

Abstract
A non-I-domain integrin, α4β1, recognizes vascular cell adhesion molecule 1 (VCAM-1) and the IIICS portion of fibronectin. To localize regions of α4 critical for ligand binding, we swapped several predicted loops within or near the putative ligand-binding site of α4 (which spans repeats 2–5 of the seven N-terminal repeats) with the corresponding regions of α5. Swapping residues 112–131 in repeat 2, or residues 237–247 in repeat 4, completely blocked adhesion to immobilized VCAM-1 and connecting segment 1 (CS-1) peptide. However, swapping residues 40–52 in repeat 1, residues 151–164 in repeat 3, or residues 282–288 (which contain a putative cation binding motif) in repeat 5 did not affect or only slightly reduced adhesion to these ligands. The binding of several function-blocking antibodies is blocked by swapping residues 112–131, 151–164, and 186–191 (which contain previously identified residues critical for ligand binding, Tyr-187 and Gly-190). These results are consistent with the recently published β-propeller folding model of the integrin α4 subunit [Springer, T. A. (1997) Proc. Natl. Acad. Sci. USA 94, 65–72], in which seven four-stranded β-sheets are arranged in a torus around a pseudosymmetric axis. The regions of α4 critical for ligand binding are adjacent to each other and are located in the upper face, the predicted ligand-binding site, of the β-propeller model, although they are not adjacent in the primary structure.
The α4β1 integrin recognizes vascular cell adhesion molecule 1 (VCAM-1) (1) and the alternatively spliced IIICS portion of fibronectin (connecting segment 1 or CS-1) (2–5). VCAM-1 is expressed on activated endothelial cells and constitutively on bone marrow stromal cells (6, 7). Mounting evidence indicates that α4β1 plays a central role in leukocyte recruitment (see ref. 8 for a review). The α4β1 integrin has been shown to initiate lymphocyte contact (“tethering”) in vitro under shear and in the absence of a selectin contribution (9, 10). Anti-α4 mAbs have been shown to have therapeutic effects in numerous animal models of disease (e.g., experimental allergic encephalomyelitis, contact hypersensitivity, nonobese diabetes, allergic lung inflammation, and inflammatory bowel disease) (see ref. 8 for a review). Therefore, ligand/α4β1 integrin interaction is a therapeutic target for many diseases. Understanding the ligand-binding mechanism and identifying ligand-binding sites are important for designing inhibitors that modulate these interactions. Very little information is available, however, on regions or residues of α4 that are critical for ligand/α4β1 interactions.
The N-terminal portion of integrin α subunits (≈440 amino acids) contains seven sequence repeats. Recently, we localized the putative ligand-binding sites of α4 (residues 108–268 of α4) (11), which span repeats 2–5 of the seven N-terminal repeats of α4, by mapping epitopes of function-blocking anti-α4 antibodies. We also identified Tyr-187 and Gly-190, which are clustered in repeat 3 of α4, as critical residues for ligand binding to α4β1 by introducing multiple mutations into the putative ligand-binding sites (12). In the present study, we localized additional critical regions for ligand binding by using another strategy, swapping the predicted loop structures (13) within or close to the putative ligand-binding sites of α4 with the corresponding regions of α5. Interestingly, swapping residues 112–131 in repeat 2 and residues 237–247 in repeat 4 completely blocked cell adhesion to immobilized ligands. The reduced affinity to ligand of these swapped mutants was not restored by activation with Mn2+. These results suggest that these predicted loops in repeats 2–4 are likely to be directly involved in α4β1/ligand interactions.
Recently, Springer (14) has proposed that these seven N-terminal sequence repeats fold into a β-propeller domain. The proposed domain contains seven four-stranded β-sheets arranged in a torus around a pseudosymmetry axis. Integrin ligands and a putative Mg2+ ion are predicted to bind to the upper face of the β-propeller. The Ca2+ binding motifs in the integrin α subunit are believed to be on the lower face of the β-propeller. The present mutagenesis data are consistent with this β-propeller model. The predicted loops, which are critical for binding to VCAM-1 and fibronectin, would be located in the upper face of the β-propeller, the predicted ligand-binding site.
MATERIALS AND METHODS
Materials.
Anti-human α4 mAbs were obtained from the following sources: B5G10 was a kind gift from M. E. Hemler (Dana–Farber Cancer Institute, Boston); HP1/3 and HP2/1 from F. Sanchez-Madrid (Hospital de la Princesa, Madrid); P4C2 from E. Wayner (University of Washington, Seattle); and SG/73 from K. Miyake (Saga Medical School, Saga, Japan). Recombinant VCAM-1/mouse Ck chain fusion protein was from D. Dottavio (Sandoz Pharmaceuticals, East Hanover, NJ); and rat serum albumin-conjugated CS-1 peptide from E. Wayner.
Construction and Transfection of Swapping Mutants of Human α4 cDNAs.
Wild-type human α4 cDNA (15) was subcloned into a pBJ-1 vector (11). The positions of the α4 swapping mutations used in this study are illustrated in Fig. 1. The expression vectors of the R1, R3b, R3c, and R4 mutants, in which residues 40–52, 181–189, 186–191, and 237–247 of α4 were swapped with the corresponding residues of α5, were made by site-directed mutagenesis using a unique site elimination method and using the NotI site in pBJ-1 (16). Other swapping mutants were constructed using the overlap extension PCR (17). Point mutation was introduced using a unique site elimination method as described above. The presence of mutation was verified by DNA sequencing. Transfection of CHO cells was carried out as described (11, 12). Typically, 20–50% of the G418-resistant cells were positive with the mAb B5G10. CHO-B2 cells expressing α4 mutants were then cloned to obtain cells expressing high levels of α4 mutants by cell sorting in FACStar (Becton Dickinson).
Figure 1.

Regions of α4 selected for swapping in this study. Integrin α subunits have seven repeats of about 60 amino acid residues each at their N terminals. We chose predicted loop structures within or close to the previously identified putative ligand-binding sites (residues 108–268) (11) for swapping with the corresponding residues of α5 (boxed regions). W1–W7 represent repeats 1–7 (14). The predicted β-strands of α4 are underlined (14). ∗, Tyr-187 and Gly-190 of α4, which are critical for ligand binding (12).
Other Methods.
The adhesion assay was carried out as described (11, 12). Immunoprecipitation and flow cytometric analyses (18) were performed as described.
RESULTS
Domain-Swapping Mutagenesis of the Putative Ligand-Binding Sites of α4.
To localize regions of α4 that are critical for ligand binding, we replaced predicted loop structures within the putative ligand-binding sites of α4 (residues 108–268, the most likely candidates for ligand-binding sites), with the corresponding regions of α5. Because the ligand-binding specificity of α5β1 (which is specific to the fibronectin cell binding domain) is distinct from that of α4β1 (which is specific to VCAM-1 and the IIICS portion of fibronectin), swapping will block the function of α4 if the function of the swapped region is specific to α4. The regions that were chosen for swapping include residues 40–52, 112–131, 151–164, 181–189, 186–191, and 237–247 within or close to the putative ligand-binding site of α4 (Fig. 1). These regions form relatively large predicted loop structures (13) in the putative ligand-binding site. We also included a region (residues 282–288 of α4) that corresponds to the second cation-binding site of αIIb, to which γ-peptide of fibrinogen has been shown to bind (19). The swapping mutants were designated R1, R2, R3a, R3b, R3c, R4, and R5, respectively (R1–R5 represent repeat 1–5) (Fig. 1). The mutant human α4 cDNA in the pBJ-1 expression vector was transfected into CHO cells that have a low hamster α5 level (the B2 variant) (20). After being selected for G418 resistance, stably expressing cells were cloned by cell sorting. The cloned cells expressed comparable levels of mutant or wild-type α4 (Fig. 2).
Figure 2.

Expression of α4 swapping mutants on CHO-B2 cells. Cloned CHO-B2 cells (20) expressing α4 swapping mutants were stained with the anti-α4 mAb B5G10 followed by fluorescein isothiocyanate-labeled goat anti-mouse IgG and analyzed by flow cytometry. (A) Parent CHO-B2 cells. (B) Wild type. (C) R1. (D) R2. (E) R3a. (F) R3b. (G) R3c. (H) R4. (I) R5. Control mouse IgG (⋅ ⋅ ⋅), B5G10 (——). Mean fluorescent intensity of α4 expression (using mAb B5G10) is 5 (background) for parent CHO-B2 cells, 246 for wild type, 104 for R1, 246 for R2, 185 for R3a, 156 for R3b, 201 for R3c, 81 for R4, and 189 for R5. The data suggest that the expression levels of the clones are comparable.
Swapping Residues 112–131 in Repeat 2 (R2) or Residues 237–247 in Repeat 4 (R4) Completely Blocks Cell Adhesion to Ligands.
Adhesion to VCAM-1 and CS-1 peptide was examined as a function of ligand concentration using cells expressing the mutant α4 (Fig. 3 a and b). Parent CHO-B2 cells did not adhere to either ligand. Adhesion to VCAM-1 of cells expressing the R1, R3a, R3b, R3c, or R5 mutant was comparable to that of cells expressing wild-type α4 at the highest ligand concentration used (Fig. 3a). Adhesion of cells expressing the R1, R3a, and R3c mutants was slightly reduced at lower ligand concentrations. In contrast, cells expressing R2 or R4 did not adhere to VCAM-1 at any ligand concentration. The effect of the R2 or R4 mutation was not reversed by activation with Mn2+ (Fig. 3c), indicating that the negative effect is not due to affinity modulation. Similar results were obtained using CS-1 peptide as a substrate (Fig. 3b). Effects of several mutations on adhesion were more obvious at suboptimal concentrations with CS-1 than with VCAM-1. The data suggest that the two predicted loop structures, R2 and R4, may be critically involved in cell adhesion to ligands.
Figure 3.

Adhesion to VCAM-1 (a) and CS-1 (b) of cells expressing α4 swapping mutants and the effects of Mn2+ (c). Wells of 96-well microtiter plates were coated with CS-1 conjugated to rat serum albumin (CS-1-RSA) at a concentration of up to 1 μg/ml in 100 μl PBS (12). For VCAM-1, wells were first coated with an anti-mouse Ck chain, and then with a VCAM-1/Ck fusion protein at a concentration of up to 1 μg/ml in 100 μl PBS (12). Clonal CHO-B2 cells expressing wild-type (wt) or mutant α4 were incubated for 1 h at 37°C with either VCAM-1 (a) or CS-1-RSA (b) immobilized to plastic wells. The adherent cells were quantified using the endogenous phosphatase assay (21). Data are expressed as means ± SD of triplicate experiments. (c) Adhesion of CHO cells to VCAM-1 was determined in the absence (filled column) and presence (hatched column) of MnCl2 (1 mM) in the assay mixture with VCAM-1 (0.5 μg/ml coating concentration) as a substrate.
To determine whether domain swapping of α4 affects the gross structure of α4β1, lysates from 125I-surface-labeled cells were immunoprecipitated with the anti-α4 mAb B5G10. Fig. 4 shows that the α4 mutants with expected sizes were detected in association with hamster β1. The α4 and β1 subunits are known to be weakly associated (23). Although the recovery of endogenous β1 subunit is lower in the R1, R2, and R4 mutants, this effect does not appear to correlate with ligand binding, because the R1 mutant shows ligand-binding function comparable with that of wt α4. Also, these mutants were expressed on the surface of CHO-B2 cells (Fig. 2), and were recognized by multiple anti-α4 antibodies (Table 1). These results suggest that the effects of swapping mutations on adhesion to ligands are probably not due to gross structural changes.
Figure 4.

Immunoprecipitation of α4 mutants. Lysates from surface 125I-labeled cells expressing wild-type (wt) or mutant α4 were immunoprecipitated with mAb B5G10; 0.3% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was used as a detergent. For R3c, polyclonal anti-α4 rabbit serum and 1% Triton X-100/0.5% Tween 20 were used instead of B5G10 and CHAPS. Immunoprecipitated materials were analyzed by SDS/PAGE (7% gel) under nonreducing conditions. A faint protein band just above α4 might be a 180-kDa form of α4 (22). Control mouse IgG did not precipitate anything from any of the cell lines used (data not shown).
Table 1.
Binding of anti-α4 mAbs to mutants of α4
| Mouse IgG | P4G9 | HP1/3 | P4C2 | SG/73 | HP2/1 | B5G10 | |
|---|---|---|---|---|---|---|---|
| CHO-B2 | 2.1 | 3.3 | 3.0 | 2.9 | 3.5 | 3.8 | 3.1 |
| wt-α4 | 2.3 | 72.9* | 98.4* | 91.3* | 96.4* | 98.4* | 99.1* |
| R1 | 1.6 | 75.3* | 1.3 | 74.9* | 97.7* | 97.2* | 98.2* |
| R2 | 1.6 | 74.4* | 96.2* | 1.5 | 1.5 | 1.6 | 97.1* |
| R3a | 2.6 | 64.3* | 98.1* | 2.1 | 2.4 | 95.3* | 98.9* |
| R3b | 0.8 | 91.3* | 96.4* | 60.7* | 92.5* | 95.5* | 95.8* |
| R3c | 4.5 | 62.3* | 98.1* | 3.1 | 96.8* | 98.4* | 99.1* |
| R4 | 3.4 | 69.2* | 81.2* | 55.0* | 80.2* | 84.7* | 86.7* |
| R5 | 1.2 | 82.3* | 99.1* | 76.9* | 98.8* | 96.3* | 99.6* |
| Y120A/G130A | 5.5 | 92.0* | 6.9 | 6.8 | 7.1 | 11.3 | 97.2* |
CHO cells stably expressing different mutants were stained by mAbs, followed by fluorescein isothiocyanate-labeled secondary anti-mouse IgG and analyzed using flow cytometry. The numbers in the table represent percentage of positive cells. wt, wild type.
Positive reactivity.
To further characterize the predicted loop spanning 112–131, we introduced multiple mutations within the region. A double mutation, Y120A/G130A, completely blocked adhesion to VCAM-1 and CS-1 peptide and binding of function-blocking mAbs (Table 1). However, other single or double mutations (H113A, R114A, W115A, K116A, N117A, F119A, Y120A, I121A, N123A, N125A, K126A, T119A, G130A, G131A, I121A/G130A, or E124A/G130A) did not significantly affect adhesion or binding of function-blocking mAbs (data not shown). Immunoprecipitation of the Y120A/G130A mutant resulted in the detection of α4, and of 80/70-kDa fragments associated with the endogenous hamster β1 subunit, indicating that the mutation did not affect the gross structure and α-β association of α4β1 (data not shown). These data suggest that the Y120A/G130A mutation induces structural changes in the predicted loop region spanning residues 112–131, which may be critically involved in adhesion to VCAM-1 and CS-1. We also introduced point mutations within the region spanning 237–247 in repeat 4. The Q244A and previously published Q241A (12) mutations did not affect adhesion to these ligands. Other α4 mutants, including H242A, E243A, and G246A, were not expressed on CHO cells, according to the flow cytometry results. Therefore, it is not conclusive whether there are any critical residues for adhesion in repeat 4.
Tyr-187 in Repeat 3 Is Replaceable with Phe and Trp, but Gly-190 Is Not.
We previously reported that mutation of Tyr-187 and Gly-190 in repeat 3 to Ala blocked ligand binding, suggesting that they are critical for ligand binding (12). These residues are also located in the predicted loop in the upper face of the proposed β-propeller model of the integrin α subunit (14). In the present study, the R3b and R3c mutations did not critically affect adhesion to either VCAM-1 or CS-1 (Fig. 3). These results indicate that residues 181–191 of α4 may be functionally replaceable with the corresponding sequence of α5. To clarify this point, we replaced Tyr-187 and Gly-190 with different amino acid residues. Cells homogeneously expressing the Y187F and Y187W mutants showed adhesion to VCAM-1, but cells expressing the Y187H, Y187S, and previously published Y187A mutants showed markedly reduced adhesion to VCAM-1 (Fig. 5a). The effects of these mutations on adhesion to CS-1 were stronger than with VCAM-1. These results suggest that Tyr-187 may be replaceable with neutral aromatic amino acid residues (e.g., Phe and Trp). In contrast, cells homogeneously expressing the G190D, G190P, G190K, or G190Y mutants did not show any affinity to either VCAM-1 or CS-1 (Fig. 5b), suggesting that Gly-190 is not replaceable. It is likely that, because both the R3b and R3c mutants retain Phe at position 187 and Gly at position 190, these mutants showed adhesion to both ligands at a level similar to that of wild-type α4.
Figure 5.
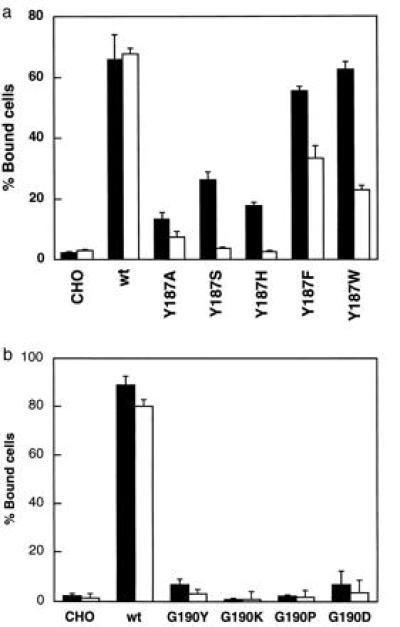
Effects on adhesion to ligands of substitution of Tyr-187 (a) and Gly-190 (b) in repeat 3. Adhesion capability of cells expressing Tyr-187 or Gly-190 mutants to VCAM-1 (solid bar) or RSA-CS-1 (open bar) (both at 1 μg/ml coating concentration) were examined as described in the legend to Fig. 3. Data are expressed as means ± SD of triplicate experiments. All of the cells expressing mutants are clonal. Mean fluorescent intensity of α4 expression with mAb B5G10 is 129 for wild-type (wt) α4, 186 for Y187A, 413 for Y187S, 309 for Y187H, 263 for Y187F, 489 for Y187W, 370 for G190Y, 221 for G190K, 231 for G190P, 277 for G190D mutants, and 4.2 (background level) for parent CHO cells.
Swapping Residues 112–131, 151–164, or 186–191 of α4 Blocks Binding of Function-Blocking Antibodies.
Some of the anti-α4 mAbs have been reported to recognize epitopes A, B (B1 and B2), and C on the α4 subunit; these epitopes have been described based on the cross-competitive cell binding and protease-sensitivity assays (24). Epitope B is related to cell attachment to fibronectin and VCAM-1, but epitopes A and C are not (or are only slightly). Epitope B has been physically mapped within residues 108–268 (11) and within residues 152–203 (25). Reactivity of function-blocking anti-α4 mAbs to the swapped α4 mutants was examined by flow cytometry. We found that function-blocking anti-α4 mAbs (P4C2, SG/73, and partially HP2/1, epitope B) do not bind to R2, R3a, and/or R3c mutants (Table 1). We have localized epitope A within residues 1–52 and epitope C within residues 269–548 (11). Consistently, mAb HP1/3 (epitope A) does not recognize the R1 mutant, while a non-function-blocking antibody, B5G10 (epitope C), reacts with all of the α4 mutants, as well as with wild-type α4. These data suggest that the three separate predicted loop structures of α4, residues 112–131 (R2), 151–164 (R3a), and 186–191 (R3c), may be related to ligand-binding function.
DISCUSSION
In the present study we located the regions of α4 that are critical for ligand binding using swapping mutagenesis. We established that the R2 or R4 mutation completely blocks adhesion to VCAM-1 and CS-1 peptide. We have also shown that the Y120A/G130A mutation in the R2 region completely blocks adhesion to these ligands. Although Tyr-187 and Gly-190 in repeat 3 are critical for ligand binding (12), we have shown that residues 181–189 or 186–191 in repeat 3 of α4 can be replaced with the corresponding residues of α5. This is probably because the swapping mutation induced a conservative change in Tyr-187 to Phe and no change in Gly-190. Consistent with the previous conclusion (12), nonconservative substitution of Tyr-187 and substitution of Gly-190 markedly affected ligand binding (Fig. 5). We have reported that mutation in the corresponding region of the α5 subunit (12) and in the αIIb subunit (26) similarly reduced or abolished ligand binding. These findings suggest that the predicted loop spanning 181–190 is ubiquitously involved in ligand binding. The predicted loops spanning 181–190 of α4 and α5 may have similar roles in ligand binding. It is possible that mutation of G190 and Y120/G130 disrupts the structure of the predicted loops. Both Gly residues are almost invariably conserved before β-strand 3 (Fig. 1). They are probably critically involved in a turn, and may have phi–psi angles that are disallowed for the other amino acids.
The present mutagenesis data are consistent with the recently proposed β-propeller model of the seven sequence repeats in the integrin α4 subunit (14). The predicted loops spanning residues 112–131, 181–190, and 237–247, which are critical for ligand interaction, are believed to be located in the upper face (the predicted ligand-binding site) of the β-propeller (Fig. 6). These critical regions are adjacent in the upper face of the β-propeller, although they are separated in the primary structure. The finding that swapping residues 282–288 in repeat 5 (R5) does not affect adhesion to ligands or binding of function-blocking antibodies might be consistent with the prediction that these residues are located in the lower face of the β-propeller. Because swapping residues 40–52 of repeat 1, which would be located in the upper face of the proposed β-propeller model, had only minimum effect on adhesion, it is possible that only some of the predicted loops in the upper face (in repeats 2–4) may be involved in ligand interaction. However, it is also possible that the effects of swapping are not detectable, because residues 282–288 and 40–52 of α4 and the corresponding α5 residues are functionally interchangeable.
Figure 6.

Predicted loops critical for adhesion to VCAM-1 and CS-1 are shown to be adjacent in the upper face of the predicted β-propeller structure of α4. Views are (a) from the top and (b) from the side. The predicted β-propeller structure was taken from (14). ∗, Predicted loops critical for adhesion. Residues 181–191 include both R3b and R3c mutants.
We have shown that the binding of several function-blocking anti-α4 antibodies is blocked by swapping residues 112–131, 151–164, and 186–191, and by the Y120A/G130A and G190A mutations (12), but is not blocked by swapping residues 40–52 or 282–288. These results support the idea that residues 112–131, 151–164, and 186–191 may be close to or within the ligand-binding site; this finding is consistent with the proposed β-propeller model. Although swapping residues 151–164 of α4 (R3a) did not affect adhesion to ligands in the present study, we recently found that swapping the corresponding regions of α3 or α6 completely blocks α3β1- or α6β4-dependent adhesion to laminin-5 (unpublished data). These results suggest that regions in the α subunits critical for ligand binding depend on integrin and/or ligand species. Consistent with the proposed β-propeller model, residues 151–164 of α4 (and the corresponding regions in α3 and α6) are located in the upper face of the β-propeller, and are adjacent to the other critical regions in repeats 2–4. Further characterization of ligand-binding sites in integrin α subunits will be required to fully substantiate the β-propeller model.
Recently, mutations of several residues in α4 have been reported, including Asn-283, Asp-346, and Asp-468 (27); Asp-489 and Asp-698 (28); Arg-89/Asp-90 (29); and Cys-278 and Cys-717 (30). These mutations partially block ligand binding, and their effects are reversed by activation. It appears that these residues are involved in affinity modulation, but are not directly involved in ligand binding. The present α4 swapping mutants, R2 and R4, the Y120A/G130A double mutant, and the previously reported G190A mutant (12) are unique in that they completely block binding of α4β1 to both VCAM-1 and CS-1 peptide, and in that the effects are not reversed by activation. This is consistent with the idea that the predicted loop structures in repeats 2–4 of α4 are directly involved in ligand binding.
Acknowledgments
We thank Drs. D. Dottavio, M. E. Hemler, K. Miyake, F. Sanchez-Madrid, and E. Wayner for reagents, and Mrs. Anna Ewers for help in preparing the manuscript. This work was supported by National Institutes of Health Grants GM47157 and GM49899. This is publication 10075-VB from The Scripps Research Institute.
ABBREVIATIONS
- VCAM-1
vascular cell adhesion molecule-1
- CS-1
connecting segment 1
References
- 1.Elices M J, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler M E, Lobb R R. Cell. 1990;60:577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- 2.Wayner E A, Garcia-Pardo A, Humphries M J, MaDonald J A, Carter W G. J Cell Biol. 1989;109:1321–1330. doi: 10.1083/jcb.109.3.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mould A P, Wheldon L A, Komoriya A, Wayner E A, Yamada K M, Humphries M J. J Biol Chem. 1990;265:4020–4024. [PubMed] [Google Scholar]
- 4.Mould A P, Komoriya A, Yamada K M, Humphries M J. J Biol Chem. 1991;266:3579–3585. [PubMed] [Google Scholar]
- 5.Guan J L, Hynes R O. Cell. 1990;60:53–61. doi: 10.1016/0092-8674(90)90715-q. [DOI] [PubMed] [Google Scholar]
- 6.Rice G E, Bevilacqua M P. Science. 1989;246:1303–1306. doi: 10.1126/science.2588007. [DOI] [PubMed] [Google Scholar]
- 7.Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, Lobb R. Cell. 1989;59:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- 8.Lobb R R, Hemler M E. J Clin Invest. 1994;94:1722–1728. doi: 10.1172/JCI117519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berlin C, Bargatze R, Campbell J, von Andrian U, Szabo M, Hasslen S, Nelson R, Berg E, Erlandsen S, Butcher E. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 10.Alon R, Kassner P D, Woldemar Car M, Finger E B, Hemler M E, Springer T A. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamata T, Puzon W, Takada Y. Biochem J. 1995;305:945–951. doi: 10.1042/bj3050945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irie A, Kamata T, Puzon-McLaughlin W, Takada Y. EMBO J. 1995;14:5542–5549. doi: 10.1002/j.1460-2075.1995.tb00242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuckwell D, Humphries M, Brass A. Cell Adhes Commun. 1994;2:385–402. doi: 10.3109/15419069409004450. [DOI] [PubMed] [Google Scholar]
- 14.Springer T. Proc Natl Acad Sci USA. 1997;94:65–72. doi: 10.1073/pnas.94.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takada Y, Elices M J, Crouse C, Hemler M E. EMBO J. 1989;8:1361–1368. doi: 10.1002/j.1460-2075.1989.tb03516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng W P, Nickoloff J A. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- 17.Horton R M, Pease L R. In: Directed Mutagenesis; A Practical Approach. McPherson M J, editor. Oxford: IRL; 1991. pp. 217–247. [Google Scholar]
- 18.Takada Y, Ylanne J, Mandelman D, Puzon W, Ginsberg M. J Cell Biol. 1992;119:913–921. doi: 10.1083/jcb.119.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Souza S, Ginsberg M H, Burke T A, Plow E F. J Biol Chem. 1990;265:3440–3446. [PubMed] [Google Scholar]
- 20.Schreiner C L, Bauer J S, Danilov Y N, Hussein S, Sczekan M, Juliano R L. J Cell Biol. 1989;109:3157–3167. doi: 10.1083/jcb.109.6.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prater C A, Plotkin J, Jaye D, Frazier W A. J Cell Biol. 1991;112:1031–1040. doi: 10.1083/jcb.112.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker C M, Pujades C, Brenner M B, Hemler M E. J Biol Chem. 1993;268:7028–7035. [PubMed] [Google Scholar]
- 23.Hemler M E, Huang C, Takada Y, Schwarz L, Strominger J L, Clabby M L. J Biol Chem. 1987;262:11478–11485. [PubMed] [Google Scholar]
- 24.Pulido R, Elices M J, Campanero M R, Osborn L, Schiffer S, Garcia-Pardo A, Lobb R, Hemler M E, Sanchez-Madrid F. J Biol Chem. 1991;266:10241–10245. [PubMed] [Google Scholar]
- 25.Schiffer S, Hemler M, Lobb R, Tizard R, Osborn L. J Biol Chem. 1995;270:14270–14273. doi: 10.1074/jbc.270.24.14270. [DOI] [PubMed] [Google Scholar]
- 26.Kamata T, Irie A, Takada Y. J Biol Chem. 1996;271:18610–18615. doi: 10.1074/jbc.271.31.18610. [DOI] [PubMed] [Google Scholar]
- 27.Masumoto A, Hemler M E. J Cell Biol. 1993;123:245–253. doi: 10.1083/jcb.123.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, Conrad P, Webb D, Blue M. J Biol Chem. 1995;270:18401–18407. doi: 10.1074/jbc.270.31.18401. [DOI] [PubMed] [Google Scholar]
- 29.Munoz M, Serrador J, Sanchez M F, Teixido J. J Biol Chem. 1996;271:2696–702. doi: 10.1074/jbc.271.5.2696. [DOI] [PubMed] [Google Scholar]
- 30.Pujades C, Teixido J, Bazzoni G, Hemler M. Biochem J. 1996;313:899–908. doi: 10.1042/bj3130899. [DOI] [PMC free article] [PubMed] [Google Scholar]