

Abstract
Amyloid β peptide (Aβ) is thought to play a central role in the pathogenesis of Alzheimer disease (AD). How Aβ induces neurodegeneration in AD is not known. A connection between AD and cholesterol metabolism is suggested by the finding that people with the apolipoprotein E4 allele, a locus coding for a cholesterol-transporting lipoprotein, have a modified risk for both late-onset AD and cardiovascular disease. In the present study we show that both Aβ and submicromolar concentrations of free cholesterol alter the trafficking of a population of intracellular vesicles that are involved in the transport of the reduced form of the tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT formazan), the formation of which is a widely used cell viability assay. Treatments that change cellular free cholesterol levels also modulate the trafficking of the MTT formazan-containing vesicles, suggesting that the trafficking of these vesicles may be regulated by free cholesterol under physiological conditions. In addition, Aβ decreases cholesterol esterification and changes the distribution of free cholesterol in neurons. These results suggest that the MTT formazan-transporting vesicles may be involved in cellular cholesterol homeostasis and that the alteration of vesicle transport by Aβ may be relevant to the chronic neurodegeneration observed in AD.
Genetic and biochemical evidence strongly suggest that amyloid β peptide (Aβ) is involved in the pathogenesis of Alzheimer disease (AD) (1–4). One of the most reproducible biological activities of Aβ is its ability to inhibit the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by living cells (5–7). MTT is a tetrazolium salt that can be reduced to purple-colored formazan and is widely used for measuring cell viability (8). Although it was widely assumed that MTT is reduced by active mitochondria, we have shown recently that mitochondria are unlikely to play a significant role in cellular MTT reduction. Instead, MTT is taken up by cells through endocytosis and reduced by a N-methylmaleimide-sensitive flavin oxidase. Reduced MTT formazan accumulates in the endosome/lysosome compartment and is then transported to the cell surface through exocytosis (9). With these new insights into the mechanism of cellular MTT reduction, we subsequently demonstrated that Aβ and other cytotoxic amyloid peptides (human amylin and calcitonin) dramatically enhance the exocytosis of the MTT formazan-transporting vesicles (10). The physiological function of the abundant MTT formazan-transporting vesicles is not known. These vesicles do not appear to be involved in the pinocytosis of horseradish peroxidase, receptor-mediated endocytosis and exocytosis of transferrin, or neurotransmitter release in PC12 cells (10). In the present study, we show that free cholesterol also regulates the transport of the MTT formazan-containing vesicles and that Aβ decreases cholesterol esterification and changes the distribution of free cholesterol in neurons. These findings suggest a connection between AD and cellular cholesterol homeostasis through the biological activity of Aβ.
MATERIALS AND METHODS
Materials and Cell Lines.
All amyloid peptides (Bachem) were dissolved in deionized water before use. Steroids were purchased from Sigma and were dissolved in ethanol or methanol. When added to cultured cells, the volume of the organic solvent was always less than 1% of the total culture media (vol/vol), and solvent vehicle controls were carried out. B12 cells are from a collection of cells made from nitrosoethylurea-induced rat brain tumors (11). Fibroblast cell lines derived from acyl-CoA:cholesterol acyltransferase (ACAT) knockout mice (12) or wild-type mice were provided by Robert V. Farese, University of California at San Francisco.
Photomicroscopy and Quantitation of MTT Formazan Exocytosis.
Cells growing on 35-mm dishes were examined and photographed with a Nikon light microscope equipped with a water-immersible object lens using Kodak EPT 64T film. The needle-like crystals on the surface of the cells incubated with MTT, which are easily visible under a light microscope, represent exocytosed MTT formazan (9). The percentage of cells exocytosing MTT formazan was determined by counting 300 cells in multiple fields. There are cells that show both some granules and some crystals or lots of crystals and no granules. Once a cell exports some MTT formazan and forms crystal needles, it exports all of them within 30 min to 1 hr. A cell is counted as exocytosing MTT formazan as long as several needle-like formazan crystals are clearly observable.
Measurement of Cellular Cholesterol Esterification and Cholesterol Synthesis.
Cholesterol esterification was determined by measuring the incorporation of [1-14C]oleic acid (51 mCi/mmol; 1 Ci = 37 GBq; DuPont/NEN) into newly synthesized cholesteryl esters (13). After evaporation to dryness under N2, [1-14C]oleic acid was resuspended in 1 mg/ml of fatty acid-free BSA solution and added to cultured cells at a final concentration of 0.5 μCi/ml. Incubation was performed for 4 hr at 37°C. The cells then were washed three times with PBS, harvested with a rubber policeman into 1 ml of 1 mM Tris⋅HCl buffer (pH 7.4), and sonicated. An aliquot (30 μl) was used for protein determination, and lipids were extracted from the rest of the cell lysate by the addition of 4 ml of chloroform/methanol (2:1, vol/vol). After the addition of 50 ng of cholesteryl oleate to serve as a carrier lipid, the lower organic phase was removed and evaporated under N2. The lipids were redissolved in 100 μl of hexane, and 20 μl was analyzed by silica gel (Whatman, LK6D, 60 Å) TLC by using a solvent system composed of hexane/ether/glacial acetic acid (90:10:1). Cholesteryl esters were identified by iodine staining and scraped into 10 ml of scintillation fluid for counting. Autoradiography of the TLC plates (overnight exposure) was performed before scraping. Cholesterol synthesis by cultured cells was measured with [1-14C]sodium acetate according to Goldstein et al. (13). [1-14C]Sodium acetate (48.9 mCi/mmol, Sigma) was introduced into the culture medium at the concentration of 10 μCi/ml, and incubation was performed for 4 hr at 37°C. Cell harvesting, lipid extraction, and analysis as well as liquid scintillation counting were carried out as described above except cholesteryl oleate was not added. Both the cholesterol band and the cholesteryl esters band were identified on TLC plate and scraped for liquid scintillation counting.
Measurement of Cellular Free Cholesterol.
Free cholesterol levels of the cultured cells were determined in cellular lipid extracts by using cholesterol oxidase (14). It is not possible to determine the free cholesterol levels of cells treated with either 25-hydroxycholesterol or 20α-hydroxycholesterol because they are also substrates of cholesterol oxidase.
Cytochemical Staining of Unesterified Cholesterol in Rat Hippocampal Neurons.
Rat hippocampal neurons were prepared from 18-day-old embryos and cultured in serum-free medium with N1 supplements (15). Cellular unesterified cholesterol was stained with filipin according to Neufeld et al. (16). Briefly, hippocampal neurons cultured in slide chambers were washed with PBS after treatments and fixed in 3% paraformaldehyde at room temperature for 30 min. After washing with PBS, the cells were incubated with 0.1% filipin in PBS overnight at room temperature. The cells were then washed, mounted, and examined with a Bio-Rad MRC 1024 confocal imaging system (9). All conditions were imaged with identical collection settings.
RESULTS
Amyloidogenic Peptides Enhance MTT Formazan Exocytosis.
MTT reduction by living cells initially is confined to intracellular formazan granules (Fig. 1A), but needle-like formazan crystals begin to appear on the cell surface after about 30 min after MTT addition (Fig. 1A, arrow). These intracellular MTT formazan granules are acidic intracellular vesicles while the needle-like MTT formazan crystals on the cell surface are exocytosed MTT formazan (9). B12 cells, a rat neural cell line (11), begin to exocytose MTT formazan after 30 min of MTT reduction (2–5% of the cells are positive; Fig. 1A, arrow), and it takes more than 3 hr for >90% of the cells to exocytose MTT formazan (Fig. 1B). Several neurotoxic amyloid peptides (Aβ25–35, Aβ1–40, Aβ1–42, human amylin, and human calcitonin) dramatically enhance MTT formazan exocytosis such that more than 95% of the cells have exocytosed MTT formazan after only 30 min of MTT reduction (Fig. 1C; also see ref. 10). This enhanced MTT formazan exocytosis is responsible for the Aβ-induced inhibition of cellular MTT reduction (10). The formazan needles induced by cytotoxic amyloid peptides look different from those of untreated cells. They are shorter, thicker, and more violet (compare Fig. 1 B with C). Nontoxic peptides (such as Aβ1–28, Aβ40–1, scrambled Aβ25–35, and rat amylin) do not enhance MTT formazan exocytosis (10). A common feature of the toxic amyloid peptides is that they all form amyloid fibrils that are rich in β-pleated sheet structure (17). Therefore, the enhancement of cellular MTT formazan exocytosis may be a common property of amyloid fibrils with a β-pleated sheet structure.
Figure 1.
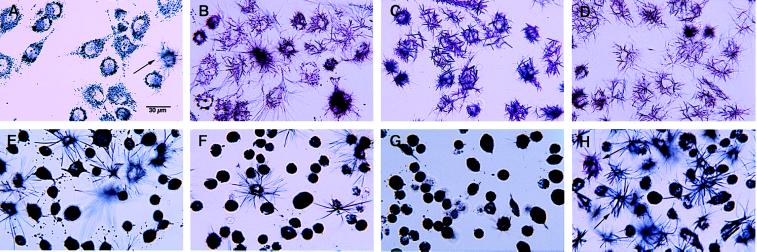
MTT formazan exocytosis: effects of Aβ, cholesterol, and cholesterol depletion. B12 cells cultured with 10% fetal calf serum (A–D and H) or with 5% lipoprotein-deprived bovine serum for 3 days (E–G) were treated as indicated below and followed by MTT reduction (0.5 mg/ml). The cells then were examined and photographed with a Nikon light microscope. (A) Control, 30 min of MTT reduction. The arrow indicates a cell with needle-like MTT formazan crystals. (B) Control, 3 hr of MTT reduction. (C) Aβ1–40 (10 μM) for 16 hr, followed by 30 min of MTT reduction. (D) Cholesterol (2 μM), immediately followed by 30 min of MTT reduction. (E) 25-Hydroxycholesterol (10 μM) for 24 hr, followed by 2-hr MTT reduction. (F) Two percent CD for 6 hr, followed by 2-hr MTT reduction. (G) 25-Hydroxycholesterol (10 μM) for 24 hr plus 2% CD for 6 hr, followed by 2-hr MTT reduction. (H) 25-Hydroxycholesterol (20 μM) for 24 hr, followed by 2-hr MTT reduction. Arrows indicate cells with formazan crystals similar to that induced by Aβ (see C). (Bar = 30 μm.)
Free Cholesterol Regulates MTT Formazan Exocytosis.
To study whether the MTT formazan-transporting vesicles are involved in cellular membrane or lipid homeostasis, the effects of various lipids on cellular MTT formazan exocytosis were examined. Remarkably, free cholesterol at concentrations as low as 1 μM enhanced MTT formazan exocytosis to a level similar to that caused by Aβ (Table 1). The morphology of the formazan crystals formed under the influence of cholesterol is, however, different from that induced by Aβ but indistinguishable from that seen in control cells (Fig. 1D, compare with B and C). The effect of cholesterol on MTT formazan exocytosis has strict structural requirements. The C3—OH group and the hydrophobicity around the C17 end of the cholesterol molecule are absolutely required for this effect since cholesterol analogs without the C3—OH group (5-cholesten-3-one, 5-cholesten, cholesterol esters) or with a hydrophilic —OH group around the C17 end of the molecule (25-hydroxycholesterol) do not have such an effect on MTT formazan exocytosis (Table 1 and Fig. 2). Cholesterol complexed (e.g., low density lipoprotein) or premixed with other lipids (e.g., phospholipids) has no effect on MTT formazan exocytosis (Table 1), indicating that free cholesterol is required. In the presence of free cholesterol (2 μM), Aβ-treated cells (10 μM Aβ25–35, 16 hr) display MTT formazan needles with a morphology that is characteristic of the effect of cholesterol, not that of Aβ. Cholesterol analogs that do not enhance MTT formazan exocytosis (e.g., 5-cholesten-3-one and 25-hydroxycholesterol) also have no effect on Aβ-enhanced MTT formazan exocytosis (data not shown). These results suggest that free cholesterol acts at a later step than Aβ in the multistep process of MTT formazan exocytosis such that it can override the effect of Aβ.
Table 1.
Effect of cholesterol on MTT formazan exocytosis
| Treatments | % of cells exocytosing MTT formazan |
|---|---|
| I. The structural specificity of cholesterol’s effect on MTT formazan exocytosis | |
| Control | 5 ± 3 |
| Aβ1-40 (10 μM, 16 hr of incubation) | 94 ± 3* |
| Cholesterol (added at the same time as MTT) | |
| 5 μM | 99 ± 1* |
| 2 μM | 98 ± 1* |
| 1 μM | 91 ± 3* |
| 0.5 μM | 84 ± 4* |
| 0.2 μM | 18 ± 3* |
| 0.1 μM | 12 ± 3* |
| Dihydrocholesterol (5 μM) | 99 ± 1* |
| 5-Cholesten-3-one (5 μM) | 7 ± 3 |
| 5-Cholesten (5 μM) | 6 ± 3 |
| Cholesteryl acetate (5 μM) | 5 ± 3 |
| Cholesterol palmitate (5 μM) | 6 ± 2 |
| 24β-Ethylcholesterol (5 μM) | 97 ± 2* |
| 25-Hydroxycholesterol (5 μM) | 6 ± 2 |
| II. Cholesterol (5 μM) complexed or mixed with other lipids | |
| Cholesterol complexed in LDL (100 μg/ml, 16 hr incubation) | 6 ± 3 |
| Mixed with egg phosphatidylcholine (20 μg/ml) | 5 ± 3 |
| Mixed with lysophosphatidylcholine (20 μg/ml) | 6 ± 3 |
| Mixed with bovine brain sphingomyelin (20 μg/ml) | 6 ± 3 |
| Mixed with ganglioside GD1a (10 μM) | 40 ± 5* |
Sterols or a mixture of cholesterol and other lipids were added to B12 cells on 35-mm dishes, followed by 30 min of MTT reduction. The needle-like crystals on the surface of the cells incubated with MTT, which are easily visible under a light microscope, represent exocytosed MTT formazan (4). The percentage of cells exocytosing MTT formazan was determined by counting 300 cells in multiple fields. Data are mean ± SD.
Significantly different from control (P < 0.05) by two-tailed Student’s t test. The experiments were repeated at least three times.
Figure 2.

The chemical structures of cholesterol and cholesterol analogs.
Since submicromolar concentrations of free cholesterol induce MTT formazan exocytosis with characteristics that are indistinguishable from those seen in control cells, it is possible that MTT formazan exocytosis is regulated by cellular-free cholesterol under physiological conditions. Treatments or conditions that are known to modulate cellular cholesterol levels were used to test this hypothesis. These include the 3-hydroxy-3-methylglutaryl CoA reductase (HMG-CoA reductase) inhibitor mevastatin, the cholesterol synthesis inhibitors 25-hydroxycholesterol and 20α-hydroxycholesterol (18, 19), the extracellular cholesterol acceptor 2-hydroxypropyl-β-cyclodextran (CD) (16), and ACAT knockout cells (12). As shown in Fig. 1 E–G and Table 2, conditions that decrease cellular-free cholesterol levels (25-hydroxycholesterol, 20α-hydroxycholesterol and CD) inhibit MTT formazan exocytosis, while cells from ACAT knockout mice, which have increased cellular-free cholesterol levels compared with that of cells from wild-type animals, have an enhanced rate of MTT formazan exocytosis (Table 2). Treatment of B12 cells with 10 μM of the HMG-CoA reductase inhibitor mevastatin did not significantly inhibit MTT formazan exocytosis, nor did it significantly decrease cellular-free cholesterol levels (Table 2). This could be because of a lower sensitivity of the HMG-CoA reductase isoform that is expressed in B12 cells to mevastatin relative to primary fibroblasts. While free cholesterol had no effect on cell division, 25-hydroxycholesterol and 20α-hydroxycholesterol slowed cell growth rate. Other reagents that slowed cell division, such as the cell cycle inhibitor aphidicolin, have no effect on MTT formazan exocytosis (data not shown), ruling out an indirect effect of these inhibitors. Curiously, 25-hydroxycholesterol at concentrations higher than 10 μM induces MTT formazan exocytosis with a formazan crystal morphology very similar to that induced by Aβ (Fig. 1H, arrows), while 20α-hydroxycholesterol at concentrations as high as 50 μM does not cause this phenomenon. These results suggest that the trafficking of the MTT formazan-transporting vesicles is regulated by cellular-free cholesterol and that these vesicles may be involved in cellular cholesterol homeostasis.
Table 2.
Modulation of cellular-free cholesterol levels alters the rate of MTT formazan exocytosis
| Treatment | % of cells exocytosing MTT formazan | μg free cholesterol/mg protein† |
|---|---|---|
| Control | 98 ± 3 | 3.04 ± 0.39 |
| Mevastatin (10 μM, 24 hr) | 88 ± 6 | 2.93 ± 0.37 |
| 25-Hydroxycholesterol (10 μM, 24 hr) | 45 ± 5* | — |
| 20α-Hydroxycholesterol (30 μM, 24 hr) | 18 ± 4* | — |
| 20α-Hydroxycholesterol (30 μM, 24 hr) plus cholesterol (20 μM, 1 hr) | 97 ± 2 | — |
| 2-Hydroxypropy-β-cyclodextran (2%, 6 hr) | 36 ± 5* | 1.52 ± 0.18* |
| 2-Hydroxypropyl,β-cyclodextran (2%, 6 hr) plus 25-hydroxycholesterol (10 μM, 24 hr) | 13 ± 4* | — |
| Fibroblasts from wild-type mouse | 52 ± 4 | 4.04 ± 0.38 |
| Fibroblasts from ACAT knockout mouse | 94 ± 3* | 5.76 ± 0.42* |
B12 cells on 35-mm dishes were cultured with 5% lipoprotein-deficient serum for 3 days, followed by the treatment with mevastatin or oxygenated cholesterols or 2-hydroxypropyl-β-cyclodextran. Fibroblasts from wild-type or ACAT knockout mouse were cultured with 10% fetal bovine serum. The cells (B12 cells or fibroblasts) were allowed to reduce MTT (0.5 mg/ml) for 2 hr. The percentage of cells exocytosing MTT formazan was determined by counting 300 cells in multiple fields. Free cholesterol levels of the cultured cells were determined in cellular lipid extracts by using cholesterol oxidase (14). Data are mean ± SD of three experiments.
Significantly different from control or from wild-type (P < 0.05) by two-tailed Student’s t test.
It is not possible to determine the free cholesterol levels of cells treated with either 25-hydroxycholesterol or 20α-hydroxycholesterol because they are also substrates of cholesterol oxidase.
Amyloid Fibrils Alter Cellular Cholesterol Homeostasis.
Since the MTT-transporting vesicles may be involved in cellular cholesterol homeostasis and their cycling is modified by Aβ, the effects of amyloid peptides on cellular cholesterol esterification and cholesterol synthesis were examined on both B12 cells and primary cultures of rat cortical neurons. Fibrillogenic amyloid peptides (Aβ25–35, Aβ1–40, Aβ1–42, and human amylin) inhibit cholesterol esterification by 15–35% while nonfibrillogenic peptides (scrambled Aβ25–35) have no effect (Fig. 3). The maximal effect of 10 μM Aβ on MTT formazan exocytosis of B12 cells is reached after 3 hr of incubation and is kept at the maximum for the next 24 hr (10). The cholesterol esterification of B12 cells treated with 10 μM Aβ for 3 hr is inhibited to an extent similar to that of B12 cells treated 10 μM Aβ for 16 hr. For example, the cholesterol esterification of B12 cells treated with 10 μM Aβ25–35 for 3 hr is inhibited to 79.7 ± 4.8% of control, while B12 cells treated with 10 μM Aβ25–35 for 16 hr is 75.3 ± 4.3%. The observation that Aβ alters cholesterol esterification similarly at 3 hr, which is before the peak of peroxide production (6), and at 16 hr demonstrates that this effect is rapid and occurs well before the small amount of cell death caused by Aβ in this system. Although the inhibition of cholesterol esterification by Aβ is not dramatic, it is highly significant and reproducible. In addition, the amyloid fibril-binding dye Congo red, which blocks Aβ neurotoxicity (20), also blocks the inhibition of cholesterol esterification induced by Aβ25–35 (Fig. 3). The effect of amyloid peptides on de novo cholesterol synthesis is less well defined. Aβ and human amylin increase cholesterol synthesis by about 20%. However, the effect is not statistically significant because of assay variability (data not shown).
Figure 3.

Effects of amyloid fibrils on cellular cholesterol homeostasis. B12 cells or primary neuronal cultures of rat cortex were treated with 10 μM of various amyloid peptides alone or plus 10 μM Congo red for 16 hr and then labeled with [1-14C]oleic acid to study the effects of amyloid peptides on cellular cholesterol esterification according to ref. 11. The first four bars of the histogram are the results of experiments with B12 cells, and only the last bar is the results of experiments with primary rat cortical neurons. SAβ25–35, scrambled Aβ25–35; H. Amylin, human amylin. Data are mean ± SD of 3–12 experiments. ∗, significantly (P < 0.05) different from control by two-tailed independent Student’s t test.
Since there may be variations within neurons in cholesterol metabolism that are difficult to detect biochemically, the effect of Aβ on cellular-free cholesterol distribution also was studied cytochemically with cultured rat hippocampal neurons by using the dye filipin, which only stains free or unesterified cholesterol (16). Aβ (20 μM, 6-hr treatment) increased the free cholesterol content in both the neurites and neuronal cell bodies (Fig. 4 A and B). Similar results were obtained when the neurons were treated with 10 μM Aβ or treated for 3 or 9 hr (data not shown). These results are consistent with the observation that Aβ inhibits cellular cholesterol esterification. CD is an efficient extracellular cholesterol acceptor that has been used to monitor the efflux of cholesterol through the plasma membrane of living cells (16). In the presence of CD, the majority of neuronal-free cholesterol was depleted within 2 hr (Fig. 4C). Aβ speeds up this depletion process so that very little filipin staining is left after 2 hr of CD treatment (Fig. 4D). This effect may be attributed to the ability of Aβ to speed up intracellular vesicle recycling through the plasma membrane, which results in increased contact between the cell membranes and CD. Human amylin has similar effects on cellular-free cholesterol distribution (data not shown).
Figure 4.

Effect of Aβ on the distribution of cellular unesterified cholesterol. Rat hippocampal neurons cultured in serum-free medium were treated with Aβ or 2-hydroxypropyl-β-cyclodextran (CD), followed by filipin staining as described in ref. 13. (A) Control. (B) Aβ1–40 (20 μM) for 6 hr. (C) Two percent CD for 2 hr. (D) Aβ1–40 (20 μM) for 3 hr, followed by 2% CD for 2 hr. (Bar = 25 μm.)
DISCUSSION
The above results show that an amyloid-initiated pathway leads to a dramatically enhanced rate of exocytosis of MTT formazan-containing vesicles and altered cellular cholesterol metabolism. How amyloid fibrils enhance MTT formazan exocytosis is not clear at present. The interaction between amyloid fibrils and the plasma membrane may involve a receptor that recognizes the β-pleated sheet structure in the fibrils, or the activity of a membrane signaling molecule may be directly altered by the fibrils, which triggers a signal transduction pathway resulting in enhanced MTT formazan exocytosis. The finding that submicromolar concentrations of free cholesterol regulate the trafficking of the MTT formazan-transporting vesicles suggests that these vesicles are involved in cellular cholesterol homeostasis. In addition, amyloid fibrils decrease cellular cholesterol esterification (Fig. 3) and increase the free cholesterol content of the membrane (Fig. 4). If the MTT formazan-containing vesicles are involved in cholesterol metabolism, then the effect of Aβ on cholesterol esterification could result from an Aβ-induced alteration in intracellular trafficking. Alternatively, since Aβ fibrils bind cholesterol preferentially to phosphatidylcholine and fatty acids (21), the membrane cholesterol bound to Aβ fibrils may be less accessible for esterification, which, in turn, results in decreased cholesterol esterification and an increased free cholesterol content of membranes.
The MTT formazan crystals on the cell surface are the result of exocytosis of intracellular formazan-containing granules (9, 10). Therefore, the percentage of cells exocytosing MTT formazan is a valid index of the rate of MTT formazan exocytosis. Because the MTT formazan-containing vesicles are not involved in the exocytosis of commonly used exocytosis markers such as horseradish peroxidase and transferrin (10), MTT formazan exocytosis cannot be used as an index of other forms of exocytosis or a general index of exocytosis. There are many types of endocytosis and exocytosis that are poorly characterized (22). We regard MTT formazan exocytosis as one of these poorly characterized types of exocytosis of unknown function. However, the finding that submicromolar concentrations of free cholesterol regulate the trafficking of the MTT formazan-transporting vesicles suggests that these vesicles may be involved in cellular cholesterol homeostasis. Besides its role in membrane structure, free cholesterol exerts a feedback regulation of cellular cholesterol metabolism by suppressing the transcription of HMG-CoA reductase and low density lipoprotein receptor and by activating ACAT (23). The above data show that free cholesterol also can regulate intracellular vesicle trafficking.
The formazan crystal morphology induced by free cholesterol is very similar to that of control cells, but Aβ-induced formazan crystals have a different color and morphology from those of control cells or those induced by free cholesterol (Fig. 1). It is possible that the direct interactions between MTT or MTT formazan and amyloid peptides or cholesterol result in the observed differences in crystal morphology. However, since some natural ceramides and 25-hydroxycholesterol induce MTT formazan crystals that are indistinguishable from those induced by Aβ (Fig. 1H and unpublished results), it is difficult to imagine that the interactions between MTT and the structurally different ceramides, amyloid peptides, and 25-hydroxycholesterol will all result in the same structure of formazan crystals. The most likely explanation is that cholesterol, amyloid peptides, or ceramides affect MTT formazan exocytosis indirectly through a signal transduction pathway. MTT formazan exocytosis is a multistep process that can be modified at different stages or sites (10). Because cholesterol and amyloid peptides act at different stages of MTT formazan exocytosis, resulting in different durations of MTT reduction and different trafficking of the MTT formazan-containing vesicles, it is not unexpected that different types of MTT formazan crystals are generated.
The Aβ-induced alteration in cellular cholesterol metabolism and vesicle trafficking potentially could contribute to the neurodegeneration observed in AD. Neurons are particularly sensitive to alterations in lipid transport since both the growth and maintenance of their elaborate neurites require directional lipid transport (24). Previous studies have found that increased membrane cholesterol inhibits the cholinergic stimulation of hippocampal pyramidal neurons (25) and that cellular membranes in AD brain have a tendency toward destabilization, which is a consequence of their abnormal membrane lipid composition (26). The Aβ-induced increase in membrane cholesterol could decrease membrane fluidity and contribute to membrane instability and the loss of cholinergic neurons. It is interesting to note that altered cholesterol homeostasis in Niemann-Pick C disease causes chronic, progressive neurodegeneration (27). A relationship between AD and cholesterol homeostasis is suggested by the finding that different subtypes of apolipoprotein E, lipoproteins involved in cholesterol homeostasis, impart different risk levels on the occurrence of late-onset AD and cardiovascular disease (28, 29). A recent study also supports this relationship by showing that cholesterol depletion inhibits the generation of Aβ in hippocampal neurons (30). Our results do not address the issue of Aβ metabolism, but do provide additional support for a role of altered cellular cholesterol homeostasis in Alzheimer disease.
Acknowledgments
We thank Dr. Jochiam Herz for advice, Dr. Robert Farese for providing acyl CoA:cholesterol acyltransferase knockout cell lines, and Dr. Pamela Maher for critically reading the manuscript. This work was supported by National Institutes of Health Grants NS09658 and NS28121 and by The Jacob Peter Hansen and Anita Charlotte Hansen Endowment Fund for Alzheimer’s Research. Yuanbin Liu is supported by a National Institutes of Health postdoctoral fellowship (5 F32 NS10279-02) and the Bundy Foundation.
ABBREVIATIONS
- Aβ
amyloid β peptide
- ACAT
acyl-CoA:cholesterol acyltransferase
- AD
Alzheimer disease
- CD
2-hydroxypropyl-β-cyclodextran
- HMG-CoA
3-hydroxy-3-methylglutaryl CoA
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
References
- 1. Yankner B A. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 3.Pike C J, Cummings B J, Cotman C W. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 4.Yankner B A, Duffy L K, Kirscher D A. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 5.Behl C, Davis J B, Cole G M, Schubert D. Biochem Biophys Res Commun. 1992;186:944–952. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 6.Behl C, Davis J B, Lesley R, Schubert D. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 7.Shearman M S, Ragan C I, Iversen L L. Proc Natl Acad Sci USA. 1994;91:1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Peterson D, Kimura H, Schubert D. J Neurochem. 1997;69:581–593. doi: 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Schubert D. J Neurochem. 1997;69:2285–2293. doi: 10.1046/j.1471-4159.1997.69062285.x. [DOI] [PubMed] [Google Scholar]
- 11.Schubert D, Heinemann S, Carlisle W, Tarika H, Kimes B, Steinbach J H, Culp W, Brandt B L. Nature (London) 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- 12.Meiner V L, Cases S, Myers H M, Sande E R, Bellosta S, Schambelan M, Pitas R E, McGuire J, Herz J, Farese R V. Proc Natl Acad Sci USA. 1996;93:14041–14046. doi: 10.1073/pnas.93.24.14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein J, Dana S E, Brown M S. Proc Natl Acad Sci USA. 1974;71:4288–4292. doi: 10.1073/pnas.71.11.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamble W, Vaughan M, Kruth H S, Avigan J. J Lipid Res. 1978;19:1068–1070. [PubMed] [Google Scholar]
- 15.Abe K, Kimura H. J Neurochem. 1996;67:2074–2078. [PubMed] [Google Scholar]
- 16.Neufeld E B, Cooney A M, Pitha J, Dawidowicz E A, Dwyer N K, Pentchev P G, Blanchette-Mackie E J. J Biol Chem. 1996;271:21604–21613. doi: 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]
- 17.Glenner G G. N Engl J Med. 1980;302:1283–1292. doi: 10.1056/NEJM198006053022305. , 1333–1343. [DOI] [PubMed] [Google Scholar]
- 18.Brown M S, Faust J R, Goldstein J L, Kaneko I, Endo A. J Biol Chem. 1978;253:1121–1128. [PubMed] [Google Scholar]
- 19.Kandutsch A A, Chen H W. Lipids. 1978;13:704–707. doi: 10.1007/BF02533749. [DOI] [PubMed] [Google Scholar]
- 20.Lorenzo A, Yankner B A. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avdulov N A, Chochina S V, Igbavboa U, Warden C S, Vassiliev A V, Wood W G. J Neurochem. 1997;69:1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- 22.Robinson M, Watt C, Zerial M. Cell. 1996;69:129–138. [Google Scholar]
- 23.Brown M S, Goldstein J L. Science. 1987;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 24.Futerman A H, Banker G A. Trends Neurosci. 1996;19:144–149. doi: 10.1016/s0166-2236(96)80025-7. [DOI] [PubMed] [Google Scholar]
- 25.Crews F T, Camacho A, Phillips M I. Brain Res. 1983;261:155–158. doi: 10.1016/0006-8993(83)91296-9. [DOI] [PubMed] [Google Scholar]
- 26.Ginsberg L, Atack J R, Rapoport S I, Gershfeld N L. Brain Res. 1993;625:355–357. doi: 10.1016/0006-8993(93)90050-w. [DOI] [PubMed] [Google Scholar]
- 27.Pentchev P G, Brady R O, Blanchette-Mackie E J, Vanier M T, Carstea G, Parker C C, Goldin E, Roff C F. Biochim Biophys Acta. 1994;1225:235–243. doi: 10.1016/0925-4439(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 28.Strittmatter W J, Saunders A M, Schmechel D, Pericak-Vance M, Enghild J, Salvesen G S, Roses A D. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stengard J H, Zerba K E, Pekkanen J, Ehnholm C, Nissinen A, Sing C F. Circulation. 1995;91:265–269. doi: 10.1161/01.cir.91.2.265. [DOI] [PubMed] [Google Scholar]
- 30.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti C G, Simons K. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]