more footslips or jumps; 1, rat falls off while walking and 0, rat falls off when placed on the pole (Mattiasson et al., 2000).
Sensorimotor assessment
The adhesive paper test (Schallert et al., 1982; Hernandez & Schallert, 1988) has been previously used successfully to assess neurologic deficits following experimental TBI and pharmacologic treatment (Riess et al., 2001). Initial tests were performed prior to brain injury or sham operation to record the baseline latencies for each animal to ensure no significant differences at baseline among the groups. The mean latency (maximum, 120 s) over three trials to remove adhesive tapes (14 × 23 mm, Avery, Pasadena, CA, USA) from the forepaws was recorded at 1, 2, 4, 6 and 8 weeks post-injury for the ipsilateral (left) and contralateral (right) side. Latencies are expressed as mean time to remove the adhesive paper in seconds over the three trials.
Evaluation of visuospatial learning at 6 and 8 weeks post-injury
Animals were introduced to the Morris water maze for the first time beginning on day 42 post-injury in order to evaluate post-traumatic anterograde amnesia using a visuospatial learning paradigm. The maze consisted of a 1.8-m diameter pool where a fixed, hidden, clear, 10-cm-diameter plexiglas platform was submerged 1 cm below the surface of water at 24 °C. Animals underwent 3 days of testing, on post-injury days 42−44, with eight trials per day (two testing blocks of four trials each) during which they learned to find the submerged platform using external visual cues, placed around the periphery of the pool. Learning trials began by placing animals into the maze and releasing them at one of four sites separated by 90° around the periphery of the maze. Each animal was given at most 60 s per trial to find the hidden platform. The animal's latency to reach the platform was recorded for each of the 24 trials and the average for each of the six testing blocks was calculated as a measure of post-injury learning. This paradigm has previously proven effective in detecting deficits in learning ability of brain-injured rats (Saatman et al., 1997; Cheney et al., 2000). On day 56 post-injury, the platform was moved from its previous location to a new location, and animals were reintroduced to the same 1.8-m maze. Learning trials were conducted in the same manner over days 56−58, as animals were released at one of four sites separated by 90° around the periphery of the maze and latency was recorded for up to 60 s for each of the 24 trials. This shift paradigm has been used successfully in detecting alterations in learning ability in rats due to many factors and conditions including FP brain injury (Blokland et al., 1999; Cirulli et al., 2004; Thompson et al., 2006).
Tissue preparation
Following cognitive evaluation at the 8-week time point, animals were killed, perfused and post-fixed with 4% paraformaldehyde as previously described (vide supra). Following overnight post-fixation, the brains from Study B were then processed for paraffin embedding. Serial coronal serial brain sections (40 μm thick) were cut on a rotary microtome (Shandon Finesse ME, Thermo Electron, Pittsburgh, PA, USA).
Hemispheric tissue loss
To quantify hemispheric tissue loss following lateral FP brain injury at 8 weeks, coronal sections (40 μm thick) were taken every 1 mm from −0.3 mm bregma to −7.3 mm bregma (Paxinos & Watson, 1994) from a random subset of brain-injured and sham-injured animals (n = 7 per group). These regions were selected based on previous studies which demonstrated that cortical and thalamic tissue damage, observed up to 1 month after lateral FP injury, occurred within this region (Soares et al., 1995; Hicks et al., 1996; Zhang et al., 1998; Bramlett et al., 1999; Bentzer et al., 2001; Bramlett & Dietrich, 2001; Marklund et al., 2001). Sections were stained with hematoxylin and eosin and imaged using a digital camera integrated with a light microscope. As previously described (Hoover et al., 2004), the peripheries of the contralateral and ipsilateral hemisphere [not including the ventricle(s)] were traced on each image by an evaluator blinded to the injury and treatment status of the animals and the area of each hemisphere was calculated using a calibrated image analysis system (IMAGE 1.62c, Scion Corp., Frederick, MD, USA). The contralateral hemisphere for each section was used to control for interanimal variation in brain size (Zhang et al., 1998). Hemispheric tissue loss was calculated as a percentage of the contralateral (uninjured) hemisphere volume (Vc) using the following formula: (Vc – Vi)/(Vc) × 100, where Vi represents the volume of the ipsilateral (injured) hemisphere. To calculate total hemispheric tissue lost, areas were integrated over the 7-mm rostro-caudal distance.
Statistical analyses
Ordinal, non-parametric neurological motor scores and rotating pole scores are presented as median values. Comparisons among groups were made using a Kruskal–Wallis anova followed by individual Mann–Whitney U-tests at each time point for these behavior tests. Continuous variables including latency to remove sticky paper and hemispheric tissue loss are presented as means + SD and were evaluated using a two-way anova followed by the Student Newman Keuls post-hoc test. Averages for each treatment group of the six testing blocks in the Morris water maze were evaluated over time using two-way repeated measures anova followed by the Student Neuman Keuls post-hoc test. A P-value of less than 0.05 was considered to be statistically significant, with corrections for multiple comparisons made where appropriate. All statistical analyses were performed using spss 12.0 for Windows (SPSS, Chicago, IL, USA).
Results
Mortality
The overall initial mortality for Studies A and B was 23.7% (14/59) similar to that reported previously in our laboratory (Hoover et al., 2004). Most animals died due to prolonged apnea immediately following TBI. An additional 12 animals, equally distributed among the treatment and injury groups, were killed during the 2-month study period due to weight loss (n = 4), infection (n = 5) or aberrant behavior (e.g. extreme lethargy, spinning; n = 3) in accordance with animal welfare guidelines. These animals were not included in the behavioral analysis.
Study A
At 72 h post-injury, anti-MAG mAb or control IgG was detectable in the majority of animals (100% of sham-injured control-treated, 60% of sham-injured anti-MAG mAb-treated, 50% of brain-injured control-treated and 100% of brain-injured anti-MAG mAb-treated animals). Penetration of the anti-MAG mAb was detected primarily in brain areas near the catheter insertion site (Fig. 1A) but diffusion was noted to more distal regions of the hippocampus and fornix (Fig. 1B) as well as the third (Fig. 1C) and fourth ventricles of some animals. The catheter was positioned correctly in the left lateral ventricle in all animals at post-mortem evaluation and pump volumes were assessed pre-insertion and post-removal to confirm antibody delivery.
Fig. 1.
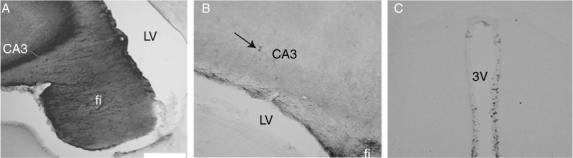
Penetration and diffusion of anti-myelin-associated glycoprotein monoclonal antibody into rat brain at 72 h post-injury in a brain-injured animal. (A) At Bregma −1.8 (Paxinos & Watson, 1994) into CA3 near catheter insertion site. (B) At Bregma −2.8 showing penetration into the ipsilateral hippocampus (CA3 arrow) and fimbria (fi). (C) At Bregma −3.6 demonstrating diffusion of the drug to the third ventricle (3V). Scale bar, 0.5 mm in A, 200 mm in B and C. LV, lateral ventricle.
Using immunohistochemistry, an increased staining profile for MAG was observed in the ipsilateral cortex, external capsule and hippocampus at 72 h post-injury in comparison to sham-injured animals (Fig. 2A–F). In the ipsilateral cortex, hippocampus and external capsule, the immunoreactivity for MAG of brain-injured anti-MAG mAb-treated animals (Fig. 2D–F) was similar to brain-injured animals that received IgG (data not shown).
Fig. 2.
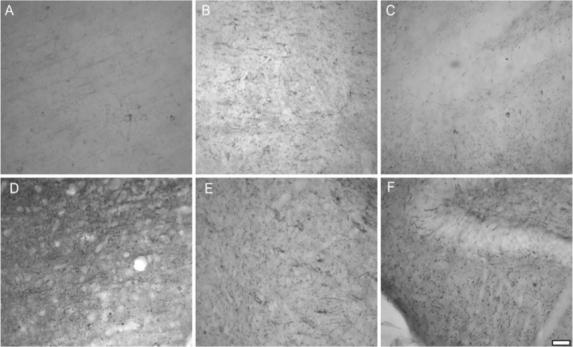
Immunohistochemistry characterizing patterns of myelin-associated glycoprotein (MAG) at 72 h post-injury. Representative sections at 72 h post-surgery from ipsilateral (A) cortex, (B) thalamus and (C) hippocampus of a sham-injured animal reveal a small amount of native MAG. Representative sections from a brain-injured anti-MAG antibody-treated animal show an increased manifestation of MAG post-injury in ipsilateral (D) cortex, (E) thalamus and (F) hippocampus in comparison to sham injury. All panels 200× magnification.
Study B
Fluid percussion brain injury induced a significant and prolonged deficit in neurologic motor function as assessed using the composite neuroscore from 48 h to 8 weeks post-injury, regardless of treatment status (Kruskal–Wallis test, d.f. = 3, P < 0.001, Fig. 3). Treatment with anti-MAG mAb resulted in a statistically significant improvement of gross neuromotor function at 6 weeks post-injury when compared with IgG treatment (Z = −2.18, P < 0.05, Fig. 3). Additionally, regardless of treatment, using the Friedman's test, brain-injured animals significantly recovered function over time (P < 0.001). On post-hoc testing (Wilcoxon sign rank) this functional recovery in injured animals was only significant from 48 h to 1 week (P < 0.05) and from 4 to 6 weeks post-injury (P < 0.01).
Fig. 3.
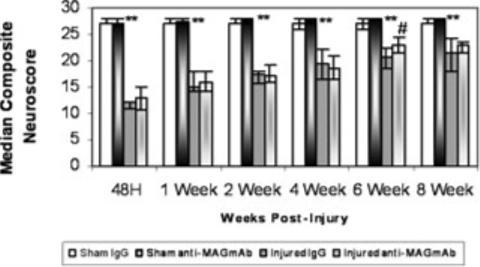
Assessment of composite neuroscore (median +25th and 75th percentile) among the treatment groups. Following injury, brain-injured IgG-treated animals had significantly lower neuroscores than sham-injured IgG-treated animals (**P < 0.01). At 6 weeks post-injury, brain-injured anti-myelin-associated glycoprotein (MAG) monoclonal antibody (mAb)-treated animals had a significantly higher neuroscore than their brain-injured IgG-treated counterparts (#P < 0.05).
Marked and prolonged post-injury deficits were also present in both IgG-treated and anti-MAG mAb-treated brain-injured animals when compared with their respective sham-injured controls in the rotating pole beginning at 1 week and continuing up to 8 weeks post-injury via Mann–Whitney U post-hoc testing (data not shown). Treatment with anti-MAG mAb did not significantly attenuate the deficits observed on the rotating pole following brain injury in comparison to IgG treatment (P = n.s.; data not shown).
Prior to injury or surgery, no differences were observed in the latency to remove sticky paper among the treatment groups (P = n.s., data not shown). Lateral FP brain injury significantly impaired sensorimotor function assessed by the adhesive paper test from 1 to 8 weeks post-injury on both the right (data not shown) and left (P < 0.01, Fig. 4) forepaws when compared with sham-injured animals. Treatment with anti-MAG mAb following brain injury resulted in a significant enhancement (decreased latency) in ipsilateral sensory function at 6 and 8 weeks post-injury in comparison to IgG treatment. Brain-injured animals receiving anti-MAG mAb were significantly faster at removing the adhesive paper from the left forepaw (P < 0.01) than those brain-injured animals that received control IgG (Fig. 4). Additionally, administration of anti-MAG mAb significantly improved the latency of the sham-injured animals to remove the adhesive paper from the left forepaw at 1 and 8 weeks post-surgery in comparison to IgG-treated, sham-injured animals (P < 0.05; Fig. 4).
Fig. 4.
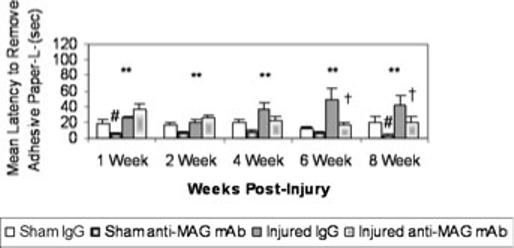
Latency to remove adhesive paper from left forepaw (mean + SEM). Following injury, brain-injured IgG-treated animals had significantly longer latencies to remove the sticky paper from the left forepaw than sham-injured IgG-treated animals (**P < 0.01). At 1 and 8 weeks post-surgery sham animals given anti-myelin-associated glycoprotein (MAG) monoclonal antibody (mAb) had significantly shorter latencies to remove the paper than their IgG-treated counterparts (#P < 0.05). At 6 and 8 weeks post-injury, brain-injured animals treated with anti-MAG mAb were able to remove the paper significantly faster than their IgG-treated counterparts (†P < 0.05).
All animals, regardless of injury and treatment status, exhibited a pattern of decreasing latencies to find the hidden platform in the Morris water maze test over the six testing blocks at 6 and 8 weeks post-injury (P < 0.001, Figs 5 and 6). In comparison to sham-injured animals, brain-injured animals showed a significant deficit in spatial learning ability on days 42−44 post-injury (F = 61.9, d.f. = 53, P < 0.001; Fig. 5). When the platform was shifted to a new location and animals were required to learn a new paradigm, brain-injured animals again showed a significant deficit in spatial learning capacity at 8 weeks post-injury (days 56−58) in comparison to sham-injured animals (F = 34.75, P < 0.001; Fig. 6). Brain-injured animals receiving anti-MAG mAb exhibited a trend towards shorter latencies to find the hidden platform at 6 weeks post-injury in comparison to IgG-treated animals but this difference was not significant (P 0.09; Fig. 5). However, by 8 weeks post-injury, brain-injured animals that received anti-MAG mAb demonstrated significantly shorter latencies to find the hidden platform than brain-injured animals receiving control IgG (F = 3.62, d.f. = 265, P < 0.05; Fig. 6).
Fig. 5.
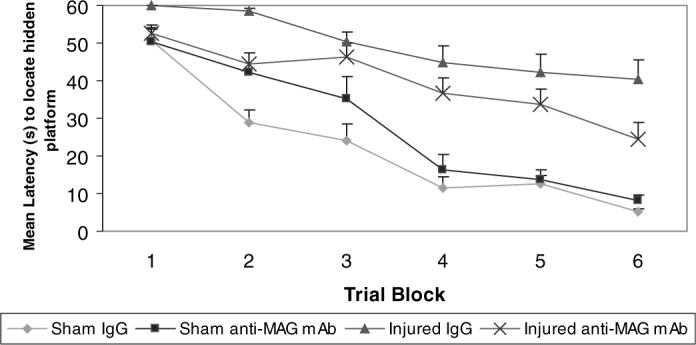
Learning at 6 weeks post-injury. Latencies to reach the hidden platform using the Morris water maze paradigm to assess visuospatial learning at 6 weeks post-injury (means + SEM). Brain-injured vehicle-treated animals had significantly longer latencies to reach the platform when compared with sham-injured, IgG-treated controls (P < 0.001). Treatment with the anti-myelin-associated glycoprotein (MAG) monoclonal antibody (mAb) following lateral fluid percussion brain injury resulted in consistently shorter latencies compared with IgG-treated brain-injured controls but this did not reach statistical significance (P = 0.094).
Fig. 6.
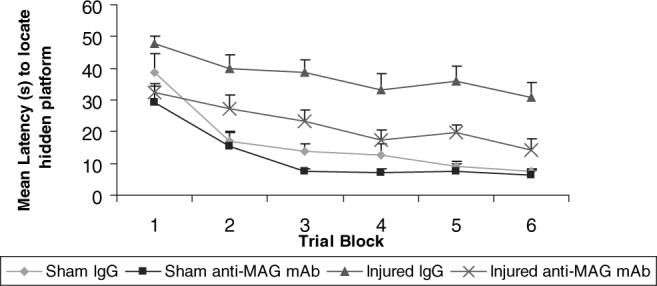
Learning at 8 weeks post-injury. At 2 weeks following the original learning paradigm, the animals were reintroduced to the Morris water maze in a paradigm shift and challenged to learn a new visuospatial task. Brain-injured vehicle-treated animals had significantly longer latencies to reach the platform when compared with sham-injured, IgG-treated controls. Treatment with the anti-myelin-associated glycoprotein (MAG) monoclonal antibody (mAb) following brain injury resulted in significantly shorter latencies to reach the platform when compared with brain-injured IgG-treated controls (P < 0.05).
At 8 weeks post-injury, all brain-injured animals regardless of treatment status lost a significant volume of tissue in the ipsilateral hemisphere in comparison to sham-injured animals (F = 99.22, d.f. = 24, P < 0.001). Additionally, there was a significant interaction effect between injury status and drug treatment (F < 6.17, d.f = 1, P < 0.05). Moreover, treatment with anti-MAG mAb following lateral FP brain injury significantly reduced the volume of tissue lost due to injury (12.5%, Fig. 7A, C and E) in comparison to control IgG treatment (18.0%, P < 0.05, Fig. 7A, B and D).
Fig. 7.
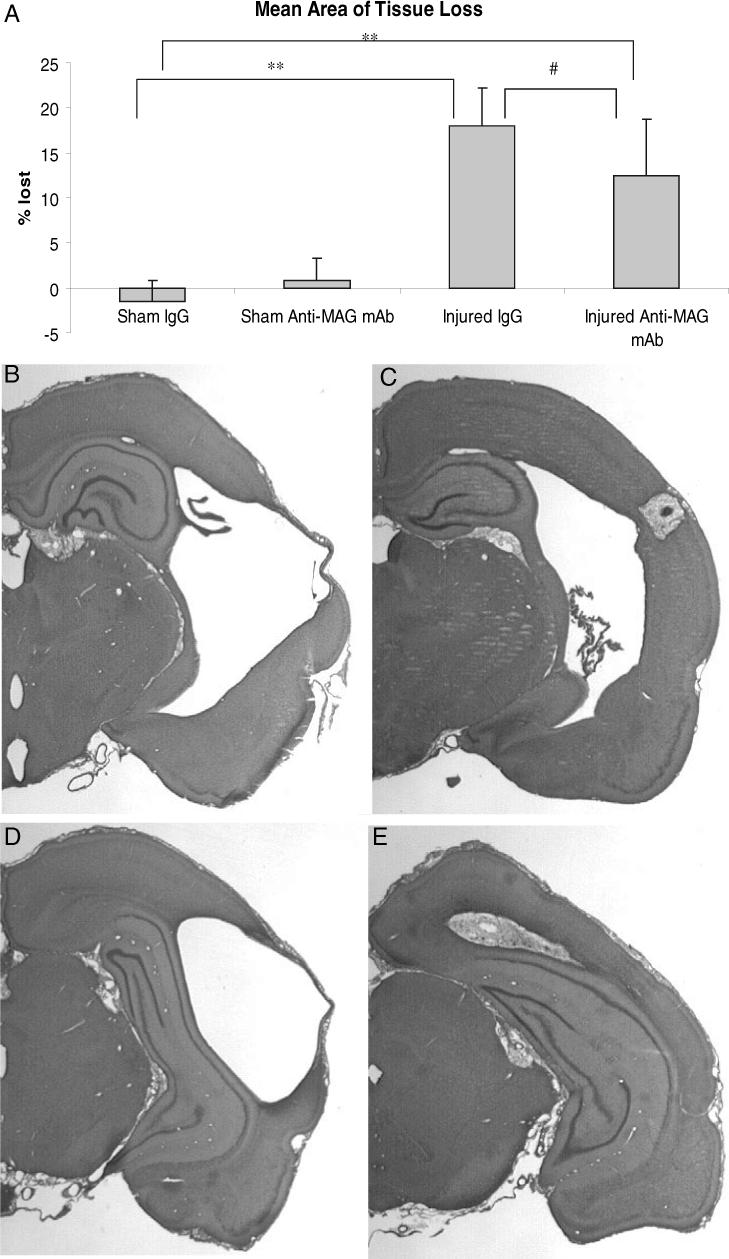
Mean percent hemispheric tissue loss + SD at 8 weeks post-injury. (A) Lateral fluid percussion (FP) brain injury resulted in significant ipsilateral hemispheric tissue loss at 8 weeks post-injury (**P < 0.001) and treatment with the anti-myelin-associated glycoprotein (MAG) monoclonal antibody (mAb) significantly reduced this tissue loss (#P < 0.05) in comparison to brain-injured animals given control IgG. (B and C) Lateral FP injury resulted in a significant loss of ipsilateral tissue at 8 weeks post-injury with visual lesions. Sections from Bregma −2.8 of (B) control IgG-treated and (C) anti-MAG mAb-treated animals and from Bregma −3.8 of (D) control IgG-treated and (E) anti-MAG mAb-treated animals demonstrating significant amelioration of tissue loss following brain injury.
Discussion
Acute treatment with a novel anti-MAG antibody for 72 h following TBI when administered intracerebroventricularly, at a dose of 0.12 μg/h, significantly improved motor, sensory and cognitive deficits as well as significantly reduced lesion volume in brain-injured animals. This is the first in vivo study supporting the beneficial effects of pharmacologic modulation of MAG on functional and histological outcomes in a model of TBI. These results are marked as it is rare that an experimental compound improves both sensorimotor and cognitive function while concomitantly reducing the amount of post-traumatic tissue injury. The neurobehavioral improvements observed were not seen until 6−8 weeks post-treatment, which is congruent with the antibody's proposed regenerative mechanism of action, and were observed in multiple areas making this a treatment strategy worthy of further investigation for clinical potential.
Our initial observations (Study A) suggest that anti-MAG mAb, when administered centrally, is distributed with a gradient from the infusion site and is capable of penetrating the injured brain. The anti-MAG mAb was found to diffuse into vulnerable brain regions, such as the ipsilateral hippocampus, which may be crucial for the plasticity observed following neutralization of MAG (Tang et al., 2001) and for the beneficial effects observed on cognitive function. We conclude that the osmotic mini-pump with intracerebroventricular infusion was an effective drug-delivery method in this model of brain injury. As diffusion of the antibody occurred mainly around the catheter insertion site and surrounding ventricle, the anti-MAG mAb should also be evaluated in models of diffuse axonal injury (e.g. impact acceleration) to confirm the efficacy of the osmotic mini-pump method vs. the need to explore an intravascular administration paradigm (Irving et al., 2005) to optimize predictive validity.
The increase in MAG at 72 h post-injury, as indicated by immunohistochemistry, was similar among the injured treatment groups and was increased in the ipsilateral (injured) cortex, hippocampus and thalamus. There were no differences in the injured groups in injury severity measures including mortality, length of apnea or seizure, indicating that differences seen between the groups are related to the actions of the anti-MAG mAb. As the anti-MAG antibody used for staining at 72 h post-injury in this study was polyclonal, it had multiple epitopes over the entire extracellular region of MAG, whereas the treatment antibody has only one epitope. Thus, it is unlikely that the treatment antibody significantly blocked the signal from the staining antibody, so the similarity in MAG staining patterns seen at 72 h post-injury served as an internal control.
We observed that treatment with anti-MAG mAb improved neurologic reflex testing using the composite neuroscore test at 6 weeks post-injury. Although this modest improvement in functional motor recovery did not persist in the treated group at 8 weeks post-injury, it is possible that the natural recovery experienced by injured animals may have diluted the treatment effect at 8 weeks.
Improvement of reflexive motor testing following experimental brain injury was not previously observed following inhibition of Nogo-A using the identical experimental lateral FP brain injury model (Marklund et al., 2004; Lenzlinger et al., 2005), although vestibulomotor improvement was reported at 1 and 4 weeks post-injury (Marklund et al., 2004). We were not able to demonstrate improvement in vestibulomotor function with administration of the anti-MAG mAb following FP brain injury. Given the proposed mechanism of action of the anti-MAG mAb in blocking the biological effects of MAG and promoting axonal outgrowth, the timing of initial improvement seen (6 weeks post-injury) is likely to be associated with a more long-term effect of this compound on plasticity. These observations validate the need to plan and conduct studies with long-term time points when evaluating compounds with similar mechanisms of action. The assessment of histopathology at intermediate time points may also be of critical importance in future study design in order to characterize the time course of therapeutic antibody presence, axonal, myelin and neuronal loss, and potential sparing of neurons and/or oligodendrocytes using anti-MAG mAb strategies. Although the optimal target to evaluate compensatory sprouting in experimental models of TBI has yet to be established, future studies of the anti-MAG mAb should also incorporate this analysis into study designs as it may be an additional mechanism of action for the compound's efficacy.
Following lateral FP brain injury, long-term deficits (up to 4 weeks) in sensorimotor function have been previously reported using the adhesive paper test (Riess et al., 2001). This study confirms and extends this work as moderately brain-injured animals showed significant sensorimotor deficits in both forelimbs up to 8 weeks post-injury. Treatment with anti-MAG mAb significantly attenuated ipsilateral sensorimotor deficits at 6 and 8 weeks post-injury in comparison to treatment with control IgG. The significantly lower latencies in sham-injured animals treated with the anti-MAG mAb are interesting given that there were no differences at baseline among the groups. Similar to human TBI, over time there is a progressive development of the injury cavity and hydrocephalus seen with the lateral FP model (Bramlett et al., 1997; Pierce et al., 1998). As treatment with the anti-MAG mAb following brain injury significantly preserved brain tissue in the ipsilateral (injured) hemisphere, including the cortex and thalamus, the anti-MAG mAb may be one mechanism for the significant differences seen in the adhesive paper test. Following glutamate toxicity, significant preservation of oligodendrocytes in vitro by the anti-MAG mAb has been previously reported (Irving et al., 2005) and this mechanism may be a component of the tissue sparing seen in the present study. It is therefore possible that the administration of the anti-MAG mAb, via oligodendrocyte-sparing mechanisms, may attenuate the progressive white matter loss observed following TBI, resulting in improved functional outcomes. However, given the current limitations of the lateral FP model for assessment of oligodendrocyte loss (Grady et al., 2003), this was not evaluated in the present study. Future studies should be planned to include additional sensorimotor evaluation as well as evaluation of the effects of the compound on axonal sprouting within the rubrospinal tract and within both control and injured animals.
In the present study we also demonstrate significant long-term improvement in cognitive function following treatment with the anti-MAG mAb. The antibody was able to diffuse into the injured (ipsilateral) hippocampus of brain-injured animals and was present up to 72 h post-injury, and the observed cognitive effects seen could be related to antagonism of MAG and/or stabilization of myelin in this vulnerable region. Improvement in memory function has recently been reported following TBI with the inhibition of Nogo-A (Marklund et al., 2004; Lenzlinger et al., 2005). Although Nogo-A and MAG both inhibit axonal regeneration, this effect does not seem to be mediated by the same receptor. Although Nogo-66 and MAG share a common receptor (NgR) (McKerracher & Winton, 2002), the receptor mediating inhibition of regeneration in response to Nogo-A is not known. Furthermore, MAG may utilize other receptors in addition to NgR (Vinson et al., 2001, 2003; Vyas et al., 2002). Thus, it is certainly possible that, although the effect of MAG and Nogo on axonal outgrowth is similar, the effect of modulating the pathways in experimental TBI may not be. Taken together, our data suggest that MAG may be involved in the development of motor and cognitive deficits post-injury and that the inhibition of MAG following experimental TBI is neuroprotective.
In the present study, the administration of the anti-MAG mAb began at 1 h post-injury and continued for 72 h; we show that there is an increased immunostaining of MAG in the ipsilateral cortex, external capsule and hippocampus at 72 h post-injury. Given the ability of dMAG to diffuse from damaged white matter (Tang et al., 1997, 2001) and the diffuse staining pattern seen after lateral FP brain injury, the diffusion of anti-MAG mAb into areas known to be vulnerable to injury as well as areas distal to the site of infusion were also important findings.
The behavioral improvements seen beginning at 6 weeks and tissue sparing following administration of the anti-MAG mAb to FP brain-injured animals support its function as a neuroprotective agent. This study confirms the findings of tissue sparing and improvement of motor function following anti-MAG mAb administration by Irving et al. (2005) in a cerebral ischemia model, and extends the previous work with the findings of sensorimotor and cognitive improvement. The discovery of the ability of MAG antagonism to exert beneficial effects across multiple behavioral endpoints is rare in the setting of FP injury (for review see Thompson et al., 2005). Thus, these novel observations suggest that MAG may be an important and differential contributor to the pathophysiology of TBI and treatment strategies targeting MAG may be suitable for further clinical evaluation.
Acknowledgements
The authors would like to thank D.J. Castelbuono, S.T. Fujimoto, J. Plevy, R. Puri and L. Tran for technical assistance and R. Armstrong for administrative support that enabled completion of this study. Thanks to Drs Rabinder Prinjha, Stephen Burbridge, Elaine Irving, Alex Harper, Andrew Parsons and John Davis for helpful discussions. This work was supported in part by grants from Glaxo Smith Kline, the National Institutes of Health (T32-NS043126, T32-NR07106, P50−08803, R01−40978 and K12RR023265) and a Merit Review Grant from the Veteran's Administration. N.M. was partly supported by a grant from the Swedish Brain Foundation.
Abbreviations
- FP
fluid percussion
- mAb
monoclonal antibody
- MAG
myelin-associated glycoprotein
- TBI
traumatic brain injury.
References
- Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J. Neurol. Neurosurg. Psychiat. 2001;71:521–524. doi: 10.1136/jnnp.71.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch U, Kirchhoff F, Schachner M. Immunohistological localization of the adhesion molecules L1, N-CAM, and MAG in the developing and adult optic nerve of mice. J. Comp. Neurol. 1989;284:451–462. doi: 10.1002/cne.902840310. [DOI] [PubMed] [Google Scholar]
- Bentzer P, Mattiasson G, McIntosh TK, Wieloch T, Grande PO. Infusion of prostacyclin following experimental brain injury in the rat reduces cortical lesion volume. J. Neurotrauma. 2001;18:275–285. doi: 10.1089/08977150151070919. [DOI] [PubMed] [Google Scholar]
- Blokland A, de Vente J, Prickaerts J, Honig W, Markerink-van Ittersum M, Steinbusch H. Local inhibition of hippocampal nitric oxide synthase does not impair place learning in the Morris water escape task in rats. Eur. J. Neurosci. 1999;11:223–232. doi: 10.1046/j.1460-9568.1999.00431.x. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J. Neurotrauma. 2001;18:891–900. doi: 10.1089/089771501750451811. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, Green EJ, Busto R. Chronic histopathological consequences of fluid-percussion brain injury in rats: effects of post-traumatic hypothermia. Acta Neuropathol. (Berl.) 1997;93:190–199. doi: 10.1007/s004010050602. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Green EJ, Dietrich WD. Exacerbation of cortical and hippocampal CA1 damage due to posttraumatic hypoxia following moderate fluid-percussion brain injury in rats. J. Neurosurg. 1999;91:653–659. doi: 10.3171/jns.1999.91.4.0653. [DOI] [PubMed] [Google Scholar]
- Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: lessons from animal models and clinical studies. Neurosurgery. 1999;45:207–217. doi: 10.1097/00006123-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- Caroni P, Savio T, Schwab ME. Central nervous system regeneration: oligodendrocytes and myelin as non-permissive substrates for neurite growth. Prog. Brain Res. 1988;78:363–370. doi: 10.1016/s0079-6123(08)60305-2. [DOI] [PubMed] [Google Scholar]
- Cheney JA, Brown AL, Bareyre FM, Russ AB, Weisser JD, Ensinger HA, Leusch A, Raghupathi R, Saatman KE. The novel compound LOE 908 MS attenuates acute neuromotor dysfunction but not cognitive impairment or cortical tissue loss following traumatic brain injury in rats. J. Neurotrauma. 2000;17:83–91. doi: 10.1089/neu.2000.17.83. [DOI] [PubMed] [Google Scholar]
- Cirulli F, Berry A, Chiarotti F, Alleva E. Intrahippocampal administration of BDNF in adult rats affects short-term behavioral plasticity in the Morris water maze and performance in the elevated plus-maze. Hippocampus. 2004;14:802–807. doi: 10.1002/hipo.10220. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling R, Hamm R, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat: Neurological, physiological, and histopathological characteristics. J. Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Emerick AJ, Neafsey EJ, Schwab ME, Kartje GL. Functional reorganization of the motor cortex in adult rats after cortical lesion and treatment with monoclonal antibody IN-1. J. Neurosci. 2003;23:4826–4830. doi: 10.1523/JNEUROSCI.23-12-04826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res. Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Montag D, Schachner M, Martini R. Crucial role for the myelin-associated glycoprotein in the maintenance of axon-myelin integrity. Eur. J. Neurosci. 1995;7:511–515. doi: 10.1111/j.1460-9568.1995.tb00347.x. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S, Graham DI. Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol. 1995;89:537–543. doi: 10.1007/BF00571509. [DOI] [PubMed] [Google Scholar]
- Grady MS, Charleston JS, Maris D, Witgen BM, Lifshitz J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological evaluation. J. Neurotrauma. 2003;20:929–941. doi: 10.1089/089771503770195786. [DOI] [PubMed] [Google Scholar]
- Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J. Neuropathol. Exp. Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- Hernandez TD, Schallert T. Seizures and recovery from experimental brain damage. Exp. Neurol. 1988;102:318–324. doi: 10.1016/0014-4886(88)90226-9. [DOI] [PubMed] [Google Scholar]
- Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. (Berl.) 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- Hoover RC, Motta M, Davis J, Saatman KE, Fujimoto ST, Thompson HJ, Stover JF, Dichter MA, Twyman R, White HS, McIntosh TK. Differential effects of the anticonvulsant topiramate on neurobehavioral and histological outcomes following traumatic brain injury in rats. J. Neurotrauma. 2004;21:501–512. doi: 10.1089/089771504774129847. [DOI] [PubMed] [Google Scholar]
- International Brain Injury Association [10.1.05];Brain injury facts. Alexandria, VA. 2005 http://www.internationalbrain.org/content.php?pages=facts.
- Irving EA, Vinson M, Rosin C, Roberts JC, Chapman DM, Facci L, Virley DJ, Skaper SD, Burbidge SA, Walsh FS, Hunter AJ, Parsons AA. Identification of neuroprotective properties of anti-MAG antibody: a novel approach for the treatment of stroke? J. Cereb. Blood Flow Metab. 2005;25:98–107. doi: 10.1038/sj.jcbfm.9600011. [DOI] [PubMed] [Google Scholar]
- Kottis V, Thibault P, Mikol D, Xiao ZC, Zhang R, Dergham P, Braun PE. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J. Neurochem. 2002;82:1566–1569. doi: 10.1046/j.1471-4159.2002.01146.x. [DOI] [PubMed] [Google Scholar]
- Laurer HL, McIntosh TK. Pharmacologic therapy in traumatic brain injury: update on experimental treatment strategies. Curr. Pharm. Design. 2001;7:1505–1516. doi: 10.2174/1381612013397285. [DOI] [PubMed] [Google Scholar]
- Lee DH, Strittmatter SM, Sah DW. Targeting the Nogo receptor to treat central nervous system injuries. Nat. Rev. Drug Discov. 2003;2:872–878. doi: 10.1038/nrd1228. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Shimizu S, Marklund N, Thompson HJ, Schwab ME, Hoover RC, Bareyre FM, Motta M, Luginbuhl A, Pape R, Clouse AK, Saatman KE, Morganti-Kossman C, McIntosh TK. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047–1056. doi: 10.1016/j.neuroscience.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Li S, Strittmatter SM. Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J. Neurosci. 2003;23:4219–4217. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI. Guidelines for head injury: their use and limitations. Neurol. Res. 2002;24:19–23. doi: 10.1179/016164102101199495. [DOI] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo–glial interactions. J. Cell Biol. 2002;156:567–577. doi: 10.1083/jcb.200111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Clausen F, McIntosh TK, Hillered L. Free radical scavenger posttreatment improves functional and morphological outcome after fluid percussion injury in the rat. J. Neurotrauma. 2001;18:821–832. doi: 10.1089/089771501316919184. [DOI] [PubMed] [Google Scholar]
- Marklund N, Fulp CT, Bareyre FM, Royo NC, Schwab ME, Puri R, Hoover RC, Soltesz K, McMillan A, Mir AK, Spangler Z, Millard M, Keck C, LeBold D, McIntosh TK. The Nogo-A neutralizing antibody 7B12 improves motor and cognitive functional outcome following traumatic brain injury in rats. Soc. Neurosci. Abstr. 2004:230.17. [Google Scholar]
- Marklund N, Fujimoto S, Morales D, Clausen F, Fulp CT, LeBold D, Hillered L, Strittmatter SM, McIntosh TK. Cognitive impairment in aged mice deficient in Nogo-A/B and Nogo-66 receptor following traumatic brain injury. J. Neurotrauma. 2005;22:1229. [Abstract] [Google Scholar]
- Mattiasson GJ, Philips MF, Tomasevic G, Johansson BB, Wieloch T, McIntosh TK. The rotating pole test: evaluation of its effectiveness in assessing functional motor deficits following experimental brain injury in the rat. J. Neurosci. Meth. 2000;95:75–82. doi: 10.1016/s0165-0270(99)00162-4. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Faden AI. Traumatic brain injury in the rat: Characterization of a lateral fluid percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- McKerracher L, Winton MJ. Nogo on the go. Neuron. 2002;36:345–348. doi: 10.1016/s0896-6273(02)01018-8. [DOI] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron. 1994;13:805–811. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- NIH Consensus Development Panel Consensus conference. Rehabilitation of persons with traumatic brain injury. NIH Consensus Development Panel on Rehabilitation of Persons with Traumatic Brain Injury. JAMA. 1999;282:974–983. [PubMed] [Google Scholar]
- Paivalainen S, Suokas M, Lahti O, Heape AM. Degraded myelin-associated glycoprotein (dMAG) formation from pure human brain myelin-associated glycoprotein (MAG) is not mediated by calpain or cathepsin L-like activities. J. Neurochem. 2003;84:533–545. doi: 10.1046/j.1471-4159.2003.01539.x. [DOI] [PubMed] [Google Scholar]
- Papadopoulos CM, Tsai SY, Alsbiei T, O'Brien TE, Schwab ME, Kartje GL. Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann. Neurol. 2002;51:433–441. doi: 10.1002/ana.10144. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1994. [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral, and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Buki A, Koiziumi H, Stone J, Okonkwo DO. Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir. (Suppl. Wien) 1999;73:15–20. doi: 10.1007/978-3-7091-6391-7_3. [DOI] [PubMed] [Google Scholar]
- Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–384. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- Quarles RH, Everly JL, Brady RO. Myelin-associated glycoprotein: a developmental change. Brain Res. 1973;58:506–509. doi: 10.1016/0006-8993(73)90022-x. [DOI] [PubMed] [Google Scholar]
- Riess P, Bareyre F, Saatman KE, Cheney JA, Lifshitz J, Raghupathi R, Grady MS, Neugebauer E, McIntosh TK. Effects of chronic, post-injury cyclosporin A administration on motor and sensorimotor function following severe, experimental traumatic brain injury. Rest. Neurol. Neurosci. 2001;18:1–8. [PubMed] [Google Scholar]
- Royo NC, Shimizu S, Schouten JW, Stover JF, McIntosh TK. Pharmacology of traumatic brain injury. Curr. Opin. Pharmacol. 2003;3:27–32. doi: 10.1016/s1471-4892(02)00006-1. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp. Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Schachner M, Bartsch U. Multiple functions of the myelin-associated glycoprotein MAG (siglec-4a) in formation and maintenance of myelin. Glia. 2000;29:154–165. doi: 10.1002/(sici)1098-1136(20000115)29:2<154::aid-glia9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Schallert T, Upchurch M, Lobaugh N, Farrar SB, Spirduso WW, Gilliam P, Vaughn D, Wilcox RE. Tactile extinction: distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol. Biochem. Behav. 1982;16:455–462. doi: 10.1016/0091-3057(82)90452-x. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr. Opin. Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Silver MA, Stryker MP. TrkB-like immunoreactivity is present on geniculocortical afferents in layer IV of kitten primary visual cortex. J. Comp. Neurol. 2001;436:391–398. doi: 10.1002/cne.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares HD, Hicks RR, Smith DH, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Woodhall RW, Shen YJ, deBellard ME, Saffell JL, Doherty P, Walsh FS, Filbin MT. Soluble myelin-associated glycoprotein (MAG) found in vivo inhibits axonal regeneration. Mol. Cell. Neurosci. 1997;9:333–346. doi: 10.1006/mcne.1997.0633. [DOI] [PubMed] [Google Scholar]
- Tang S, Qiu J, Nikulina E, Filbin MT. Soluble myelin-associated glycoprotein released from damaged white matter inhibits axonal regeneration. Mol. Cell. Neurosci. 2001;18:259–269. doi: 10.1006/mcne.2001.1020. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J. Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, LeBold DG, Marklund NM, Morales DM, Hagner AP, McIntosh TK. Cognitive evaluation of traumatically brain injured rats using serial testing in the Morris water maze. Rest. Neurol. Neurosci. 2006;24:109–114. [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Andrews SB, Wong A, O'Connell M, Griffin JW. Co-localization of the myelin-associated glycoprotein and the microfilament components, F-actin and spectrin, in Schwann cells of myelinated nerve fibres. J. Neurocytol., 1989;18:47–60. doi: 10.1007/BF01188423. [DOI] [PubMed] [Google Scholar]
- Venkatesh KF, Chivatakarn OF, Lee HF, Joshi PS, Kantor DB, Newman BA, Mage RF, Rader CF, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J. Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson M, Strijbos PJ, Rowles A, Facci L, Moore SE, Simmons DL, Walsh FS. Myelin-associated glycoprotein interacts with ganglioside GT1b. A mechanism for neurite outgrowth inhibition. J. Biol. Chem. 2001;276:20 280–20 285. doi: 10.1074/jbc.M100345200. [DOI] [PubMed] [Google Scholar]
- Vinson M, Rausch O, Maycox PR, Prinjha RK, Chapman D, Morrow R, Harper AJ, Dingwall C, Walsh FS, Burbidge SA, Riddell DR. Lipid rafts mediate the interaction between myelin-associated glycoprotein (MAG) on myelin and MAG-receptors on neurons. Mol. Cell. Neurosci. 2003;22:344–352. doi: 10.1016/s1044-7431(02)00031-3. [DOI] [PubMed] [Google Scholar]
- Vyas AA, Patel HV, Fromholt SE, Heffer-Lauc M, Vyas KA, Dang J, Schachner M, Schnaar RL. Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc. Natl Acad. Sci. U.S.A. 2002;99:8412–8417. doi: 10.1073/pnas.072211699. [See comment.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002a;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002b;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Weiss MD, Hammer J, Quarles RH. Oligodendrocytes in aging mice lacking myelin-associated glycoprotein are dystrophic but not apoptotic. J. Neurosci. Res. 2000;62:772–780. doi: 10.1002/1097-4547(20001215)62:6<772::AID-JNR3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Wong EV, David S, Jacob MH, Jay DG. Inactivation of myelin-associated glycoprotein enhances optic nerve regeneration. J. Neurosci. 2003;23:3112–3117. doi: 10.1523/JNEUROSCI.23-08-03112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Raghupathi R, Saatman KE, Smith DH, Stutzmann J-M, Wahl F, McIntosh TK. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J. Neurosci. Res. 1998;52:342–349. doi: 10.1002/(SICI)1097-4547(19980501)52:3<342::AID-JNR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]