

Abstract
The signal transducer and activator of transcription, STAT5b, has been implicated in signal transduction pathways for a number of cytokines and growth factors, including growth hormone (GH). Pulsatile but not continuous GH exposure activates liver STAT5b by tyrosine phosphorylation, leading to dimerization, nuclear translocation, and transcriptional activation of the STAT, which is proposed to play a key role in regulating the sexual dimorphism of liver gene expression induced by pulsatile plasma GH. We have evaluated the importance of STAT5b for the physiological effects of GH pulses using a mouse gene knockout model. STAT5b gene disruption led to a major loss of multiple, sexually differentiated responses associated with the sexually dimorphic pattern of pituitary GH secretion. Male-characteristic body growth rates and male-specific liver gene expression were decreased to wild-type female levels in STAT5b−/− males, while female-predominant liver gene products were increased to a level intermediate between wild-type male and female levels. Although these responses are similar to those observed in GH-deficient Little mice, STAT5b−/− mice are not GH-deficient, suggesting that they may be GH pulse-resistant. Indeed, the dwarfism, elevated plasma GH, low plasma insulin-like growth factor I, and development of obesity seen in STAT5b−/− mice are all characteristics of Laron-type dwarfism, a human GH-resistance disease generally associated with a defective GH receptor. The requirement of STAT5b to maintain sexual dimorphism of body growth rates and liver gene expression suggests that STAT5b may be the major, if not the sole, STAT protein that mediates the sexually dimorphic effects of GH pulses in liver and perhaps other target tissues. STAT5b thus has unique physiological functions for which, surprisingly, the highly homologous STAT5a is unable to substitute.
Growth hormone (GH) has diverse physiological actions and regulates the tissue-specific expression of numerous genes involved in growth, metabolism, and differentiation (1, 2). Several of the effects of GH on somatic growth and gene expression are sex-dependent and are regulated by pituitary GH secretory patterns, which are sexually differentiated. The resultant sex differences in plasma GH profiles are particularly striking in rodents (for example, intermittent GH pulses in male rats vs. continuous plasma GH in female rats) and are the major determinant of sex differences in body growth rates and the sexually dimorphic expression in liver of numerous genes, including the major urinary proteins (MUP) (3), prolactin receptors (3, 4), and several cytochrome P450 (CYP) enzymes (5–7). Altering the temporal pattern of plasma GH by GH infusion or by hypophysectomy with GH replacement causes changes in body growth rate and in the patterns of liver gene expression that directly reflect the changes in plasma GH profiles (3, 5, 8). In mice, GH secretion is pulsatile in both males and females; however, the interval between pulses is longer in males (9), and this period of low or no plasma GH is thought to be an important determinant of the male-specific pattern of liver gene expression (8).
Recently, STAT5, a latent cytoplasmic transcription factor, has been shown to be activated in rat liver by the male pulsatile pattern of GH stimulation but not by the more continuous GH pattern that occurs in females (10). This activation, which involves GH pulse-induced STAT5 tyrosine phosphorylation followed by serine or threonine phosphorylation (11, 12), induces nuclear translocation of STAT5 and has been proposed to be a key event in mediating the effects of GH pulses on male-specific liver gene expression (10). Most of this STAT5 appears to be STAT5b (11, 13), which belongs to a gene family, the signal transducers and activators of transcription, whose members are resident in the cytosol but become activated to nuclear DNA-binding proteins following the binding of cytokines and growth factors to their cognate cell-surface receptors. STAT5 was originally identified as a prolactin-inducible transcription factor in lactating mammary gland (14–16). Subsequently, two highly homologous STAT5 genes (17, 18), STAT5a and STAT5b, were found to be expressed in a wide range of tissues (17–20). These STATs can be activated by many growth factors and cytokines, including prolactin (21), GH (10, 22, 23), interleukins (20, 24), epidermal growth factor (25, 26), and erythropoietin (27). The high (≈96%) sequence similarity between STAT5a and STAT5b suggests that there may be redundancy in their signaling pathways. However, subtle differences in the tissue distribution of STAT5a and STAT5b mRNAs suggest that they may have distinct functions (19). Targeted disruption of STAT genes, either individually or in combinations, is now allowing elucidation of the specific functions of each STAT (28–34). We have targeted the STAT5b gene to define its function and, in particular, to assess its proposed role in the GH pulse-regulated sexual dimorphism of liver gene expression.
EXPERIMENTAL PROCEDURES
Target Vector and Gene Disruption.
Clones containing the murine STAT5b gene were isolated from a 129 mouse genomic DNA library (Stratagene) and used to construct a positive/negative selection vector for targeting the gene. The vector was constructed by inserting a neomycin resistance (neor) cassette at the BamHI site interrupting the codon for amino acid 181. It contained 0.9 kb of 5′ DNA, 12 kb of 3′ DNA, and the herpes simplex virus thymidine kinase gene to allow negative selection (Fig. 1A). Embryonic stem cells were electroporated and selected, and targeted clones were injected into blastocysts to obtain chimaeric mice that transmitted the disrupted locus through the germ line. A 1-kb ApaI/EcoRV fragment was used for Southern blot analysis. BglII restriction fragments were 8 kb for the wild-type allele and 4.7 kb for the targeted allele (Fig. 1B). Electroporation of embryonic stem cells, cell selection, and cell and mouse screening have been described (35). Experiments described in this report were carried out using littermates obtained from 129 × BALB/c outcrossed mice.
Figure 1.
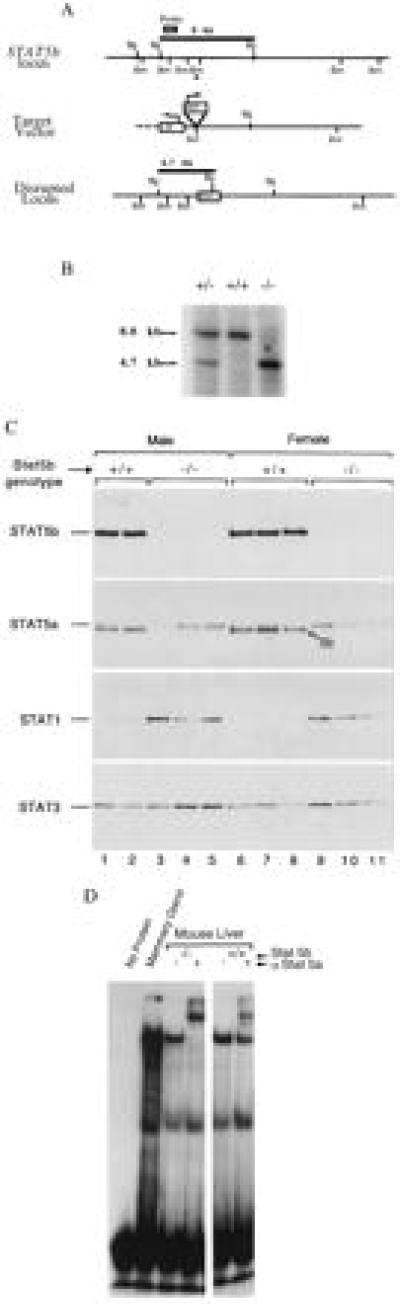
Targeted disruption of the murine STAT5b gene. (A) Targeting vector and recombination at STAT5b locus, with restriction sites for BamHI (Bm) and BglII (Bg). Two contiguous STAT5b gene BamHI fragments (0.9 kb, 12 kb) were used to construct the positive/negative selection vector. The neor cassette was inserted at a BamHI site (∗), and the HSV tk gene was used for negative selection. The probe used for Southern blot analyses (1-kb ApaI/EcoRV fragment) and the expected BglII restriction fragments are indicated. (B) Southern blot of wild-type (+/+), heterozygous (+/−), and STAT5b−/− (−/−) DNA using probe shown in A. (C) Western blot probed sequentially for the indicated STAT proteins in liver cytosols of STAT5b−/− and wild-type mice. Small arrow on second panel points to STAT5b, which apparently cross-reacts with anti-STAT5a at the extended exposure required to detect STAT5a in these liver samples. No cross-reactivity between STATs 1, 3, and 5 is detected using these antibodies (11). (D) Gel-shift analysis of liver nuclear STAT5 DNA-binding activity from wild-type and STAT5b−/− mice in the absence or presence of a supershifting anti-STAT5a antibody (α STAT5a). Lane 2, lactating goat mammary gland extract, a positive control for activated STAT5.
Western Blot and Gel Shift Analyses.
Liver cytosolic proteins prepared from 8- to 9-week-old wild-type and STAT5b−/− mice by ultracentrifugation were analyzed on Western blots probed sequentially with anti-STAT antibodies (11). Western blots of mouse liver microsomes (25 μg per lane) were probed with anti-rat CYP3A monoclonal antibody 2–3-2 (36) or anti-mouse CYP2D [anti-C-P45016α, provided by M. Negishi, National Institute on Environmental Health Sciences (NIEHS), Research Triangle Park, NC (5)] as described (37). Gel shift assays used mouse liver nuclear extracts (38) and a rat β-casein gene STAT5 binding site probe (10) in the absence or presence of a supershifting anti-STAT5a antibody (sc-1081x, Santa Cruz Biotechnology).
Northern Blot Analyses.
Total liver RNA (5 μg) was run on a 0.8% agarose/formaldehyde gel, transferred to nylon, and sequentially hybridized and stripped with cDNA probes for CYP15α/2A4 (M. Negishi, NIEHS), the short form of the prolactin receptor (P. Kelly, Institut National de la Santé et de la Recherche Médicale, Paris), and an oligonucleotide for 28S rRNA. Relative RNA band intensities were measured using the image 1.57 program (NIH). STAT5b mRNA (5.6 kb) and STAT5a mRNA (4.2 kb) (19) in wild-type and STAT5b−/− mouse tissues were analyzed by Northern blotting (10 μg RNA) using a 1.1-kb 5′-end STAT5b cDNA isolated from a mouse 129 brain cDNA library (Stratagene).
Other Methods.
Mice were weighed regularly from birth to 42 days of age (n = 194 individual mice), and the body weight means were adjusted for small variations between litters. genstat 5 software was used for statistical analysis. Plasma IGF-I levels (39) and plasma GH levels at sacrifice (rat GH RIA kit, Amersham) were measured by standard methods. Serum progesterone was measured by RIA (Diagnostic Products, Los Angeles) using tail vein blood samples collected every 3 days of pregnancy. To analyze MUP protein levels, morning and afternoon urines were combined and centrifuged (1,000 g × 3 min). The supernatant (0.5 μl) was analyzed on a 12% SDS–polyacrylamide gel stained with Coomassie blue. Testosterone hydroxylase activity was measured in mouse liver microsomes prepared by differential centrifugation (37).
RESULTS
STAT5b Gene Disruption.
Matings of STAT5b+/− mice (129 × BALB/c outcross) produced STAT5b−/− offspring that were close to the expected Mendelian frequency. Northern blot analyses failed to detect STAT5b mRNA in liver, spleen, mammary gland, thymus, kidney, or skeletal muscle of STAT5b−/− mice, and Western blotting of protein extracts of these same tissues detected STAT5b protein in wild-type but not STAT5b−/− mice. By contrast, STAT5a mRNA (data not shown) and protein were present in both wild-type and STAT5b−/− mice at levels similar to those in wild-type mice (Fig. 1C; data not shown). This STAT5a was functionally active in a DNA-binding gel-shift assay using a β-casein gene STAT5 binding-site probe (Fig. 1D). Supershift analysis using anti-STAT5a antibodies revealed a complete supershifting of the STAT5–DNA complex in STAT5b−/− liver nuclear extracts (lane 4) but an incomplete supershift in STAT5b+/+ samples. Presumably, the residual, unshifted band represents STAT5b homodimers bound to the DNA probe. Liver STAT3 protein levels were unchanged or slightly increased by the STAT5b gene knockout, whereas liver STAT1 protein levels were increased more substantially (Fig. 1C).
Decreased Growth of STAT5b−/− Males.
Examination of two independent lines of STAT5b−/− mice revealed striking decreases in body weight gain for STAT5b−/− males that were not seen in STAT5b−/− females (Fig. 2). STAT5b−/− males grew at the lower rate, which is characteristic of wild-type female mice, thereby abolishing the striking sexual dimorphism in body growth rate normally evident from 3 weeks of age. This sex difference in body growth is determined by the sex-specific pattern of pituitary GH secretion (8, 40, 41), which first emerges at this stage in development (1). At 4 to 5 weeks of age, STAT5b−/− males were 27% lighter than wild-type males, and this difference persisted beyond puberty (Fig. 2). X-rays and alizarin red staining showed that the skeletons of STAT5b−/− males were smaller than wild-type males but otherwise appeared normal (data not shown). There were no obvious histological defects or weight differences in abdominal or thoracic organs, although the livers in some STAT5b−/− mice were pale and enlarged. These phenotypic effects of STAT5b disruption, as well as the effects on reproduction (see below), were observed for two independent lines of STAT5b−/− mice.
Figure 2.
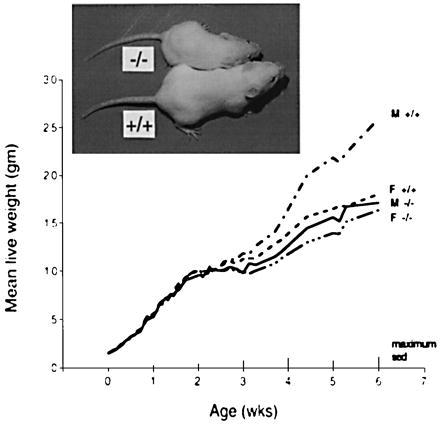
Growth curves of wild-type (+/+) and STAT5b−/− (−/−) mice, and 42-day-old wild-type and STAT5b−/− males from the same litter (Insert). Body weights of wild-type males were significantly different from STAT5b−/− males (M) and females (F) (P ≤ 0.001). No significant differences were seen between STAT5b−/− males, wild-type females, and STAT5b−/− females. Standard errors of differences (SED) are indicated on the x axis.
Analysis of circulating hormone levels revealed normal to elevated plasma GH levels in STAT5b−/− males at the time of sacrifice (46 ± 4 ng/ml for wild-type vs. 89 ± 33 ng/ml for STAT5b−/− males, reflecting elevated GH in three of the six knockout males). No significant plasma GH differences were seen in females [51 ± 15 (wild type) vs. 54 ± 13 ng/ml (STAT5b−/− females)]. Plasma IGF-I concentrations were significantly lower in STAT5b−/− mice (197 ± 13 ng/ml) than in STAT5b+/+ mice (282 ± 15 ng/ml) (P < 0.001), with no sex differences apparent. GH rapidly stimulates the transcriptional activation of IGF-I in liver, but the role of STAT proteins in this response is unknown.
Loss of GH Pulse-Regulated, Sexually Dimorphic Liver Gene Expression.
To determine whether STAT5b is required for expression of sex-dependent differences in liver gene expression, we compared the amounts of MUP in the urine of STAT5b−/− and wild-type mice. MUP is a family of α2-microglobulin-related liver secretory proteins that comprise a major protein component of mouse urine. MUP protein is excreted in male mouse urine at levels that are ≈3-fold higher than in female urine (3). Induction of MUP by pulsatile plasma GH, a characteristic of males, has been demonstrated using GH-deficient Little mice (3). Little mice have very low levels of MUP, and GH replacement by intermittent injection (to mimic the male pulsatile GH pattern) specifically increases MUP to normal male levels (3). We found MUP levels in urine from STAT5b−/− males similar to the amounts in female urine and considerably less than in wild-type males (Fig. 3). A simple interpretation of our data, consistent with published findings on the pulsatile GH activation of liver STAT5 (10), is that STAT5b is required for the male-specific pulsatile GH induction of MUP and, consequently, STAT5b−/− males are unable to respond to the male pulsatile GH secretory pattern. We also noted lower levels of MUP when STAT5b−/− females were compared with wild-type females (Fig. 3), indicating that there are also some STAT5b-dependent effects on MUP gene expression in females. This latter finding may relate to the multihormonal control reported for MUP protein expression (42), and is analogous to the lower MUP levels that female Little mice have compared with wild-type females (3).
Figure 3.
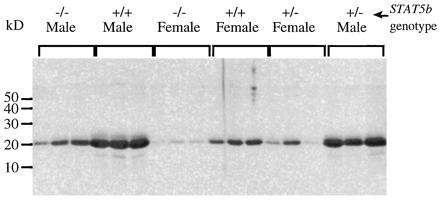
SDS gel analysis of MUP in urine of wild-type (+/+), STAT5b+/− (+/−), and STAT5b−/− (−/−) mice stained with Coomassie blue. Densitometric analysis revealed statistically significant differences between the wild type and knockout for both male genotypes (P < 0.005 for +/+ vs. +/− and +/+ vs. −/−) and female genotypes (P < 0.02 for +/+ vs. −/−). Other comparisons within each sex were not significant (P > 0.05; two-tailed Student’s t test).
Liver expression of several CYP enzymes and other liver gene products is sex-specific and regulated by the plasma GH pattern (5–8). P45015α hydroxylase (CYP2A4) and prolactin receptors are both expressed predominantly in female mice, and their expression is known to be primarily regulated by GH (3, 5) with some influences from other hormones such as thyroxine. Northern blots therefore were used to compare mRNA levels of these CYP enzymes, and of prolactin receptors, in the livers of STAT5b−/− and wild-type mice. Although there was variation between animals, the expression of P45015α mRNA was elevated in STAT5b−/− males to a level somewhat lower than that in wild-type females (Fig. 4A). The same trend of increased expression of prolactin receptors was seen in STAT5b−/− males (Fig. 4B). The individual animal variation seen in these samples may be due to the random genetic contribution from the 129 × BALB/c outcrossed background, because these two strains are known to differ in their patterns of expression of these proteins (43). Similarly, STAT5b gene disruption in males increased expression of the female-dominant liver CYP3A proteins and their associated microsomal testosterone 6β-hydroxylase activity to near-female levels (Fig. 5). By contrast, no effect was seen on liver microsomal testosterone 2α-hydroxylase, a sex-independent mouse liver P450 activity. STAT5b−/− males also exhibited a selective loss of the steroid 16α-hydroxylase protein CYP2D9 (C-P45016α, Fig. 5B Lower, band b), which is expressed in a male-specific fashion in mouse liver under the positive influence of plasma GH pulses (5).
Figure 4.
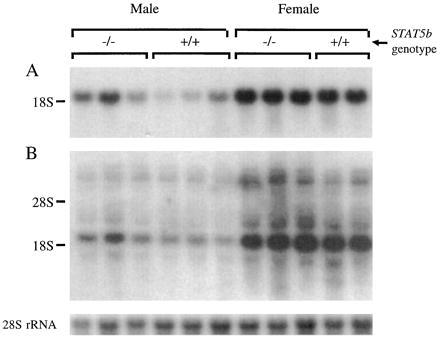
Northern blots showing relative levels of CYP15α/2A4 (A) and prolactin receptor mRNA (B), and 28S rRNA (Bottom). The mean ± SEM relative mRNA levels (normalized using 28S rRNA) for STAT5b−/− males, wild-type males, STAT5b−/− females, and wild-type females were, respectively, 1.04 ± 0.26, 0.30 ± 0.13, 2.13 ± 0.11, and 1.92 ± 0.15 for CYP15α, and 1.10 ± 0.20, 0.42 ± 0.10, 2.47 ± 0.17, and 2.36 ± 0.08 for the 2.5-kb short form of prolactin receptor.
Figure 5.
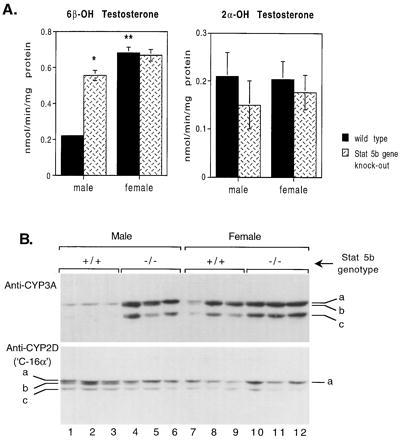
CYP-catalyzed testosterone hydroxylase enzyme activity (A) and CYP protein levels (B) in liver microsomes from STAT5b wild-type and knockout mice. (A) Specific activities are mean ± SEM values for n = 5–6 individual mice per group. ∗, significant difference from wild-type at P ≤ 0.05; ∗∗, significant difference from male at P ≤ 0.05 (two-tailed Student’s t test). Error bar is too small to be seen for the first sample in A. (B) Western blot of CYP3A and CYP2D proteins. Shown at Top are three CYP3A-immunoreactive proteins detected using anti-rat CYP3A antibody; bands marked b and c are both female-dominant and are increased in male STAT5b−/− mice. Shown at Bottom are three CYP2D-immunoreactive proteins detected using anti-mouse C-P45016α antibody; band b exhibits the male predominance reported for the C-P45016α (5) and is decreased to female levels in male STAT5b−/− mice.
Other Phenotypes of STAT5b−/− Mice.
In addition to the phenotypic effects of STAT5b gene disruption on body growth and liver gene expression, we observed effects on lactation, reproduction, fat deposition, and hair growth. Preliminary analyses indicated that mammary gland development is impaired in STAT5b−/− females, and although milk protein genes are expressed (Northern and Western blot analyses), there is insufficient milk to feed pups (data not shown). STAT5b−/− females consistently aborted between day 8 and 17 of pregnancy, with no obvious maternal, placental, or fetal defects. Heterozygous females were also affected, with higher numbers of pups dying perinatally than with wild-type females, and these deaths were not associated with the genotype of the pups. Subcutaneous delivery of progesterone (about 4 mg; s.c. implantation on day 5 to 7 of pregnancy of a 5 × 1.5 × 1.5-mm segment of a controlled intravaginal drug release device; CIDR, InterAg, Hamilton, New Zealand) maintained pregnancy in STAT5b−/− females. In the absence of progesterone implants, serum progesterone declined precipitously by day 12 of pregnancy in STAT5b−/− mice, while in STAT5b+/+ mice progesterone remained high until parturition (data not shown).
The effects of STAT5b disruption on body fat deposition were marked, with significantly less adipose tissue in both STAT5b−/− females and males compared with wild-type mice. Weights of sublumbar adipose tissue showed that the effect was greater in females (reduced to 36% of wild-type) than males (69% of wild-type). In addition, although there were low amounts of fat in young adult STAT5b−/− mice, obesity became apparent in some of the mice beginning at about 9 weeks of age, and these individuals had large testicular fat pads and greater amounts of abdominal fat. Numerous cells had no lipid, suggesting that adipocyte differentiation might indeed be impaired (data not shown). GH induces the differentiation of preadipocytes to adipocytes (44) and can activate STAT5 in 3T3-F442A mouse preadipocytes in culture (22), indicating a potential role for STAT5 in adipocyte differentiation.
Examination of the hair of STAT5b−/− mice revealed no structural abnormalities, but the pelage of some animals was sparse. To observe new hair growth, the hair was dyed when the mice were 25 days old. This revealed a delay of at least 2 weeks in the commencement of second-generation hair growth in STAT5b−/− mice. Further study is required to ascertain whether these latter effects of STAT5b gene disruption reflect an insensitivity of target tissues to GH, or whether they result from loss of responsiveness to other cytokines and growth factors that also signal via STAT5b.
DISCUSSION
We have shown that STAT5b gene disruption leads to a major loss of multiple, sexually differentiated responses associated with the sexually dimorphic pattern of pituitary GH secretion. Male-characteristic body growth rates and male-specific liver gene expression are decreased to wild-type female levels in STAT5b−/− males (MUP, CYP2D9), while female-predominant liver gene products are increased in males to a level intermediate between wild-type male and female levels (CYP15α/2A4, CYP6β/3A, and prolactin receptors). These responses are very similar to those observed in GH-deficient Little mice and support our hypothesis (10) that STAT5b is the major, if not the sole, STAT protein that mediates the sexually dimorphic effects of GH on the liver and perhaps other target tissues. The closely related STAT5a [96% sequence identity; (17, 18)] is a minor STAT5 form in rat liver (11, 13). However, the present studies indicate that although STAT5a can be detected in an active form in the liver (Fig. 1D), it alone is insufficient to maintain normal sexually dimorphic GH responses. Liver STAT1 and STAT3 also respond to GH (11, 45, 46); however, neither of these STATs exhibits the striking discrimination between intermittent and continuous GH stimulation seen with STAT5 (11), and the present study suggests that neither is likely to play an important role in the sexually dimorphic effects of GH. Interestingly, STAT1 protein expression was up-regulated in both male and female STAT5b−/− mice in what may be an attempt at a compensatory response to the loss of STAT5b expression; however, the significance of this STAT1 increase and its possible contribution to the observed STAT5b−/− phenotype are unclear.
The phenotype of STAT5b gene disruption in males contrasts with the lack of a major effect on liver gene expression or body growth rates in STAT5b−/− females. A modest decrease in growth rate is seen in STAT5b−/− females, a finding that could relate to the lower circulating IGF-I levels seen in these animals. This finding is in accord with reports on GH-deficient Little mice, where loss of circulating GH has a much less pronounced effect in females than males (5), and further supports the proposal that, in the mouse model, male GH pulses can both positively regulate male-specific liver gene expression and can negatively regulate female-predominant gene expression (5). In contrast to these STAT5b-dependent effects of GH pulses, studies in the rat model demonstrate that continuous GH can positively regulate female-specific liver gene products, such as CYP2C12, by activation of a novel signaling pathway that appears to be independent of STAT factors (47).
The mechanism underlying the loss of sexually dimorphic body growth rates and liver gene expression in STAT5b−/− mice is uncertain but could reflect either of two possibilities. The simplest explanation is that STAT5b is necessary for (and perhaps directly mediates) the physiological effects of male plasma GH pulses on body growth rates and liver gene expression. This possibility is strongly supported by the direct response of liver STAT5b, but not other STAT forms, to the male pattern of plasma GH pulsation (10, 11), which previously has been established as being both necessary and sufficient to achieve male body growth rates (8, 40, 41) and male liver gene expression (5–8). One prediction of this hypothesis is that at least some of the sexually dimorphic liver gene products whose expression is altered in STAT5b−/− males (Figs. 3, 4, 5) will contain functional STAT5b-binding DNA response elements in their genes. Indeed, at least two male-specific, GH pulse-regulated liver CYP genes in other rodent species contain functional STAT5 binding sites in their 5′-flanking DNA (S.-H.P. and D.J.W., unpublished data; ref. 48). An alternative possibility is that the apparent feminization of body growth rates and liver enzyme patterns in STAT5b−/− mice is a secondary response to a general loss of circulating GH or perhaps a loss of the male pattern of pulsatile pituitary GH secretion. Although the phenotype of the STAT5b−/− mouse does have important similarities to that of the GH-deficient Little mouse, as noted above, STAT5b−/− mice are, in fact, not GH-deficient, as demonstrated by the presence of circulating GH at sacrifice in both males and females at levels at least comparable to those of corresponding wild-type mice. It is possible, however, that the temporal pattern of pituitary GH release is perturbed in STAT5b−/− mice in a manner such that female growth rates and a liver enzyme phenotype result (compare apparent elevation in plasma GH levels in three of six individual STAT5b−/− males). Further experimentation, including a direct analysis of the pulsatility of circulating GH profiles in STAT5b−/− mice, is required to address this point. It should be noted, however, that in Laron-type dwarfism, a human GH-resistance syndrome most commonly associated with GH receptor defects, changes in plasma GH patterns, including elevated peak GH levels, are observed (49). Thus, if the STAT5b−/− mouse is indeed a GH pulse-resistant animal model, then any alteration of circulating GH patterns in these animals would still leave unanswered the question of whether such a change is a secondary response, as is seen with other examples of GH pulse resistance, or whether the altered GH profile is a primary consequence of the STAT5b gene disruption, which, in turn, causes the growth rate and liver enzyme phenotypes described in this report. The apparent GH resistance of our STAT5b−/− mice, as well as the dwarfism, elevated plasma GH, lower IGF-I, and the development of obesity that we have seen are all characteristics of Laron-type dwarfism (49). This raises the interesting possibility that cases of nonclassical Laron-type dwarfism, which are associated with post-GH receptor defects (50, 51), may include examples of STAT5b defects.
Although our findings establish that STAT5b is needed for the plasma GH-regulated sexual dimorphism of liver gene expression, additional factors are also likely to be required for this response. This is indicated by the rapid (within 10 min) activation of liver STAT5 by a physiological GH pulse given in vivo (10, 11), which contrasts to the much slower response of male-specific liver CYP gene transcription (52) and mRNA expression (5) to GH pulsation, and by the relatively modest stimulatory effect demonstrated for a STAT5 response element when analyzed in cultured cell transfection studies of a GH-regulated male-specific hamster liver CYP gene (48). Further study is required to identify these other factors and to establish the underlying mechanisms through which STAT5b would appear to regulate the male pattern of long bone growth that is characteristic of many species, including humans.
Finally, comment should be made on the evolution of the STAT gene family. Recent work suggests that STATs 1–4 and STAT6 arose by a series of chromosome duplications from an ancestral STAT5 gene (53, 54). Presumably, each of these genes diverged and subsequently evolved unique and essential roles in one or more cytokine signaling pathways. Indeed, this conclusion is supported by gene-disruption experiments involving STATs 1, 4, and 6 (28–33). One would likewise predict that the STAT5a and STAT5b genes would have properties that are similar to each other, but quite distinct from STATs 1–4 and STAT6, given their high homology (53). Although this may be the case for some STAT5-dependent phenotypes, such as prolactin-dependent responses of mammary gland (cf. this study with ref. 34), our data clearly show that in the liver, endogenous STAT5a expression cannot substitute for a global STAT5b gene deficiency. Whether this results from intrinsic differences in the activities of STAT5a and STAT5b or from differences in their patterns of expression within the liver or other critical tissues cannot presently be determined. Further studies, including analysis of liver gene expression patterns in STAT5a gene knockout mice, either alone (34) or in combination with STAT5b gene disruption, may help answer these questions.
Acknowledgments
We thank Ric Broadhurst for assistance with animal work, Tony Craven for help with the hair analysis, Brendan Haigh for contribution to the Northern blotting and gel-shift assays, Jim Napier for the IGF-I RIAs, Harold Henderson for statistical analyses, and Masahiko Negishi and Paul Kelly for kindly sharing antibodies and cDNA probes. This work was supported by the New Zealand Foundation for Research Science and Technology (G.B.U., R.P.T., R.G.S., R.J.W., H.W.D.) and by Grant DK33765 from the National Institutes of Health (D.J.W.).
ABBREVIATIONS
- GH
growth hormone
- STAT
signal transducer/activator of transcription
- CYP
cytochrome P450
- MUP
major urinary protein
- IGF-I
insulin-like growth factor I
References
- 1.Isaksson O G, Eden S, Jansson J O. Annu Rev Physiol. 1985;47:483–499. doi: 10.1146/annurev.ph.47.030185.002411. [DOI] [PubMed] [Google Scholar]
- 2.Carter-Su C, Schwartz J, Smit L S. Annu Rev Physiol. 1996;58:187–207. doi: 10.1146/annurev.ph.58.030196.001155. [DOI] [PubMed] [Google Scholar]
- 3.Norstedt G, Palmiter R. Cell. 1984;36:805–812. doi: 10.1016/0092-8674(84)90030-8. [DOI] [PubMed] [Google Scholar]
- 4.Kelly P A, Ali S, Rozakis M, Goujon L, Nagano M, et al. Recent Prog Horm Res. 1993;48:123–164. doi: 10.1016/b978-0-12-571148-7.50009-9. [DOI] [PubMed] [Google Scholar]
- 5.Noshiro M, Negishi M. J Biol Chem. 1986;261:15923–15927. [PubMed] [Google Scholar]
- 6.Westin S, Tollet P, Strom A, Mode A, Gustafsson J A. J Steroid Biochem Mol Biol. 1992;43:1045–1053. doi: 10.1016/0960-0760(92)90332-D. [DOI] [PubMed] [Google Scholar]
- 7.Waxman D J. J Steroid Biochem Mol Biol. 1992;43:1055–1072. doi: 10.1016/0960-0760(92)90333-E. [DOI] [PubMed] [Google Scholar]
- 8.Waxman D J, Pampori N A, Ram P A, Agrawal A K, Shapiro B H. Proc Natl Acad Sci USA. 1991;88:6868–6872. doi: 10.1073/pnas.88.15.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacLeod J N, Pampori N A, Shapiro B H. J Endocrinol. 1991;131:395–399. doi: 10.1677/joe.0.1310395. [DOI] [PubMed] [Google Scholar]
- 10.Waxman D J, Ram P A, Park S H, Choi H K. J Biol Chem. 1995;270:13262–13270. doi: 10.1074/jbc.270.22.13262. [DOI] [PubMed] [Google Scholar]
- 11.Ram P A, Park S H, Choi H K, Waxman D J. J Biol Chem. 1996;271:5929–5940. doi: 10.1074/jbc.271.10.5929. [DOI] [PubMed] [Google Scholar]
- 12.Gebert C A, Park S H, Waxman D J. Mol Endocrinol. 1997;11:400–414. doi: 10.1210/mend.11.4.9904. [DOI] [PubMed] [Google Scholar]
- 13.Ripperger J A, Fritz S, Richter K, Hocke G M, Lottspeich F, Fey G H. J Biol Chem. 1995;270:29998–30006. doi: 10.1074/jbc.270.50.29998. [DOI] [PubMed] [Google Scholar]
- 14.Wakao H, Schmitt-Ney M, Groner B. J Biol Chem. 1992;267:16365–16370. [PubMed] [Google Scholar]
- 15.Wakao H, Gouilleux F, Groner B. EMBO J. 1994;13:2182–2191. doi: 10.1002/j.1460-2075.1994.tb06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gouilleux F, Wakao H, Mundt M, Groner B. EMBO J. 1994;13:4361–4369. doi: 10.1002/j.1460-2075.1994.tb06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mui A L F, Wakao H, O’Farrell A M, Harada N, Miyajima A. EMBO J. 1995;14:1166–1175. doi: 10.1002/j.1460-2075.1995.tb07100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azam M, Erdjument-Bromage H, Kreider B L, Xia M, Quelle F, Basu R, Saris C, Tempst P, Ihle J N, Schindler C. EMBO J. 1995;14:1402–1411. doi: 10.1002/j.1460-2075.1995.tb07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Robinson G W, Gouilleux F, Groner B, Hennighausen L. Proc Natl Acad Sci USA. 1995;92:8831–8835. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou J, Schindler U, Henzel W J, Wong S C, McKnight S L. Immunity. 1995;2:321–329. doi: 10.1016/1074-7613(95)90140-x. [DOI] [PubMed] [Google Scholar]
- 21.Dajee M, Kazansky A V, Raught B, Hocke G M, Fey G H, Richards J S. Mol Endocrinol. 1996;10:171–184. doi: 10.1210/mend.10.2.8825557. [DOI] [PubMed] [Google Scholar]
- 22.Silva C M, Lu H, Day R N. Mol Endocrinol. 1996;10:508–518. doi: 10.1210/mend.10.5.8732682. [DOI] [PubMed] [Google Scholar]
- 23.Smit L S, Meyer D J, Billestrup N, Norstedt G, Schwartz J, Carter-Su C. Mol Endocrinol. 1996;10:519–533. doi: 10.1210/mend.10.5.8732683. [DOI] [PubMed] [Google Scholar]
- 24.Fujii K, Nakagawa Y, Schindler U, Kawahara A, Mori H, Gouilleux F, Groner B, Ihle J N, Minami Y, Miyazaki T, Taniguchi T. Proc Natl Acad Sci USA. 1995;92:5482–5486. doi: 10.1073/pnas.92.12.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruff-Jamison S, Chen K, Cohen S. Proc Natl Acad Sci USA. 1995;92:4215–4218. doi: 10.1073/pnas.92.10.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai C F, Ripperger J, Morella K K, Wang Y, Gearing D P, Horseman N D, Campos S P, Fey G H, Baumann H. J Biol Chem. 1995;270:23254–23257. doi: 10.1074/jbc.270.40.23254. [DOI] [PubMed] [Google Scholar]
- 27.Gouilleux F, Pallard C, Dusanter-Fourt I, Wakao H, Haldosen L A, Norstedt G, Levy D, Groner B. EMBO J. 1995;14:2005–2013. doi: 10.1002/j.1460-2075.1995.tb07192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan M H, Sun Y L, Hoey T, Grusby M J. Nature (London) 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 29.Thierfelder W E, van Deursen J M, Yamamoto K, Tripp R A, Sarawar S R, Carson R T, Sangster M Y, Vignali D A, Doherty P C, Grosveld G C, Ihle J N. Nature (London) 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Nature (London) 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 31.Shimoda K, van Deursen J, Sangster M Y, Sarawar S R, Carson R T, Tripp R A, Chu C, Quelle F W, Nosaka T, Vignali D A, Doherty P C, Grosveld G, Paul W E, Ihle J N. Nature (London) 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 32.Durbin J E, Hackenmiller R, Simon M C, Levy D E. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 33.Meraz M A, White J M, Sheehan K C, Bach E A, Rodig S J, Dighe A S, Kaplan D H, Riley J K, Greenlund A C, Campbell D, Carver-Moore K, Du Bois R N, Clark R, Aguet M, Schreiber R D. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Robinson G W, Wagner K U, Garrett L, Wynshaw-Boris A, Hennighausen L. Genes Dev. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 35.Udy G B, Evans M J. BioTechniques. 1994;17:887–894. [PubMed] [Google Scholar]
- 36.Waxman D J, Lapenson D P, Park S S, Attisano C, Gelboin H V. Mol Pharmacol. 1987;32:615–624. [PubMed] [Google Scholar]
- 37.Waxman D J. Methods Enzymol. 1991;206:462–476. doi: 10.1016/0076-6879(91)06115-j. [DOI] [PubMed] [Google Scholar]
- 38.Happ B, Groner B. J Steroid Biochem Mol Biol. 1993;47:21–30. doi: 10.1016/0960-0760(93)90053-y. [DOI] [PubMed] [Google Scholar]
- 39.Hua K M, Hodgkinson S C, Bass J J. J Endocrinol. 1995;147:507–516. doi: 10.1677/joe.0.1470507. [DOI] [PubMed] [Google Scholar]
- 40.Clark R G, Robinson I C. Nature (London) 1985;314:281–283. doi: 10.1038/314281a0. [DOI] [PubMed] [Google Scholar]
- 41.Pampori N A, Agrawal A K, Shapiro B H. Acta Endocrinol. 1991;124:283–289. doi: 10.1530/acta.0.1240283. [DOI] [PubMed] [Google Scholar]
- 42.Kulkarni A B, Gubits R M, Feigelson P. Proc Natl Acad Sci USA. 1985;82:2579–2582. doi: 10.1073/pnas.82.9.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Negishi M, Burkhart B, Aida K. Methods Enzymol. 1991;206:267–273. doi: 10.1016/0076-6879(91)06096-l. [DOI] [PubMed] [Google Scholar]
- 44.Morikawa M, Nixon T, Green H. Cell. 1982;29:783–789. doi: 10.1016/0092-8674(82)90440-8. [DOI] [PubMed] [Google Scholar]
- 45.Gronowski A M, Zhong Z, Wen Z, Thomas M J, Darnell J E, Jr, Rotwein P. Mol Endocrinol. 1995;9:171–177. doi: 10.1210/mend.9.2.7776967. [DOI] [PubMed] [Google Scholar]
- 46.Gronowski A M, Rotwein P. J Biol Chem. 1994;269:7874–7878. [PubMed] [Google Scholar]
- 47.Waxman D J, Zhao S, Choi H K. J Biol Chem. 1996;271:29978–29987. doi: 10.1074/jbc.271.47.29978. [DOI] [PubMed] [Google Scholar]
- 48.Subramanian A, Teixeira J, Wang J, Gil G. Mol Cell Biol. 1995;15:4672–4682. doi: 10.1128/mcb.15.9.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laron Z. Curr Opin Pediatr. 1993;5:474–480. doi: 10.1097/00008480-199308000-00018. [DOI] [PubMed] [Google Scholar]
- 50.Postel-Vinay M C. Rev Prat. 1994;44:1281–1285. [PubMed] [Google Scholar]
- 51.Laron Z, Klinger B, Eshet R, Kaneti H, Karasik A, Silbergeld A. Isr J Med Sci. 1993;29:757–763. [PubMed] [Google Scholar]
- 52.Sundseth S S, Alberta J A, Waxman D J. J Biol Chem. 1992;267:3907–3914. [PubMed] [Google Scholar]
- 53.Copeland N G, Gilbert D J, Schindler C, Zhong Z, Wen Z, et al. Genomics. 1995;29:225–228. doi: 10.1006/geno.1995.1235. [DOI] [PubMed] [Google Scholar]
- 54.Xianyu S A, Melnick M B, Perrimon N. Cell. 1996;84:411–419. doi: 10.1016/s0092-8674(00)81286-6. [DOI] [PubMed] [Google Scholar]