

Abstract
Vascular endothelial growth factor (VEGF) is a homodimeric member of the cystine knot family of growth factors, with limited sequence homology to platelet-derived growth factor (PDGF) and transforming growth factor β2 (TGF-β). We have determined its crystal structure at a resolution of 2.5 Å, and identified its kinase domain receptor (KDR) binding site using mutational analysis. Overall, the VEGF monomer resembles that of PDGF, but its N-terminal segment is helical rather than extended. The dimerization mode of VEGF is similar to that of PDGF and very different from that of TGF-β. Mutational analysis of VEGF reveals that symmetrical binding sites for KDR are located at each pole of the VEGF homodimer. Each site contains two functional “hot spots” composed of binding determinants presented across the subunit interface. The two most important determinants are located within the largest hot spot on a short, three-stranded sheet that is conserved in PDGF and TGF-β. Functional analysis of the binding epitopes for two receptor-blocking antibodies reveal different binding determinants near each of the KDR binding hot spots.
Keywords: cystine knot, mutagenesis, receptor dimerization
Vascular endothelial growth factor (VEGF) is a potent mitogen in embryonic and somatic angiogenesis with a unique specificity for vascular endothelial cells (1–4). The central role of VEGF in embryonic angiogenesis was exemplified in recent studies showing that heterozygote knock-out mice suffer fatal deficiencies in vascularization (5, 6). VEGF also plays an important role in the pathogenesis of cancer, proliferative retinopathy, and rheumatoid arthritis (7). Antibodies against it have shown therapeutic potential as agents capable of suppressing in vivo tumor growth by inhibiting tumor-induced angiogenesis (8). VEGF binds to cell-surface receptors of the tyrosine kinase family, the kinase domain receptor (KDR), and fms-like tyrosine kinase (Flt-1). Flt-1 binds VEGF with 50-fold higher affinity than KDR (9); however, KDR dominates the angiogenic response (10) and is therefore of greater therapeutic interest. VEGF is a homodimer and exists in four different isoforms. Proteolytic removal of the C-terminus has shown that the binding domain for KDR resides within the 110 N-terminal residues (11). VEGF is a member of the cystine-knot family of growth factors, with highest sequence identity to platelet-derived growth factor (PDGF) BB and less similarity to transforming growth factor β2 (TGF-β). Crystal structures of PDGF (12) and TGF-β (13, 14) have been determined before this work.
Here, we present the 2.5 Å crystal structure of the receptor binding domain of human VEGF as well as the results of a comprehensive mutagenesis study to elucidate the KDR binding site. VEGF is proposed to engage KDR receptors using two symmetrical binding sites located at opposite ends of the molecule; each site is composed of two “hot spots” for binding that consist of residues presented from both subunits of the homodimer. Residues important for binding two receptor-blocking antibodies are outside but near the KDR binding hot spots.
MATERIALS AND METHODS
Crystal Structure Determination.
The receptor binding domain of VEGF (residues 8–109) was overexpressed in Escherichia coli inclusion bodies, purified, refolded, and crystallized in space group P21 (15), with two dimers per asymmetric unit. The crystals diffract very anisotropically, beyond 2 Å in the a*,c*-plane but only to 2.5 Å in the b* direction. Molecular replacement using the structure of PDGF (12) as a search model failed, and heavy atom soaking experiments resulted in severe nonisomorphism, with variations in the b axis between 57 and 61 Å. Therefore, an artificial heavy atom binding site was introduced by mutating Pro-53 to cysteine (15). This mutant was also crystallized in P21, with a = 55.9 Å, b = 60.8 Å, c =77.6 Å, β = 90.0° (“native 2” in Table 1). Diffraction data were collected at 18°C from a native mutant crystal and from mutant crystals soaked overnight in 1 mM lead acetate or mercury iodide. All data were recorded on a MAR-Research imaging plate system using a Rigaku rotating anode generator with CuKα radiation, and processed with program xds (16). Heavy atom positions were identified with the program heavy (17) and refined using program mlphare (18). Both derivatives share the same four noncrystallographic symmetry (NCS)-related heavy atom sites. The NCS operators relating the four monomers were clear from the heavy atom binding sites and from a solvent-flattened multiple isomorphous replacement (MIR) map. Pairs of monomers are related by 180° rotations and form biological dimers with the dimer axes parallel to the b axis. Both dimers lie in the same a, c plane, with a pseudo p21221 arrangement within these layers. Density modification at 3.5 Å resolution using the program dm (18) allowed for complete model building (program O, ref. 19). All refinement was done with x-plor (20), after a subset of 1,755 reflections was set aside for monitoring the free R-value. Initially, the 3 Å cysteine mutant data set was used in refinement; later a 2.5 Å data set was collected from a flash-frozen wild-type crystal (a = 56.19 Å, b = 59.81 Å, c = 77.52 Å, β = 90.0°; “native 1”), and corrected for the anisotropy in diffraction (Table 1). NCS restraints were applied to most of the structure throughout refinement. The final R-factor for all data between 10 and 2.5 Å was 20.4% (Rfree = 29.7%), after applying a solvent correction. Only His-86 of one monomer lies in a “disallowed region” (21) of the Ramachandran plot. This residue is part of a segment (residues 83–88) with high B-factors, which is involved in crystal packing contacts.
Table 1.
Structure determination and refinement statistics
| Diffraction data and MIR phasing | Resolution, Å | No. reflections (% completeness) | 〈I/σI〉 | Mult. | Rmerge | Riso | Phasing power | Cullis R |
|---|---|---|---|---|---|---|---|---|
| Native 1 | 2.5 | 17,684 (97†) | 56 | 3.6 | 0.041 | |||
| Native 2 | 3.0 | 9,707 (92) | 22 | 2.0 | 0.067 | 0.42 | ||
| Pb(CH3COO)2 | 3.5 | 5,828 (87) | 15 | 2.0 | 0.062 | 0.23 | 2.4 | 0.49 |
| HgI2 | 3.5 | 6,341 (95) | 21 | 2.6 | 0.084 | 0.23 | 2.0 | 0.63 |
| Refined model | ||||||||
| Resolution10–2.5 Å | Disordered residues8-13, 108-109 | |||||||
| R-value (no. reflections)20.4% (15,187) | No. residues in asymmetric unit376 | |||||||
| Rfree (no. reflections)29.7% (1,755) | No. solvent molecules111 | |||||||
| rms deviation in bonds0.013 Å | Total no. non-H atoms3,235 | |||||||
| rms deviation in angles1.71° | Anisotropic correction | |||||||
| rms deviation in temperature | (a*; b*; c*)18 Å2; −34 Å2; 16 Å2 | |||||||
| factors of bonded atoms1.9 Å2 | Average B-factor:47 Å2 | |||||||
84% complete between 2.6 and 2.5 Å.
Phage ELISA.
Phagemid vector pB2105 was produced by PCR amplification of the cDNA encoding for residues 1–109 of human VEGF, using primers that allowed subsequent ligation as a NsiI/XbaI restriction fragment into the phagemid vector, phGHam-g3 (22). This introduced an amber codon immediately following residue 109, and fused this DNA to the C-terminal half of gene III encompassing residues 249 through 406. Phage production in a suppressor strain of E. coli (Stratagene, XL1-blue) allowed for the expression of both the VEGF-gIII fusion protein and the free VEGF 1–109 protein. The ability of the phagemid to bind tightly to KDR-IgG indicated that a functional VEGF dimer, with one subunit of each of these forms, was displayed on the phage surface. Serial dilutions of competing KDR-IgG and a subsaturating concentration of phagemid were added to KDR-IgG-coated microtiter plates (Nunc, Maxisorp). After equilibrium, phagemid bound to the plate were stained with anti-phage mAb horseradish peroxidase conjugate (Pharmacia) and assayed. Affinities (EC50) were calculated as the concentration of competing receptor that resulted in half-maximal phagemid binding.
Analysis of Alanine Mutations.
Binding affinities were determined by an RIA that measured the displacement of radio-iodinated VEGF 8–109 from KDR-IgG by serial dilutions of cold mutant or wild-type VEGF 1–109. Binding buffer consisted of PBS with 0.5% BSA and 0.1% Tween 20. Bound label was captured by incubating equilibrated binding solutions with immobilized anti-IgG in microtiter plate wells for 40 min. Individual VEGF mutant proteins were isolated as refractile bodies from shake flask cultures of E. coli (27C7) harboring the phagemid vector described in Fig. 3, refolded as previously described (23), and quantitated by densitometry measurements of PAGE-resolved sample bands relative to those of known standards.
Figure 3.

KDR-IgG binding affinities of VEGF 1–109 displaying phagemid, and the two N-terminal deletion mutants, VEGF 8–109 and VEGF 11–109, were determined by phage ELISAs (33), giving EC50 values of 4.6, 4.1, and 4.4 nM, respectively. The roughly 100-fold weaker affinity we observe for phagemid binding relative to free VEGF (Table 3) may be partially attributable to the gene III fusion blocking an avidity component present in the binding of the free hormone to the bivalent KDR-IgG fusion protein.
RESULTS AND DISCUSSION
Crystal Structure.
A construct of residues 8–109 of VEGF, which binds KDR with wild-type affinity, was used for the structure determination. The structure was solved by multiple isomorphous replacement and 4-fold map averaging after substitution of residue Pro-53 with a free cysteine to bind heavy metal atoms (Table 1 and Fig. 1). The structure shows VEGF forms an antiparallel homodimer covalently linked by two disulfide bridges between Cys-51 and Cys-60. The dominant feature within the monomer is the cystine knot motif (Figs. 1 and 2), which is found in other growth factors (25, 26). The knot consists of an eight-residue ring formed by the disulfide bridges Cys-57–Cys-102 and Cys-61–Cys-104, with a third disulfide bond (Cys-26–Cys-68) passing through it. From this motif extends a central four-stranded β-sheet (labeled β1, β3, β5, and β6 in Fig. 1A). As in the other cystine knot growth factors, only a single hydrogen bond is present between β3 and β5, making the four-stranded sheet highly irregular. The segment connecting β1 and β3 contains a single turn of α-helix as well as a short β-strand (labeled α2 and β2, respectively, in Fig. 1A). This strand, together with the end of β5 and the beginning of β6, forms a short three-stranded β-sheet at the opposite end of the molecule from the cystine knot. Residues from this sheet, helix α2, and loop regions β1-β3 and β5-β6, together with residues from the N-terminal helix of the other monomer, form a small hydrophobic core. This presumably provides additional stabilization to the four-stranded central sheet, which is solvent accessible from both sides. In the VEGF dimer, the 2-fold axis is perpendicular to the sheet, resulting in an antiparallel orientation of the monomers with the two four-stranded sheets side by side (Fig. 1B).
Figure 1.

Ribbon representations of the receptor binding domain of VEGF, generated with program molscript (24), showing the monomer in a, with the secondary structural elements labeled, and the dimer in b. Disulfide bonds are rendered as black lines. Helix α1 consists of residues 16 to 24. The central four-stranded β-sheet is formed by β1 (residues 27 to 34), β3 (51 to 58), β5 (73 to 83) and β6 (89 to 99), with the characteristic cystine knot motif (25) on one end of the molecule and a short three-stranded β-sheet (strands β2 (residues 46 to 49), β5 and β6) on the other. The four monomers in the asymmetric unit are very similar, and a core region of 2,302 of 3,094 protein atoms can be superimposed pair-wise with rms differences of 0.13 Å. The remaining residues superimpose with rms differences of about 1.25 Å and are part of the loop regions β1-β2, β3-β4, and β5-β6. Although these residues have distinct crystallographic environments, they have very similar main chain conformations in all monomers and differ mainly by their temperature factors.
Figure 2.

Comparison of the monomer of VEGF (Left) with PDGF (Center) and TGF-β (Right). PDGF and TGF-β were aligned with VEGF using 71 and 45 Cα positions, respectively, in the cystine knot region of the molecules. The short three-stranded sheet at the opposite end of the molecules from the cystine knot is shown in red. The bend in the middle of the four-stranded sheet of TGF-β has the effect of twisting this sheet compared with the orientation observed in VEGF and PDGF. In the VEGF dimer, residues from this short sheet and the N-terminal helix from the other monomer that packs against it contain the functional determinants for KDR binding. Note that residues 28 to 36 of PDGF are disordered and not shown.
Comparison to Other Cystine-knot Growth Factors.
Both the side-by-side dimer association and the monomer topology of VEGF are similar to those observed in PDGF (12), despite a low sequence homology of only 19%. The VEGF and PDGF monomers can be superimposed with an rms difference of 1.6 Å for 71 equivalent Cα positions using a distance cut-off of 3.5 Å (Fig. 2). The most significant differences between the monomers are that PDGF has an extended, rather than α-helical, N terminus, a disordered segment between β1 and β2, and a three-residue insertion in loop β5-β6. VEGF and PDGF have the same mode of subunit association, allowing the dimers to be superimposed with a difference of only 1.8 Å (131 Cαs). In both dimers, the adjacent strands β1 and β1′ do not hydrogen-bond to each other. The total area buried at the VEGF dimer interface is 2,750 Å2 compared with 2,200 Å2 in PDGF (program areamol, ref. 18). The difference results from the larger area buried with the N-terminal helix in VEGF (990 Å2) compared with the extended segment in PDGF (540 Å2).
In contrast to the side-by-side orientations of VEGF and PDGF dimers, TGF-β dimers form with their β-sheets face-to-face. Large contributions to this 2,600-Å2 dimer interface are made by an α-helix inserted between the third and fourth cysteine of the knot (26); this helix is absent in VEGF and PDGF. Furthermore, the superposition of VEGF and TGF-β monomers is poor (13, 14), because of a bend in the middle of the central four-stranded β-sheet of VEGF. However, a more detailed examination reveals substantial structural similarities (Fig. 2). Good superposition can be obtained if one considers the cystine knot and the first half of the central β-sheet (rms difference 1.62 Å, 45 Cαs) separately from the second half of the sheet and loop regions β1-β3 and β5-β6 (rms difference 2.06 Å, 43 Cαs). The α-helical conformation of residues near the N terminus of VEGF is also found in TGF-β and in the closely related osteogenic protein-1 (OP-1) (27), but not in PDGF. However, unlike in VEGF and PDGF, where the N terminus is involved in interactions with the other monomer, the helix in TGF-β2 and OP-1 folds back against the central β-sheet of the same subunit. The conformation of loop β1-β3, which is disordered in PDGF, is conserved between VEGF and TGF-β, including the short helix α2. The short three-stranded sheet formed by β2, β5, and β6 is present in VEGF, PDGF, TGF-β, and OP-1, but absent in nerve growth factor (28, 29) and chorionic gonadotropin (30, 31). This conserved region, which presents a large hydrophobic patch, has been proposed to be important for PDGF and VEGF receptor recognition (12, 32).
Mapping the KDR Binding Site.
Introduction of a glycosylation group at position 84 in loop β5-β6 blocks binding to KDR (32). Based on this finding, we produced 50 single alanine mutants that cover all exposed side chains found within a radius of about 25 Å of residue 84. This region is highly discontinuous in primary sequence and contains residues on both faces of the molecule, presented from helices α1 and α2, strands β2, β5 (C-terminal half), β6 (N-terminal half) and β7, and loops α1-β1, α2-β2, and β3-β4. To screen rapidly for KDR binding activity of these mutants, we used the Phage ELISA method (33). The dimer of VEGF 1–109 was displayed in single-copy format on the surface of filamentous phagemid particles. These phagemid particles bound a dimeric form of KDR (KDR-IgG) with high affinity (EC50 = 4 nM, Fig. 3). Because deletion studies show that the 10 N-terminal residues do not contribute to KDR binding (Fig. 3), we did not further analyze this region.
Phage ELISAs on the 50 alanine mutants identified nine single mutations that affected KDR binding by more than 3-fold compared with wild type (Table 2). Soluble proteins corresponding to these and nearby mutant (K84A) were analyzed using an RIA that measured the ability of each mutant to displace radio-iodinated VEGF 8–109 from KDR-IgG (Table 3). The five side chains most important for binding were Ile-46 from strand β2, Ile-83 from strand β5, Glu-64 from loop β3-β4, Phe-17 from helix α1, and Gln-79 from strand β5; when mutated to alanine affinity was reduced by 330, 250, 110, 90, and 89-fold, respectively. If one assumes the effects are additive (34), these residues together would account for 70% of the total disruption in binding free energy observed for all the alanine mutations. The next three most important residues were Ile-43 from loop β1-β2, and Lys-84 and Pro-85 from strand β5, which would account for ≈20% of the decrease in binding energy.
Table 2.
Relative binding affinities as measured by phage ELISAs for 50 single alanine mutants of VEGF 1–109 to bind KDR-IgG or either of the two anti-VEGF mAbs, A4.6.1 and 3.2E3.1.1
| VEGF residue | Exposed surface area, Å2 | Relative affinity (EC50 mut/EC50 wt)
|
||
|---|---|---|---|---|
| KDR | A4.6.1 | 3.2E3.1.1 | ||
| WT | — | 1.0 | 1.0 | 1.0 |
| H11 | — | 2.5 | 1.7 | 1.2 |
| H12 | — | 1.8 | 1.1 | 1.0 |
| E13 | — | 0.8 | 1.3 | 0.9 |
| V14 | 26 | 1.8 | 1.5 | 0.8 |
| V15 | 17 | 0.8 | 1.1 | 0.8 |
| K16 | 112 | 1.1 | 1.2 | 0.6 |
| F17 | 20 | •NB | 1.2 | 0.9 |
| M18 | 95 | •5.0 | 1.2 | •24.0 |
| D19 | 64 | 0.6 | 0.7 | 0.9 |
| Y21 | 52 | 2.8 | 2.6 | •74.0 |
| Q22 | 96 | 2.2 | 1.4 | •47.0 |
| R23 | 94 | 1.5 | 1.3 | 0.7 |
| Y25 | 81 | 1.7 | 1.2 | •NB |
| F36 | 41 | 1.7 | 1.2 | 2.8 |
| Q37 | 61 | 1.5 | 1.0 | 2.0 |
| E38 | 20 | 0.5 | 0.7 | 1.2 |
| Y39 | 80 | 1.3 | 1.5 | 1.5 |
| P40 | 26 | 0.6 | 2.0 | 1.0 |
| D41 | 87 | 1.5 | 1.9 | 2.6 |
| E42 | 42 | 0.7 | 1.4 | 1.3 |
| I43 | 69 | •5.6 | 1.2 | 1.5 |
| E44 | 45 | 0.7 | 1.3 | 2.3 |
| Y45 | 2 | 1.6 | 2.2 | 1.7 |
| I46 | 86 | •NB | 2.1 | 0.9 |
| F47 | 1 | •3.6 | •NB | 0.8 |
| K48 | 60 | 0.7 | 0.7 | 1.7 |
| N62 | 1 | 2.0 | 2.2 | 2.1 |
| D63 | 66 | 0.5 | 0.9 | 1.2 |
| E64 | 114 | •8.5 | 1.7 | 0.7 |
| G65 | — | 1.3 | 1.7 | 2.5 |
| L66 | 57 | 0.3 | 1.6 | 0.7 |
| E67 | 56 | 0.3 | 1.6 | 1.5 |
| V69 | 47 | 0.6 | 1.4 | 0.8 |
| Q79 | 50 | •NB | 0.7 | 1.1 |
| M81 | 42 | 2.0 | •NB | 0.8 |
| R82 | 79 | 0.6 | •3.3 | 1.5 |
| I83 | 31 | •100.0 | 1.0 | 0.9 |
| K84 | 73 | 2.2 | 1.8 | 1.1 |
| P85 | 49 | •5.0 | 1.0 | 1.9 |
| H86 | 138 | 1.3 | 1.9 | 1.4 |
| Q87 | 85 | 1.8 | 1.3 | 1.3 |
| G88 | — | 1.0 | •NB | 0.7 |
| Q89 | 48 | 2.5 | •NB | 1.4 |
| H90 | 92 | 1.7 | 1.8 | 1.1 |
| I91 | 66 | 1.0 | 1.2 | 2.1 |
| G92 | — | 1.2 | •8.5 | 1.7 |
| E93 | 85 | 0.5 | •6.6 | 1.0 |
| M94 | 14 | 2.8 | •NB | 0.9 |
| E103 | 72 | 1.1 | 2.3 | 1.2 |
| R105 | 106 | 0.8 | 1.4 | 1.2 |
The exposed surface area is the side chain area beyond the Cβ atom removed by the alanine mutation. Relative EC50 values are calculated as EC50 mutant/EC50 wild-type; numbers greater than one indicate reductions in binding affinity. Any variant with a 3-fold or greater reduction in EC50 is marked with a bullet. Because phage ELISAs require substantial binding of the mutant phagemid to its protein target to generate a signal for measurement, nonbinders (NB) cannot be precisely quantitated but may be interpreted to have a greatly reduced binding affinity. Typical losses observed for nonbinders are in the range of greater than 100-fold, but vary considerably due to differences in expression.
Table 3.
Relative IC50 values (IC50 mutant/IC50 wild-type) measured for VEGF 1–109 alanine mutants
| VEGF residue | Exposed surface area, Å2 | Relative affinity (Kd mut/Kd wt) |
|---|---|---|
| WT | 1 (13 pM) | |
| F17 | 20 | 90.0 |
| M18 | 95 | 3.3 |
| I43 | 69 | 19.0 |
| I46 | 86 | 330.0 |
| F47 | 1 | 9.1 |
| E64 | 114 | 110.0 |
| Q79 | 50 | 89.0 |
| I83 | 31 | 250.0 |
| K84 | 73 | 9.5 |
| P85 | 49 | 35.0 |
The Kd measured for VEGF 1-109 was 13 pM. The exposed surface is calculated as the total solvent-accessible surface of atoms beyond the Cβ atom.
When the eight binding determinants are mapped on the structure of the VEGF dimer they define two symmetrical binding surfaces, one located at each pole of the molecule (Fig. 4A). Within each of these surfaces the eight side chains cluster into two nearby patches or “hot spots,” exposed on the same face of the molecule. Both hot spots are surrounded by residues that could be mutated to alanine with little effect on affinity, suggesting that the mutational analysis has been comprehensive. Remarkably, even though the hot spots are highly localized, each consists of side chains contributed from both subunits. The largest hot spot consists of the two most important residues (Ile-46 and Ile-83) and three other residues of lesser importance (Ile-43, Lys-84, and Pro-85), along with one residue of moderate importance from the opposite subunit (Glu-64). The smaller hot spot contains one residue of moderate importance from each subunit (Gln-79 and Phe-17). The side chains of these eight residues are solvent accessible (Table 3) with exposed areas that range from 20 Å2 to 114 Å2. The side chains in the larger hot spot comprise about 500 Å2 of solvent-accessible area, while the smaller hot spot exposes some 70 Å2. The total accessible area of these functionally important side chains in VEGF (570 Å2) is comparable to that of the functionally important side chains that interact between hGH (≈400 Å2) (34) and its receptor (≈450 Å2) (37).
Figure 4.
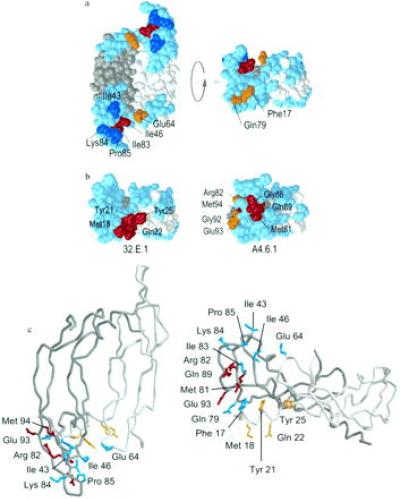
Space-filling and tube representations (35, 36) of the binding determinants on the structure of VEGF 8–109. (a) Functional maps of KDR binding site. (Left) The VEGF dimer with one monomer in white and the other in gray. All residues that were alanine-scanned are colored: light blue (<1.0 kcal/mol impact on binding free energy), blue (1.0–2.0 kcal/mol), yellow (2.0–3.0 kcal/mol), and red (>3.0 kcal/mol). The locations of hydrophobic hot spot residues Phe-17, Ile-43, Ile-46, Ile-83, and Pro-85, as well as the three important polar residues, Glu-64, Gln-79, and Lys-84, are noted. (Right) An end-on view generated by rotating the molecule up by 90°, showing the determinants associated with helix α1, Phe-17, and Gln-79. (b) Functional epitopes for the KDR-blocking mAbs, 3.2E3.1.1 and A4.6.1, as identified by alanine-scanning phage ELISA, oriented similarly to the end-on view of the KDR site. (c) Main-chain representations including the side chains of the binding determinants for KDR (cyan) and both antibodies (red for A4.6.1 and yellow for 3.2E3.1.1).
Recent biophysical and cell biology experiments have provided direct evidence that two molecules of KDR bind a single VEGF dimer (G. Fuh, B.C.C., and J.A.W., unpublished results). The clustering of the hot spots on the VEGF homodimer strongly suggests that KDR dimerization is achieved by binding a receptor at each pole (Fig. 4A). Therefore, we propose that VEGF possesses two identical symmetrical KDR binding sites each defined by strands β2 (Ile-46) and β5 (Gln-79, Ile-83, Pro-84, Lys-85) and loop α2-β2 (Ile-43) of one monomer, together with the N-terminal helix (Phe-17) and loop β3-β4 (Glu-64) of the second monomer.
Mapping the Epitopes for Two Neutralizing Antibodies.
We used phage ELISAs to study the mechanism by which two receptor-blocking anti-VEGF antibodies function (Table 2). This analysis revealed two small clusters of discontinuous residues that caused greater than 10-fold disruptions in antibody affinity, and mapped to sites that were distinct from the KDR binding site (Fig. 4B). The four mutants that most disrupt binding to mAb A4.6.1 were F47A from β2, M81A on β5, G88A and Q89A on loop β5-β6, and M94A on β6. Among these, Phe-47 is not surface exposed and probably acts indirectly. The five mutants that disrupted binding to mAb 3.2E3.1.1 were M18A, Y21A, and Q22A on helix α1, and Y25A on the following loop. The mAb A4.6.1 epitope lies adjacent to the larger KDR hot spot, and that for mAb 3.2E3.1.1 next to the smaller one. Thus, these antibodies do not block receptor binding by direct competition for the same binding determinants, but rather by sterically blocking access to the KDR binding surface.
Because the antibodies and KDR bind nearby sites on VEGF, the antibodies can be used as local probes for the structures of the alanine mutants. For example, the three alanine mutants most disrupted in binding to KDR (I46A, I83A, and E64A) bind with near wild-type affinity to both antibodies (Table 2). Similarly, the most disruptive mutants in each of the antibodies do not affect binding to the other antibody or KDR. Thus, these results suggest that the conformation of the alanine mutants is similar to wild-type VEGF.
Implications for Ligand–Receptor Interactions.
The functional KDR binding site of VEGF consists of binding determinants that are contributed by both subunits in the homodimer. Subunit-overlapping receptor binding sites have also been deduced from mutational analyses of nerve growth factor (38) and neurotrophin-3 (39). Structural evidence for ligand-receptor contacts overlapping the subunits of oligomeric ligands is seen in the x-ray structures of trimeric tumor necrosis factor (TNF)-β bound to three molecules of the TNF-R55 receptor (40), as well as in dimeric interferon-γ bound to two molecules of its high-affinity receptor (41). This suggests that in these cases, too, the functional epitope may lie across the subunit interface. It remains to be seen whether oligomeric hormones that oligomerize their receptors will generally use subunit-overlapping modes of binding.
To date only fragmentary mutational data is available for other cystine-knot hormones (25, 26, 42), so it is unclear whether these bind their receptors in a topologically equivalent manner to VEGF. The limited number of residues identified for PDGF (12) (Leu-38, Arg-73, and Ile-77) map onto strands β2 and β5 in an analogous region to the large hot spot on VEGF. If PDGF binds receptor similarly to VEGF, then its N terminus may contain determinants analogous to VEGF’s small hot spot. For TGF-β recent experimental evidence shows that residues in loop β5-β6 are important (43). Therefore, TGF-β, too, may bind its receptor with the structurally conserved region that corresponds to the large hot spot in VEGF. In that case, because the face-to-face homodimer association of TGF-β places the second subunit away from this putative receptor-binding surface, the binding site would not overlap the subunits and each monomer subunit would contain a complete receptor binding site.
Recently, we have developed minimization strategies for the production of small, high-affinity polypeptides from proteins with discontinuous binding epitopes. For atrial natriuretic factor the hormone was reduced from 28 residues to just 14 (33), and for the Z-domain of protein A a reduction from 59 to 33 residues was obtained (44). In both cases, success was achieved by using structure-function information to guide the application of phage display methods. Because we have shown that only a small number of VEGF residues are important for binding to KDR, it also may be feasible to generate small mimics of VEGF. The reduced size of such mimics should make them more amenable to translation into organic molecules and thus could provide a rational route to produce orally available drugs.
Acknowledgments
We thank Dot Reilly and Sylvia Wong for fermentations, Greg Bennet and Wai Lee Wong for radioiodinated VEGF, Jin Kim and Ji Lu for antibodies, Bruce Keyt for protein and help with purification during the early stages of the structural work, Kay Diederichs for discussions on possible twinning, and Tony Kossiakoff and Melissa Starovasnik for critical reading of the manuscript.
ABBREVIATIONS
- VEGF
vascular endothelial growth factor
- KDR
kinase domain receptor
- PDGF
platelet-derived growth factor
- TGF-β
transforming growth factor β2
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Brookhaven National Laboratory, Upton, NY 11973 (reference 1VPF).
References
- 1.Ferrara N. Breast Cancer Res Treat. 1995;36:127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 3.Dvorak H F, Brown L F, Detmar M, Dvorak A M. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 4.Neufeld G, Tessler S, Gitay-Goren H, Cohen T, Levi B Z. Prog Growth Factor Res. 1994;5:89–97. doi: 10.1016/0955-2235(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea K S, Powell-Braxton L, Hillian K J, Moore M W. Nature (London) 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Andras N. Nature (London) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 7.Aiello L P, Avery R L, Arrigg P G, Keyt B A, Jampel H D, Shah S T, Pasquale L R, Thieme H, Iwamoto M A, Park J E, Nguyen H V, Aiello L M, Ferrara N, King G. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 8.Kim J K, Li B, Winer J, Armanini M, Gillet N, Phillips H S, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 9.de Vries C, Escobedo J A, Ueno H, Houck K A, Ferrara N, Williams L T. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 10.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin C-H. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 11.Keyt B A, Berleau L T, Nguyen H V, Chen H, Heinsohn H, Vandlen R, Ferrara N. J Biol Chem. 1996;271:7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 12.Oefner C, D’Arcy A, Winkler F K, Eggimann B, Hosang M. EMBO J. 1992;11:3921–3926. doi: 10.1002/j.1460-2075.1992.tb05485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlunegger M P, Grutter M G. J Mol Biol. 1993;231:445–458. doi: 10.1006/jmbi.1993.1293. [DOI] [PubMed] [Google Scholar]
- 14.Daopin S, Li M, Davies D R. Proteins. 1993;17:176–192. doi: 10.1002/prot.340170207. [DOI] [PubMed] [Google Scholar]
- 15.Christinger H W, Muller Y A, Berleau L, Keyt B A, Cunningham B C, Ferrara N, de Vos A M. Proteins. 1996;26:353–357. doi: 10.1002/(SICI)1097-0134(199611)26:3<353::AID-PROT9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Kabsch W. J Appl Crystallogr. 1988;21:916–924. [Google Scholar]
- 17.Terwilliger T, Kim S-H, Eisenberg D. Acta Crystallogr A. 1987;43:1–5. [Google Scholar]
- 18.Collaborative Computational Project, No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 19.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Brünger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 21.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 22.Lowman H B, Wells J A. J Mol Biol. 1993;234:564–578. doi: 10.1006/jmbi.1993.1612. [DOI] [PubMed] [Google Scholar]
- 23.Coa Y, Chen H, Zhou L, Chiang M-K, Anand-Apte B, Weatherbee J A, Wang Y, Fang F, Flanagan J G, Tsang M L-S. J Biol Chem. 1996;271:3154–3162. doi: 10.1074/jbc.271.6.3154. [DOI] [PubMed] [Google Scholar]
- 24.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 25.Murray-Rust J, McDonald N Q, Blundell T L, Hosang M, Oefner C, Winkler F, Bradshaw R A. Structure. 1993;1:153–159. doi: 10.1016/0969-2126(93)90029-g. [DOI] [PubMed] [Google Scholar]
- 26.Sun P D, Davies D R. Annu Rev Biophys Biomol Struct. 1995;24:269–291. doi: 10.1146/annurev.bb.24.060195.001413. [DOI] [PubMed] [Google Scholar]
- 27.Griffith D L, Keck P C, Sampath T K, Rueger D C, Carlson W D. Proc Natl Acad Sci USA. 1996;93:878–883. doi: 10.1073/pnas.93.2.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald N Q, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell T L. Nature (London) 1991;354:411–414. doi: 10.1038/354411a0. [DOI] [PubMed] [Google Scholar]
- 29.Holland D R, Cousens L S, Meng W, Matthews B W. J Mol Biol. 1994;239:385–400. doi: 10.1006/jmbi.1994.1380. [DOI] [PubMed] [Google Scholar]
- 30.Lapthorn A J, Harris D C, Littlejohn A, Lustbader J W, Canfield R E, Machin K J, Morgan F J, Isaacs N W. Nature (London) 1994;369:455–461. doi: 10.1038/369455a0. [DOI] [PubMed] [Google Scholar]
- 31.Wu H, Lustbader J W, Liu Y, Canfield R E, Hendrickson W A. Structure. 1994;2:545–558. doi: 10.1016/s0969-2126(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 32.Keyt B A, Nguyen H V, Berleau L T, Duarte C M, Park J, Chen H, Ferrara N. J Biol Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Tom J Y K, Oare D, Yen R, Fairbrother W J, Wells J A, Cunningham B C. Science. 1995;270:1657–1660. doi: 10.1126/science.270.5242.1657. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham B C, Wells J A. J Mol Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 35.Nicholls A J, Sharp K, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 36.Huang C C, Pettersen E F, Klein T E, Ferrin T E, Langridge R. J Mol Graphics. 1991;9:230–236. doi: 10.1016/0263-7855(91)80016-s. [DOI] [PubMed] [Google Scholar]
- 37.Clackson T, Wells J A. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 38.Bradshaw R A, Murray-Rust J, Ibanez C F, McDonald N Q, Lapatto R, Blundell T L. Protein Sci. 1994;3:1901–1913. doi: 10.1002/pro.5560031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urfer R, Tsoulfas P, Soppet D, Escandon E, Parada L F, Presta L G. EMBO J. 1994;13:5896–5909. doi: 10.1002/j.1460-2075.1994.tb06935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banner D W, D’Arcy A, Janes W, Gentz R, Schoenfeld H J, Broger C, Loetscher H, Lesslauer W. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 41.Walter M R, Windsor W T, Nagabhushan T L, Lundell D J, Lunn C A, Zauodny P J, Narula S K. Nature (London) 1995;376:230–235. doi: 10.1038/376230a0. [DOI] [PubMed] [Google Scholar]
- 42.McDonald N Q, Hendrickson W A. Cell. 1993;73:421–424. doi: 10.1016/0092-8674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- 43.Qian S W, Burmester J K, Tsang M L-S, Weatherbee J A, Hinck A P, Ohlsen D J, Sporn M B, Roberts A B. J Biol Chem. 1996;271:30656–30662. doi: 10.1074/jbc.271.48.30656. [DOI] [PubMed] [Google Scholar]
- 44.Braisted A C, Wells J A. Proc Natl Acad Sci USA. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]