

Abstract
The gene encoding 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase (MHPCO; EC 1.14.12.4) was cloned by using an oligonucleotide probe corresponding to the N terminus of the enzyme to screen a DNA library of Pseudomonas sp. MA-1. The gene encodes for a protein of 379 amino acid residues corresponding to a molecular mass of 41.7 kDa, the same as that previously estimated for MHPCO. MHPCO was expressed in Escherichia coli and found to have the same properties as the native enzyme from Pseudomonas sp. MA-1. This study shows that MHPCO is a homotetrameric protein with one flavin adenine dinucleotide bound per subunit. Sequence comparison of the enzyme with other hydroxylases reveals regions that are conserved among aromatic flavoprotein hydroxylases.
2-Methyl-3-hydroxypyridine-5-carboxylic acid oxygenase (MHPCO; EC 1.14.12.4) is a flavin adenine dinucleotide (FAD)-containing enzyme involved in the degradation of vitamin B6 (pyridoxine) by the soil bacterium Pseudomonas sp. MA-1 (P-MA1) (1). Degradation of vitamin B6 proceeds via an oxidative pathway that is induced when these bacteria are grown on pyridoxine or pyridoxamine as their sole source of carbon and nitrogen (2, 3). The pathway consists of a series of oxidative, hydrolytic, and decarboxylation reactions that convert pyridoxine to metabolites readily assimilated for growth. MHPCO catalyzes an oxygenation reaction and a ring cleavage of its substrate, MHPC, to yield α-(N-acetylaminomethylene)succinic acid as shown in Eq. 1 (1).
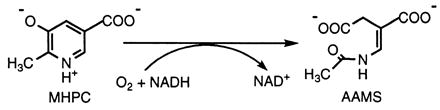 |
Although the reaction catalyzed by MHPCO is formally a dioxygenation, MHPCO belongs to the external aromatic flavoprotein monooxygenase (or aromatic flavoprotein hydroxylase) class (4, 5). The reductive half-reaction (4) shows roles of the aromatic substrate and pyridine nucleotide in the reaction mechanism similar to those found with conventional aromatic flavoprotein hydroxylases, and the oxidative half-reaction (5) involves C(4a)-hydroperoxy-flavin and C(4a)-hydroxy-flavin intermediates, which are common to all other aromatic flavoprotein hydroxylases. Studies of the reaction with a substrate analog, 5-hydroxynicotinic acid, also show that the enzymatic reaction of MHPCO consists of two parts: hydroxylation and a subsequent ring cleavage reaction (5).
Previous studies with MHPCO were unable to firmly establish if the enzyme has a structure of α4 or α2β2. The stoichiometry of 2 FAD per holoenzyme was reported by Sparrow et al. (1) and Kishore et al. (6). Molecular mass of the holoenzyme was determined to be about 166 kDa (1, 6), and each subunit has a size of 43 kDa as shown by SDS/PAGE (6). The subunits of the enzyme appeared to be identical, because they migrate as a single band in SDS/PAGE. It was therefore thought that the enzyme had an α4 subunit-composition with two FAD bound per tetramer or an α2β2 composition with the α and β subunits being very similar in size. Therefore, we decided to clone and express the enzyme, which would help to clarify the subunit composition and provide information that should be valuable in future structure–function relationship studies.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Reagents.
P-MA1 and MHPCO were prepared according to ref. 4. P-MA1 genomic DNA was purified by using a DNA purification column (Qiagen). The pGEM-T vector and Escherichia coli JM109 were obtained from Promega. The pBluescript II SK(+) was from Stratagene. The pET-11a vector and E. coli BL21(DE3) were from Novagen. Restriction enzymes, BamHI, HindIII, EcoRI, NotI, PstI, and EagI were from Boehringer Mannheim, and BssHII was from New England Biolabs. PCR was performed by using Ampli Taq polymerase from Perkin–Elmer. Isopropyl β-d-thiogalactopyranoside and 5-bromo-4-chloro-3-indolyl β-d-galactoside were purchased from GIBCO/BRL. Digoxigenin-11-dUTP (Dig-11-dUTP), anti-digoxigenin-alkaline phosphatase conjugate, 5-bromo-4-chloro-3-indolyl phosphate, and nitroblue tetrazolium salt were from Boehringer Mannheim. Vent DNA polymerase was from New England Biolabs. Oligonucleotide primers for PCR and DNA sequencing were synthesized on automated instruments from Applied Biosystems (Biopolymers Core DNA Synthesis, University of Michigan). BCA and Bradford reagent were from Pierce.
PCR and Probe Labeling.
PCR was performed with a Perkin–Elmer Cetus GeneAmp PCR system 9600. The reaction conditions were as follows: primers (each at 1 μM), P-MA1 DNA (0.3 μg), dNTPs (each at 200 μM), MgCl2 (2 mM) in 10 mM Tris⋅HCl, 50 mM KCl, pH 8.3 (total reaction volume 100 μl). The reaction was started by adding 2.5 units of Taq polymerase in the reaction mixture at 95°C. Cycling parameters were as follows (for 30 cycles): denaturing at 95°C for 45 sec, annealing at 50°C for 1 min, extending at 72°C for 90 sec. For probe labeling reactions, where we wanted to incorporate Dig-11-dUTP instead of dTTP in the PCR product, the conditions for PCR were the same as above except a mixture of 130 μM dTTP and 200 μM Dig-11-dUTP was used instead of 200 μM dTTP. All PCR products were purified by using QIAquick-spin columns (Qiagen). The concentration of Dig-11-dUTP-labeled probe was quantified according to methods described in the Boehringer Mannheim Genius system user’s guide, version 3.0 (7).
Library Screening and Gene Cloning.
The general procedures for library screening and gene cloning were carried out as described in ref. 8. P-MA1 DNA was partially purified to isolate a fraction that contains a high concentration of the MHPCO gene. Briefly, genomic P-MA1 DNA was digested with PstI and run on 0.5% agarose gel. The digested DNA was transferred from the gel to a nylon membrane by the turboblotter transfer system (Schleicher & Schuell). The membrane was hybridized with Dig-11-dUTP labeling probe (probe concentration 20 ng/ml) according to the method described in ref. 7. Hybridization and the final wash were performed at 65–68°C. The probe was hybridized with a 3.5-kb fragment of DNA. The PstI-cut DNA near this 3.5-kb region was isolated from agarose gel by using a QIAquick-spin column.
P-MA1 DNA from the above procedure was ligated to the PstI site of pBluescriptII SK(+) and transformed to E. coli JM109. White colonies, which contain the DNA insert in the cloning vector, were subjected to the in situ hybridization procedure as described in detail in ref. 7.
DNA Sequencing.
PCR products and genomic DNA fragments were sequenced with an Applied Biosystems DNA Sequencer model 373A using the PRISM Ready Reaction Dye Deoxy Terminator Sequencing Kit (DNA Sequencing Core Facility, University of Michigan). T7 and SP6 primers were used to sequence the PCR product. T7, T3, and sequence specific primers were used to sequence both strands of the genomic DNA fragment. The Wisconsin Sequence Analysis package (Genetics Computer Group, Madison, WI) version 8.1 was used for sequence analysis.
N-Terminal and Internal Sequence of MHPCO.
N-terminal protein sequence analysis by automated Edman degradation was performed using standard procedures on a pulse liquid phase sequenator (Applied Biosystems model 473). Internal sequence information was obtained from N-terminal sequencing of an isolated fragment of MHPCO after cyanogen bromide cleavage. Both N-terminal and internal sequence analysis of MHPCO were done by the Protein and Carbohydrate Structure Core Facility, University of Michigan.
Steady-State Kinetics.
MHPCO assay reaction progress was monitored at 340 nm using Δ ɛ340 = 7.03 mM−1·cm−1 (Δ ɛ340 of NADH = 6.22; Δ ɛ340 of 5-hydroxynicotinic acid = 0.81 mM−1·cm−1). Initial rate measurements were made at 25°C, 50 mM sodium phosphate, pH 8.0, using the stopped-flow spectrophotometer (Hi-Tech Scientific SF-61), similar to the method described in ref. 9. One reaction syringe of the stopped-flow spectrophotometer contained an anaerobic solution of MHPCO (0.7 μM). The other reaction syringe contained appropriate concentrations of NADH, 5-hydroxynicotinic acid, and oxygen. The concentration of oxygen in the substrate syringe was achieved by equilibration of the solution with standardized oxygen/nitrogen mixtures. A minimum of three or four estimations were performed at each concentration of substrate.
RESULTS
N-Terminal and Internal Sequence of MHPCO and Synthesis of DNA Probe.
The N-terminal sequence (residues 1–30) of MHPCO was determined to be ANVNKTPGKARRAEVAGGGFAGLTAAIALK. The sequence of 14 residues of the internal fragment obtained by cyanogen bromide cleavage was determined to be HNKSVLKETFNGLP. From this sequence information, two degeneracy primers were designed to use in PCR to amplify the DNA fragment flanking both amino acid sequences. The first primer, designed to account for residues 2–10 (NVNKTPGKA) of the N-terminal sequence, had the following 5′ to 3′ sequence: (T/C)GTIAA(T/C)AA(A/G) ACICCIGG(A/T/G/C)AA(A/G) GC. The second primer (antisense strand), designed to account for sequence HNKSVLKE of the internal sequence, had the following 5′ to 3′ sequence: TC(T/C)TTIA(G/A)IACI(G/C)(T/A)(T/C)TT(A/G) TT(A/G)TG.
PCR procedures using both primers and the reaction conditions as described in Materials and Methods resulted in a DNA product of 270 nucleotides. This PCR product was purified from agarose gel and ligated to a pGEM-T vector. E. coli JM109 was used as a host for transformation of this PCR product-inserted plasmid. Sequencing showed that the PCR product had a nucleotide sequence consistent with the N-terminal 30 amino acid at one end and the internal sequence of HNKSVLKE at the other end, confirming that this PCR product is the N-terminal part of the MHPCO gene. We then used this PCR product as a template for generating the probe for library screening as described in Materials and Methods.
Library Screening and DNA Mapping.
The partial P-MA1 DNA library constructed as described in Materials and Methods was screened with the 270-nt probe by the colony hybridization procedure as described in ref. 7. One colony of the total of 150 colonies showed a strong positive signal with the probe, implying that that colony contained the insert that has the MHPCO gene.
The plasmid (pBluMH) from the positive colonies was purified and mapped to locate the position of the probe in the insert. The plasmid was digested separately with EagI and BssHII, restriction enzymes that each would cut the probe sequence at only one site (Fig. 1). The plasmid also was double-digested with EagI + PstI, and BssHII + PstI. The digested plasmid then was run on 0.8% agarose gel and Southern-hybridized with the probe. The results are shown in Fig. 2.
Figure 1.
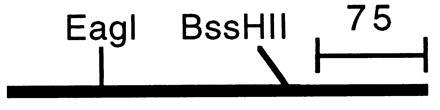
Location of restriction enzyme EagI and BssHII sites in the probe sequence. The EagI site is located 66 nucleotides and BssHII site is located 192 nucleotides from the 5′ end. The mark indicates the length representing 75 nucleotides.
Figure 2.
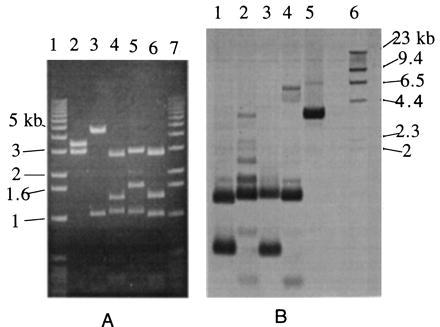
DNA mapping of pBluMH. (A) Molecular mass markers (lanes 1 and 7). pBluMH digested with PstI (lane 2), EagI (lane 3), BssHII (lane 4), EagI + PstI (lane 5), and BssHII + PstI (lane 6). (B) Southern hybridization of the gel in A. pBluMH digested with BssHII + PstI (lane 1), EagI + PstI (lane 2), BssHII (lane 3), EagI (lane 4), PstI (lane 5), and DNA molecular mass markers (lane 6).
In Fig. 2A, lane 3, EagI-digested pBluMH results in 4.5-, 1.1-, 0.4-, and 0.2-kb fragments (the last two fragments are very faint). Because pBluescript vector has a size of 3 kb and has only one restriction site for EagI, which is close to the T3 promoter, the finding of a 4.5-kb fragment implies that the EagI sites responsible for this 4.5-kb fragment are the EagI site of pBluescript and the site located approximately midway between the PstI sites of T7 and T3 (i.e., about 1.5 kb from the PstI cutting site near T7 promoter) (see Fig. 3). From Southern hybridization (Fig. 2B, lane 4) results of pBluMH digested with EagI, we can see that the probe hybridizes to the fragments of 0.2 kb and 1.1 kb, implying that these two fragments contained the probe sequence and were next to each other in the pBluMH map. Because the signal is more intense in the 1.1-kb fragment than in the 0.2-kb fragment, it implied that the 0.2-kb contains the sequence of 5′ side of the probe (66 nucleotides) and the 1.1-kb fragment contains the sequence of 3′ region of the probe (204 nucleotides) as illustrated in Fig. 3A. In Fig. 2A, lane 5, pBluMH that has been double-digested with EagI + PstI resulted in 3.0-, 1.6-, 1.1-, 0.4-, and 0.2-kb fragments and also two light bands of 2.0 and 1.3 kb resulting from incomplete digestion, implying that the 0.4-kb fragment is next to the 1.6-kb fragment in the pBluMH map. From the Southern hybridization (Fig. 2B, lane 2), the incomplete digestion fragment of 0.6 kb also was observed, implying that the 0.2-kb fragment is next to the 0.4-kb fragment in pBluMH map. The results from this EagI + PstI double-digestion and the results obtained from EagI digestion suggested that the probe region was located about 1.3 kb from the T3 promoter side with the orientation shown in Fig. 3A.
Figure 3.
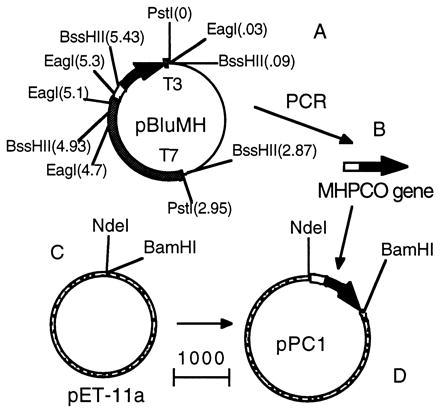
Construction of pPC1 expression plasmid. (A) Plasmid pBluMH. The thin line indicates the plasmid region derived from pBluescript vector, the empty box indicates the probe area. Number in parenthesis indicates the length of DNA (in kb) with reference to the PstI site near T3 promoter as the origin point. MHPCO gene (B) was isolated from pBluMH by PCR and ligated to pET-11a (C), resulting in the expression plasmid pPC1 (D). The mark indicates the length representing 1,000 nucleotides.
In Fig. 2A, lane 4, pBluMH digested with BssHII resulted in 2.9-, 1.15-, 1.4-, 0.5-, 0.3-, and 0.3-kb fragments with those of 1.1 and 0.5 kb containing the probe as shown by the Southern hybridization results (Fig. 2B, lane 3), implying that the probe region is located in these 1.1- and 0.5-kb fragments. Because the signal of 0.5-kb fragment is more intense than that of the 1.1-kb fragment, it implied that the 0.5-kb fragment contains the 5′ side of the probe (192 nucleotides), and the 1.1-kb fragment contains the 3′ side (78 nucleotides). According to the pBluescript map, we would expect to see the DNA fragment next to the pBluescript vector at the T3 promoter site in pBluMH to be about 0.1 kb smaller when double-digested with BssHII + PstI than when digested with BssHII alone. The Southern hybridization results show that the 1.1-kb fragment from BssHII digestion corresponds to a 1-kb fragment in the BssHII + PstI double-digestion. Therefore, the probe region should be located about 1.3 kb from the T3 promoter, as shown in Fig. 3A, in good agreement with the results obtained from the EagI and EagI + PstI digestions.
Nucleotide Sequencing Analysis.
pBluMH is sequenced in both strands. The nucleotide sequence and the derived primary structure of MHPCO are presented in Fig. 4. Residues 2–31 of the deduced protein sequence corresponded to the N-terminal sequence of purified MHPCO. The ORF predicts a 379-aa protein with a calculated molecular mass of 41.7 kDa, consistent with the subunit molecular mass of MHPCO determined by SDS/PAGE. Therefore, the entire MHPCO gene was located in pBluMH as illustrated in Fig. 3A.
Figure 4.

Nucleotide sequence of MHPCO gene, including upstream and downstream flanking regions and the predicted MHPCO sequence.
MHPCO Expression.
The MHPCO gene was isolated from pBluMH by using PCR with proofreading DNA polymerase (Vent DNA polymerase). Two primers with the following 5′ to 3′ sequence: AGGATAGCATATGGCCAATGTAAA (sense strand) and CTTCTGCGGATCCAATTGATCTGT (antisense strand) were used to amplify the MHPCO gene, and at the same time introduce the restriction sites for NdeI and BamHI at the beginning and the end of the gene. The MHPCO gene from the PCR reaction was isolated from agarose gel and digested with NdeI and BamHI.
The expression plasmid pPC1 was constructed by digesting the expression vector pET-11a with NdeI and BamHI and ligating with NdeI and BamHI double-digested MHPCO gene. The plasmid pPC1 was transformed into JM109 for amplification. Plasmid DNA was isolated and sequenced, showing that pPC1 contained the correct MHPCO gene sequence without any error introduced by PCR. pPC1 then was transformed to the expression host, BL21(DE3). The construction of the expression plasmid is summarized as in Fig. 3.
Protein Purification.
BL21(DE3) that contained pPC1 plasmid was grown in Luria–Bertani media plus ampicillin (50 μg/ml) at 37°C. When OD600 of cells was about 0.8, the culture was cooled to 23°C, and 1 mM isopropyl β-d-thiogalactopyranoside was added to induce protein expression. Cells were allowed to grow at this temperature until OD600 reached about 4.5. The cells were sonicated and centrifuged. The supernatant was assayed and found to have MHPCO activity. BL21(DE3) without the plasmid pPC1 also was grown as a control. The control cells do not contain any MHPCO activity.
MHPCO from the induced E. coli was purified according to the method described in ref. 4. About 50 mg of pure MHPCO was obtained from 10 L of cell culture (43 g of cell paste). The recombinant MHPCO shows the same visible spectrum as does P-MA1 MHPCO, with wavelength maxima of 382, 454 nm. Both enzymes have the same specific activity under the assay conditions.
Stoichiometry of FAD/Subunit.
The amount of FAD in cloned and P-MA1 MHPCO solution was quantified by using the determined ɛ454 of 13.11 mM−1·cm−1 (1). The amount of protein was quantified by both the bicinchoninic acid (BCA) method (10) and the Coomassie dye binding (Bradford) method (11). The enzyme used in this procedure was in buffer without DTT to eliminate interference of DTT with the BCA method. BSA was used as a standard to quantify the amount of protein. Because protein assays are known to have protein-to-protein variability, we also used p-hydroxybenzoate hydroxylase (PHBH) as another standard. The stoichiometry of FAD/subunit was calculated in both cloned and P-MA1 MHPCO. The results are shown in Table 1.
Table 1.
The amount of FAD per subunit
| Type of MHPCO | Method used
|
||
|---|---|---|---|
| BCA (BSA as a standard) | BCA (PHBH as a standard) | Bradford (BSA as a standard) | |
| Cloned | 1.0 ± 0.1 | 0.94 ± 0.06 | 0.93 ± 0.04 |
| P-MA1 | 1.03 ± 0.03 | — | 1.03 ± 0.03 |
The results in Table 1 show that both cloned and P-MA1 MHPCO have 1 FAD/subunit. Because MHPCO is a tetrameric protein (1, 6), the enzyme has four FAD/tetramers. This result differs from that of Sparrow et al. (1), who reported that MHPCO has two FAD/tetramers. Because only a single gene product of 42 kDa is encoded, it can be concluded that the enzyme has an α4 structure, not an α2β2 structure as suggested by Kishore et al. (6).
Table 1 shows that BSA can be used as a protein standard for protein assays for both MHPCO and PHBH, because the protein quantitation using BSA as a standard gives the same result as when PHBH is used as a standard.
Steady-State Kinetics.
A general equation that describes the steady-state kinetics of MHPCO is shown in Eq. 2 (4, 5, 12, 13).
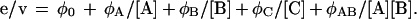 |
2 |
The steady-state parameters, kcat and Km values for 5-hydroxynicotinic acid, NADH, and O2, were derived from the values of 1/φo, φA/φo, φB/φo, φC/φo, respectively. The values of φo, φA, φB, and φC were evaluated according to the graphical method described in ref. 12. The kinetic constants Km of cloned and P-MA1 MHPCO are summarized in Table 2. Km values of cloned and P-MA1 are all similar within the experimental uncertainty, implying that the recombinant MHPCO has the same properties as the native MHPCO from P-MA1. Cloned MHPCO has kcat value of 552 min−1. P-MA1 MHPCO is assumed to have the same kcat value as in cloned MHPCO, because both enzymes have the same specific activity under the assay conditions (4).
Table 2.
Km values, μM
| Type of MHPCO | 5HN | NADH | O2 |
|---|---|---|---|
| Cloned | 65 ± 5 | 180 ± 22 | 148 ± 33 |
| P-MA1 | 68 ± 16 | 205 ± 20 | 126 ± 27 |
5HN, 5-hydroxynicotinic acid.
Homology with Other Hydroxylases.
The amino acid sequence comparison between MHPCO and other flavoprotein hydroxylases [PHBH (14, 15), phenol hydroxylase (16), salicylate hydroxylase (17), 2–4-dichlorophenol hydroxylase (18), pentachlorophenol hydroxylase (19), and 4-aminobenzoate hydroxylase (20)] shows that three amino acid sequence regions are very conserved among these hydroxylases (Fig. 5). The first region (region A; residues 18–34 in MHPCO) is a consensus sequence associated with an ADP binding site. This ADP binding fingerprint forms a characteristic secondary structure, called a βαβ-fold or Rossmann fold (21, 22). In PHBH, this region is a binding site for the ADP portion of FAD (23).
Figure 5.

Alignment of MHPCO with other aromatic flavoprotein hydroxylases. Black areas indicate amino acid residues conserved in all six hydroxylases, and the gray boxes indicate amino acid residues conserved in two or more hydroxylases. (A) Conserved region A: residues 18–34 in MHPCO or 9–21 in PHBH. (B) Conserved region B: residues 154–157 in MHPCO, residues 157–160 in PHBH. (C) Conserved region C: residues 281–295 in MHPCO or residues 279–293 in PHBH. The underlined sequence is the conserved region C1, and the nonunderlined sequence is the conserved region C2.
The second region (region B; residues 154–160 in MHPCO) is very conserved among the aromatic flavoprotein hydroxylases. The third conserved region (region C; residues 281–295 in MHPCO) contains the first part of a sequence that was reported as a fingerprint for the binding site of the ribityl moiety of FAD (24) (region C1; the underlined sequence). However, the second part of the third conserved region (region C2) has not previously been included as a fingerprint for an FAD binding region.
When the SwissProt and Genpept protein databases were scanned using the conserved region B and C with space between 10–300 residues, only aromatic flavoprotein hydroxylases and putative FAD-binding proteins were extracted (Table 3). We examined the derived sequences of these unknown proteins and found that two of them have all three conserved regions A-C as in aromatic flavoprotein hydroxylases. The protein with accession code U29897 lacking N-terminal sequence data contains the conserved regions B and C. It is possible that the region A is in the lacking N-terminal sequence. It is possible that these hypothetical proteins are also aromatic flavoprotein hydroxylases.
Table 3.
Proteins extracted from SwissProt and Genpept Database using sequence G-[AC]-D-G-X-X-[SG]-X(10-300)-G-X(5)-D-A-X-H
| Proteins |
|---|
| Known |
| PHBH |
| Phenol hydroxylase |
| Salicylate hydroxylase |
| 2,4-Dichlorophenol hydroxylase |
| Pentachlorophenol hydroxylase |
| 4-Aminobenzoate hydroxylase |
| Hypothetical |
| 52.4 kDa protein in ATP-ROX3, intergenic region (GenBank |
| accession code P38169) |
| Partial sequence of FAD binding gene (GenBank |
| accession code U29897) |
| Putative oxygenase from biosynthesis and oxygenation |
| pathway of urdamycin A (GenBank accession code |
| X87093) |
Database searching used the PatternFind server from the Bioinformatics Group at the Swiss Institute for Experimental Cancer Research (http://ulrec3.unil.ch:80/software).
DISCUSSION
This study reports the gene cloning, sequence determination, and enzyme expression of MHPCO, a flavoprotein monooxygenase. Characterization of the cloned enzyme clearly shows that all the properties of the enzyme so far examined are the same as those of the native enzyme from P-MA1. These properties include the subunit molecular mass, specific activities with MHPC and 5-hydroxynicotinic acid, Km for each substrate, and stoichiometry of FAD/subunit. Because MHPCO is a tetrameric protein and the cloned MHPCO was expressed as a single polypeptide of 379 amino acids, it clearly demonstrates that MHPCO is a homotetrameric protein with one flavin bound to each subunit.
Sequence comparison of MHPCO with PHBH shows that the two enzymes have about 26% identity. The identity between PHBH with individual enzymes in the flavoprotein hydroxylase class [phenol hydroxylase (16), salicylate hydroxylase (17), 2,4-dichlorophenol hydroxylase (18), pentachlorophenol hydroxylase (19), and 4-aminobenzoate hydroxylase (20)] is 23–33%. With this level of identity, it may be assumed that all of the flavoprotein hydroxylases have a similar core structure (25). Currently, the only enzyme in the class where the three-dimensional structure is known is PHBH (26).
From all the proteins listed in the SwissProt and Genpept protein databases, the only proteins that have all of the three conserved regions shown in Fig. 5 are the flavoprotein hydroxylases themselves and the hypothetical proteins where only DNA sequences were identified (Table 3). These hypothetical proteins have lengths of 460, 495, and >287 amino acid residues, and thus are in the range of the known aromatic flavoprotein hydroxylases, which all have molecular mass of 40–55 kDa per subunit. It is likely that these hypothetical proteins are also aromatic flavoprotein hydroxylases.
These highly conserved amino acid sequences in aromatic flavoprotein hydroxylases may have some importance in the catalytic functions of the enzymes. Region A, residues 18–34 in MHPCO or 9–21 in PHBH, is clearly involved in the binding of the ADP moiety of FAD (23–24), as shown by its sequence and location in PHBH. Region C1, residues 281–288 in MHPCO or residues 279–286 in PHBH, was proposed to be a fingerprint for the binding of FAD (24). However, region C2 (residues 289–295 in MHPCO or residues 287–293 in PHBH), which is not included in the fingerprint proposed by Eggink et al. (24), is very conserved among the aromatic flavoprotein hydroxylases, but not in other flavoproteins. In the PHBH structure, the location of this sequence is in the loop close to the protein surface and also close to the region B (residues 154–157 in MHPCO, residues 157–160 in PHBH). It can be envisaged that these two regions may have importance in enzyme function.
One of the common properties among the aromatic flavoprotein hydroxylases is that the enzymes require three substrates: aromatic substrate, NAD(P)H, and O2. It is known that the hydroxylation reaction takes place in the catalytic pocket, which is the binding site for the aromatic substrate, the isoalloxazine ring of FAD, O2, and the nicotinamide ring of NADPH. However, it is not known where the AMP moiety of the pyridine nucleotide binds to the enzyme. Because there is space between the catalytic pocket and the loops of regions B and C, it is possible that the area around these two loops is the recognition site for the AMP moiety of pyridine nucleotide. To test this possibility, we modeled the NADPH molecule into PHBH in a way that the nicotinamide ring was almost parallel to the isoalloxazine ring of the flavin so that the hydride equivalent can be transferred from the pro-R side of the nicotinamide ring to the re-side of the flavin (27), and also similar to the arrangement described in ref. 28. With this configuration, it is possible to have the AMP moiety of NADPH docking around the loop of 287–289, region C2 (Fig. 6), which is near to region B.
Figure 6.
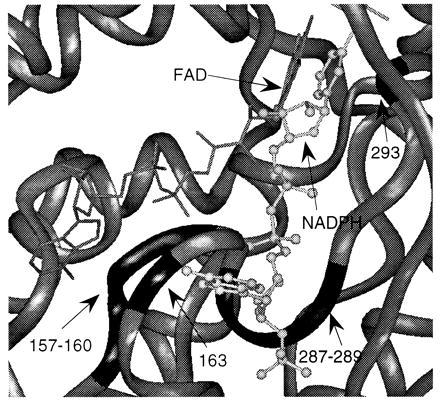
The postulated model of binding of NADPH molecule in PHBH. FAD is indicated as the molecule on the left (line drawing), and NADPH is indicated as the molecule on the right (ball and stick drawing). The PHBH molecule is represented as gray ribbon. Residues 157–160 and 163 are indicated as the black loop on the left. Residues 287–289 and 293 are indicated as the black loop on the right.
The information obtained so far cannot determine if the arrangement of Fig. 6 is correct. Wierenga et al. (28) proposed that the pyrophosphate group of NADPH can form salt bridges with R166, R269, or H162 in PHBH. R166 and H162 are located near the conserved region B. Suzuki et al. (29) reported that K165 in salicylate hydroxylase (corresponding to H162 in PHBH) is important for the binding of NADH as shown by chemical modification studies. This information is consistent with the mode of binding postulated in Fig. 6. However, Seibold et al. (30) suggest that helix H2 (residues 35–42) in PHBH is involved in the binding of pyridine nucleotide. This is based on the results of chemical modification studies that implicated Y38 in the binding of pyridine nucleotide and the result of sequence comparison between PHBH from Pseudomonas fluorescens and PHBH from Pseudomonas sp. CBS3 (30). PHBH from P. fluorescens uses NADPH as a substrate while PHBH from Pseudomonas sp. CBS3 uses NADH (30). The sequence difference between the two enzymes around helix H2 leads to the postulation that helix H2 is involved in determining the pyridine nucleotide specificity. It should be noted, however, that there are other regions besides helix H2 that differ in sequence. Clearly, this latter proposal is inconsistent with the model of NADPH binding of Fig. 6. Therefore, we cannot rule out the possibility that the second and third conserved regions are not involved in the binding of pyridine nucleotide, but rather are involved in some other role such as structural stabilization.
In conclusion, MHPCO has been cloned, sequenced, and expressed in E. coli. We have shown that the enzyme is a homotetrameric protein with one FAD per subunit. The sequence comparison of MHPCO and aromatic flavoprotein hydroxylases show several conserved sequence regions.
Acknowledgments
We thank Mr. Witoon Tirasophon and Dr. Betty Jo Brown for valuable suggestions during the gene cloning process. We thank Ms. Mariliz Ortiz-Maldonado for providing PHBH used for a standard on protein determination, and Dr. Brian Guenther for suggestions about protein database searching. This work was supported by the U.S. Public Health Service, Grants GM 20877 (D.P.B.) and GM 11106 (V.M.), and the Development and Promotion of Science and Technology Talent Project, Thailand (P.C.).
ABBREVIATIONS
- MHPCO
2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase
- P-MA1
Pseudomonas sp. MA-1
- Dig-11-dUTP
digoxigenin-11-dUTP
- PHBH
p-hydroxybenzoate hydroxylase
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF001965).
References
- 1.Sparrow L G, Ho P P K, Sundaram T K, Zach D, Nyns E J, Snell E E. J Biol Chem. 1969;244:2590–2600. [PubMed] [Google Scholar]
- 2.Burg R W, Rodwell V W, Snell E E. J Biol Chem. 1960;235:1164–1169. [PubMed] [Google Scholar]
- 3.Sundaram T K, Snell E E. J Biol Chem. 1969;244:2577–2585. [PubMed] [Google Scholar]
- 4.Chaiyen P, Brissette P, Ballou D P, Massey V. Biochemistry. 1997;36:2612–2621. doi: 10.1021/bi962325r. [DOI] [PubMed] [Google Scholar]
- 5.Chaiyen, P., Brissette, P., Ballou, D. P. & Massey, V. (1997) Biochemistry, in press. [DOI] [PubMed]
- 6.Kishore G M, Snell E E. J Biol Chem. 1981;256:4234–4240. [PubMed] [Google Scholar]
- 7.Boehringer Mannheim Co. (1995) Genius System User’s Guide for Membrane Hybridization, Version 3.0.
- 8.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 9.Husain M, Massey V. J Biol Chem. 1979;254:6657–6666. [PubMed] [Google Scholar]
- 10.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 11.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 12.Dalziel K. Biochem J. 1969;114:547–556. doi: 10.1042/bj1140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cleland W W. Biochemistry. 1975;14:3220–3224. doi: 10.1021/bi00685a029. [DOI] [PubMed] [Google Scholar]
- 14.Weijer W J, Hofsteenge J, Beintema J J, Wierenga R K, Drenth J. Eur J Biochem. 1983;133:109–118. doi: 10.1111/j.1432-1033.1983.tb07435.x. [DOI] [PubMed] [Google Scholar]
- 15.Entsch B, Nan Y, Weaich K, Scott K F. Gene. 1988;71:279–291. doi: 10.1016/0378-1119(88)90044-3. [DOI] [PubMed] [Google Scholar]
- 16.Nurk A, Kasak L, Kivisaar M. Gene. 1991;102:13–18. doi: 10.1016/0378-1119(91)90531-f. [DOI] [PubMed] [Google Scholar]
- 17.You I-S, Ghosal D, Gunsalus I C. Biochemistry. 1991;30:1635–1641. doi: 10.1021/bi00220a028. [DOI] [PubMed] [Google Scholar]
- 18.Perkin E J, Gordon M P, Caceres O, Lurquin P F. J Bacteriol. 1990;172:2351–2359. doi: 10.1128/jb.172.5.2351-2359.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orser C S, Lange C C, Xun L, Zahrt T C, Schneider B J. J Bacteriol. 1993;175:411–416. doi: 10.1128/jb.175.2.411-416.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuji H, Oka T, Kimoto M, Hong Y M, Natori Y, Ogawa T. Biochim Biophys Acta. 1996;1309:31–36. doi: 10.1016/s0167-4781(96)00131-5. [DOI] [PubMed] [Google Scholar]
- 21.Rossmann M G, Moras D, Olsen K W. Nature (London) 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 22.Wierenga R K, Terpstra P, Hol W G J. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]
- 23.Wierenga R K, Drenth J, Schulz G E. J Mol Biol. 1983;167:725–739. doi: 10.1016/s0022-2836(83)80106-5. [DOI] [PubMed] [Google Scholar]
- 24.Eggink G, Engel H, Vriend G, Terpstra P, Witholt B. J Mol Biol. 1990;212:135–142. doi: 10.1016/0022-2836(90)90310-I. [DOI] [PubMed] [Google Scholar]
- 25.Sander C, Schneider R. Proteins Struct Funct Genet. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 26.Wierenga R K, de Jong R J, Kalk K H, Hol W G J, Drenth J. J Mol Biol. 1979;131:55–73. doi: 10.1016/0022-2836(79)90301-2. [DOI] [PubMed] [Google Scholar]
- 27.Manstein D J, Pai E F, Schopfer L M, Massey V. Biochemistry. 1986;25:6807–6816. doi: 10.1021/bi00370a012. [DOI] [PubMed] [Google Scholar]
- 28.Wierenga R K, Kalk K H, van der Lann J M, Drenth J, Hofsteenge W J W, Jekel P A, Beintema J J, Muller F, van Berkel W J H. In: Flavins and Flavoproteins. Massey V, Williams C H, editors. New York: Elsevier/North–Holland; 1982. pp. 11–18. [Google Scholar]
- 29.Suzuki K, Mizuguchi M, Gomi T, Itagaki E. J Biochem. 1995;117:579–585. doi: 10.1093/oxfordjournals.jbchem.a124747. [DOI] [PubMed] [Google Scholar]
- 30.Seibold B, Matthes M, Eppink M H M, Lingens F, Van Berkel W J H, Muller R. Eur J Biochem. 1996;239:469–478. doi: 10.1111/j.1432-1033.1996.0469u.x. [DOI] [PubMed] [Google Scholar]