

Abstract
Multiprotein bridging factor 1 (MBF1) is a transcriptional cofactor that bridges between the TATA box-binding protein (TBP) and the Drosophila melanogaster nuclear hormone receptor FTZ-F1 or its silkworm counterpart BmFTZ-F1. A cDNA clone encoding MBF1 was isolated from the silkworm Bombyx mori whose sequence predicts a basic protein consisting of 146 amino acids. Bacterially expressed recombinant MBF1 is functional in interactions with TBP and a positive cofactor MBF2. The recombinant MBF1 also makes a direct contact with FTZ-F1 through the C-terminal region of the FTZ-F1 DNA-binding domain and stimulates the FTZ-F1 binding to its recognition site. The central region of MBF1 (residues 35–113) is essential for the binding of FTZ-F1, MBF2, and TBP. When the recombinant MBF1 was added to a HeLa cell nuclear extract in the presence of MBF2 and FTZ622 bearing the FTZ-F1 DNA-binding domain, it supported selective transcriptional activation of the fushi tarazu gene as natural MBF1 did. Mutations disrupting the binding of FTZ622 to DNA or MBF1, or a MBF2 mutation disrupting the binding to MBF1, all abolished the selective activation of transcription. These results suggest that tethering of the positive cofactor MBF2 to a FTZ-F1-binding site through FTZ-F1 and MBF1 is essential for the binding site-dependent activation of transcription. A homology search in the databases revealed that the deduced amino acid sequence of MBF1 is conserved across species from yeast to human.
During development, many genes are expressed under the precise temporal and spatial control. This is accomplished by a proper combination of cis-regulatory elements (enhancers or silencers) and regulatory proteins that bind to them in a sequence-specific fashion (1, 2). Later studies revealed that another class of transcription factors termed coactivators, adaptors, or mediators exist that play a crucial role by interconnecting the regulatory proteins and the basal transcription machinery (3, 4). Recent studies illustrate the importance of this new class of transcription factors for gene expression in various systems (5–10).
The fushi tarazu (ftz) gene of Drosophila melanogaster is expressed in a seven-striped manner during blastoderm stage of embryos that is required for proper development of the corresponding body segments (11, 12). Analyses using transgenic flies identified a binding site for the nuclear hormone receptor FTZ-F1 located 280 bp upstream of the transcription initiation site as a major cis-element required for the striped expression of ftz (13–15). FTZ-F1 has been implicated in the activation of ftz during embryogenesis (15, 16) and other genes at metamorphosis (17, 18).
In our in vitro transcription studies, aimed at analyzing the mechanism of transactivation by FTZ-F1, we found that BmFTZ-F1, a silkworm counterpart of FTZ-F1 (19, 20), can activate transcription of the ftz gene by binding to the FTZ-F1 site in posterior silk gland extracts of the silkworm Bombyx mori (21). This transcriptional activation requires two additional factors, MBF1 and MBF2 (for multiprotein bridging factors 1 and 2), that do not directly bind to DNA. MBF2 is a positive cofactor that activates transcription through its contact with TFIIA (22). MBF1 is a bridging molecule that interconnects FTZ-F1 (or BmFTZ-F1), MBF2, and TATA box-binding protein (TBP) (21). To learn more about MBF1, we have isolated a cDNA coding for the factor and examined function of the recombinant MBF1. Our results suggest that MBF1 is an evolutionarily conserved transcriptional coactivator, essential for bridge formation between certain sequence-specific regulators and TBP.
MATERIALS AND METHODS
Isolation of a cDNA Encoding MBF1.
MBF1 was purified from the posterior silk glands of B. mori as described (21). The purified factor was electrophoresed on a SDS/12.5% polyacrylamide gel. After staining with Coomassie brilliant blue, the 18-kDa band of MBF1 was cut from the gel, treated with V8 protease within the gel slices, and run on an SDS/15% polyacrylamide gel as described (23). Discrete peptide bands were transferred to polyvinylidene difluoride membranes and subjected to amino acid sequence analyses on an Applied Biosystem model 477A protein sequencer. Based on the amino acid sequence TEELRHEKIPLDLGKLIMQG, oligonucleotides P1 (5′-PyTN A/CGNCAPy GAPuAAPuAT Py/A CC-3′) and P2 (5′-CCPyTGCAT T/Pu ATNAPuPyTTNCC-3′) were prepared and served as primers in PCR (24). The first strand cDNA was synthesized on poly(A)+ RNA isolated from the posterior silk glands of B. mori at the fourth larval molting stage and used as PCR template. A resulting 50-bp PCR product was then used to screen the molting stage cDNA library as described before (20). One of the positive clones with the longest insert was sequenced on both strands by the dideoxy-termination method (25).
Protein Expression and Purification.
To produce MBF1 bearing six consecutive histidine residues (his-tag MBF1) in Escherichia coli, an NdeI–BamHI tagged open reading frame (nucleotide positions 101–541 in Fig. 1) was subcloned into 6HisT–pET11d to give pMBF1. To produce the glutathione S-transferase (GST)–MBF1 fusion protein, and its deletion derivatives, each BamHI–XhoI tagged open reading frame was inserted between the BamHI and XhoI sites of pGEX-4T-3 (Pharmacia). Recombinant MBF2 (rMBF2) and recombinant human TBP (rhTBP) were expressed from their corresponding cDNAs inserted into 6HisT–pET11d. MBF2Δ70–92 was expressed as described (22). For preparation of 32P-labeled rMBF2 and rhTBP, the 18-bp sequence encoding cyclic AMP-dependent protein kinase phosphorylation site (26) was inserted in-frame at the NdeI site. The histidine-tagged recombinant proteins were purified using Ni-immobilized resin (Novagen) followed by Mono S column chromatography. GST and GST fusion proteins were purified using glutathione-Sepharose 4B (Pharmacia). Protein labeling with [γ-32P]ATP was performed as described (27). Purification of BmFTZ-F1 (21) and preparation of FTZ622 and its derivatives (27) were done as described.
Figure 1.

Sequence of MBF1 cDNA. Nucleotide sequence of the longest cDNA insert is shown. The predicted amino acid sequence of the factor is presented in single-letter code. The experimentally determined amino acid sequence is underlined. The asterisk represents the putative stop codon.
GST Interaction Assays.
Protein–protein interactions through GST fusion proteins were analyzed as described (22), except that the protein-bound beads were washed with PBB (20 mM Hepes-KOH, pH 7.9/20% glycerol/0.5 mM EDTA/60 mM KCl/6 mM MgCl2/0.1% Nonidet P-40/5 mM 2-mercaptoethanol/1 mM phenylmethylsulfonyl fluoride). In the experiments shown in Fig. 5A, FTZ622 and its derivatives were detected by immunoblot using an anti-FTZ-F1 polyclonal antibody (18) and the Vistra fluorescence western blotting kit (Amersham), then quantitated on a Molecular Dynamics FluorImager SI. To examine the recovery of the GST fusion proteins, a portion of each eluate was subjected to Western blot analysis using an antibody against GST. In all cases tested, >95% of the input fusion protein were recovered in the eluate.
Figure 5.
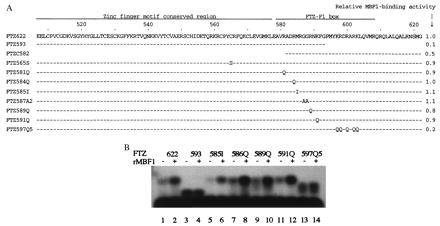
(A) MBF1-binding activities of FTZ622 mutants. Numbers on the top line represent amino acid positions within FTZ-F1 (16). Bars indicate sequences identical to those of FTZ-F1. Positions of amino acid substitutions are indicated by letters representing the substituted amino acids. Each peptide has a methionine at the N terminus (data not shown). GST assays for the binding of rMBF1 to FTZ622 or its mutants were carried out as described in Fig. 4B except that nonradioactive polypeptides were used in place of [32P]FTZ622. For each deletion mutant, the amount of input polypeptide was adjusted according to its molecular mass. Band intensities of bound FTZ622 or its derivatives were quantitated as described in Materials and Methods and divided by the corresponding input intensity. This value for each mutant in relation to that for FTZ622 is shown as relative MBF1-binding activity. (B) Interactions between MBF1 and FTZ622 mutants inferred from the MBF1-mediated increase of the DNA binding. The binding of FTZ622 or its mutants to the [32P]FTZ-F1-binding site probe was analyzed as described in Fig. 4A. Where indicated, the binding mixture contained 25 ng of his-tag MBF1.
In Vitro Transcription.
Transcription reactions were performed as described (21). Plasmid DNA of pE(HU)5-N and pFLBH (each 50 ng) were used for transcription of the ftz gene and adenovirus 2 major late promoter (Ad2MLP), respectively. Transcripts were analyzed by a modified S1 nuclease method (21) and quantitated using a Fuji BAS-2000II bioimage analyzer.
Other Methods.
Southern and Northern blot hybridizations were carried out as described (24). SDS/PAGE was performed according to the method of Laemmli (28). For the analyses of short polypeptides, such as FTZ622 and its derivatives, the Tricine buffer system (29) was employed. Electrophoresis mobility shift assays were performed as described (21) except that 60 mM KCl was used instead of 100 mM NaCl for the binding of FTZ-F1 or its derivatives.
RESULTS
MBF1 Sequence Is Evolutionarily Conserved.
MBF1 was purified from the posterior silk glands of B. mori and after partial digestion with V8 protease, subjected to microsequence analyses. A partial amino acid sequence TEELRHEKIPLDLGKLIMQG was obtained and used to design oligonucleotides for PCR amplification of the corresponding cDNA, which then served as a probe for screening of a B. mori posterior silk gland cDNA library (see Materials and Methods). From 1.2 × 105 plaques, 6 positive clones were isolated. Restriction analyses showed that all of them were derived from a single species of mRNA (data not shown). The longest clone was sequenced in its entirety.
The cDNA sequence contained a single long open reading frame of 146 amino acids (Fig. 1). The deduced amino acid sequence of the factor predicts a basic protein (pI = 10.7) with a molecular mass of 16.2 kDa and contains the peptide we had sequenced (underlined in Fig. 1). Homology search in the databases showed that the MBF1 sequence is conserved across species from yeast to human (Fig. 2). The silkworm MBF1 appears most similar to the human counterpart (62% amino acid identity). There is also a marked degree of sequence conservation among three plant species (A. thaliana, R. communis, and Z. mays).
Figure 2.

Evolutional conservation of the MBF1 sequence. The predicted amino acid sequence of MBF1 is compared with the homologous sequences in the databases. The amino acid sequences have been conceptually translated from the nucleotide sequences with accession numbers T38224 (Saccharomyces cerevisiae), T74845 (Homo sapiens), Z29020 (Arabidopsis thaliana), Z49698 (Ricinus communis), T14752 (Zea mays), and X15385 (Dictyostelium discoideum). Identical or similar amino acids are shadowed. Bars represent gaps.
Southern blot hybridization of EcoRI-, HindIII-, or BamHI-digested genomic DNA indicated that there is a single gene for the factor within the Bombyx genome (data not shown). Northern blot analyses of poly(A)+ RNA isolated from the posterior silk glands revealed a single hybridizing RNA of 2 kb (data not shown).
Central Region of MBF1 Is Required for the Binding of MBF2, TBP, and FTZ-F1.
We have previously shown that natural MBF1 forms a heterodimer with MBF2, interacts with FTZ-F1, and stabilizes the FTZ-F1⋅DNA complex, and that it also makes a direct contact with TBP (21). We identified the regions of MBF1 required for these protein–protein interactions using a series of bacterially expressed rMBF1 truncated proteins. To test interactions between MBF1 and MBF2, [32P]rMBF2 was incubated with the GST–MBF1 fusion protein and then allowed to bind glutathione-Sepharose beads. The radioactive rMBF2 was recovered from the beads after it had been incubated with GST–MBF1 (Fig. 3A, lane 4) but not after incubation with buffer or GST alone (Fig. 3A, lanes 2 and 3). These results show that rMBF1 binds directly to rMBF2. The ability of the rMBF1-truncated proteins to bind rMBF2 was tested using the GST pull-down assay. As shown in Fig. 3A, deletion of the first 34 N-terminal amino acids did not affect the binding of rMBF2 (lane 8), but deletion extended to residue 66 abolished the binding (lane 9). Deletion mutant lacking residue 114 to C terminus showed a reduced but significant level of binding (lane 7). However, no binding of rMBF2 was observed with deletion from C terminus to residue 67 (lane 6). These results suggest that the central region of the factor (residues 35–113) is important for the binding of MBF2.
Figure 3.
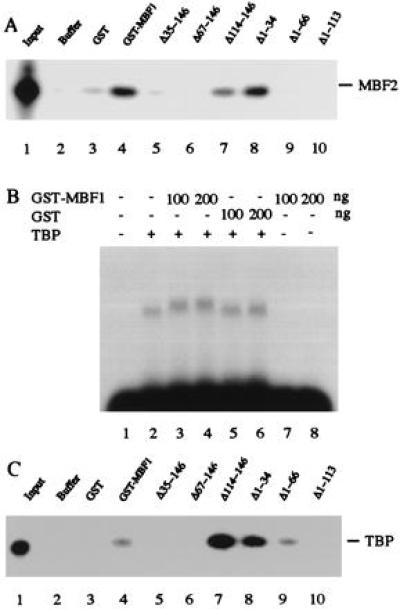
Protein–protein interactions through MBF1. (A) GST assay for the binding of rMBF1 to rMBF2. Approximately 20 ng of [32P]rMBF2 was incubated with buffer alone (lane 2), 200 ng of GST (lane 3), or each 200 ng of GST–MBF1 or its deletion derivative (lanes 4–10). The bound rMBF2 was collected using glutathione-Sepharose beads and analyzed by 12 % SDS/PAGE (Tricine buffer system). Lane 1, input [32P]rMBF2. (B) Electrophoresis mobility shift assay for the binding of rMBF1 to rTBP. 32P-labeled double-stranded oligonucleotide bearing the TATA element of the ftz gene was incubated with GST or GST–MBF1 fusion protein in the presence or absence of 6 ng of rhTBP. The specific protein⋅DNA complexes were resolved by electrophoresis on a 1% agarose gel containing 2 mM MgCl2. (C) GST assay for the binding of rMBF1 to rTBP. The assay was performed as in A except that [32P]rhTBP was used in place of [32P]rMBF2 and that electrophoresis was performed on an SDS/10% polyacrylamide gel. Lane 1, input [32P]rhTBP
Next, interactions between MBF1 and TBP were analyzed by adding the GST–MBF1 fusion protein to electrophoresis mobility shift assay mixtures including rhTBP and a probe DNA carrying the TATA element of the ftz gene (Fig. 3B). In the absence of TBP, GST–MBF1 did not form a complex with the probe DNA (lanes 7 and 8). When the probe DNA was incubated with TBP, we observed a TBP⋅DNA complex (lane 2). Further addition of GST–MBF1 resulted in a small but significant retardation of the TBP–TATA element complex (lanes 3 and 4). Such a retardation did not result from addition of the GST moiety alone (lanes 5 and 6). If we added antibody against GST in the presence of TBP and GST–MBF1, supershift of the retarded complex was observed suggesting the presence of GST–MBF1 in the shifted complex (data not shown). These results indicate that rMBF1 makes a direct contact with TBP. This interaction was confirmed by a GST pull-down assay (Fig. 3C), in which [32P]TBP was recovered from the beads incubated with GST–MBF1 but not from those incubated with buffer or GST only (lanes 2–4). Deletion of the N-terminal region (residues 1–34) or the C-terminal region of rMBF1 (residues 114–146) resulted in enhanced binding of TBP compared with the intact rMBF1 (lane 4 vs. lane 7 or 8), suggesting that these regions are inhibitory to the binding of TBP. Deletion analysis of MBF1 also revealed that the central region (residues 35–113) is required for the binding of TBP (lanes 5–10). Within the central region, residues 66–113 appear to contribute mainly because deletion from N terminus to residue 66 still retains significant binding ability (lane 9).
To examine interactions between MBF1 and FTZ-F1, we added his-tag MBF1 to electrophoresis mobility shift assay mixtures containing the FTZ622 polypeptide bearing the DNA-binding domain of FTZ-F1 and a probe DNA carrying the FTZ-F1-binding site. Although addition of rMBF1 did not result in further retardation of the FTZ622⋅DNA complex, significantly larger amounts of the complex were formed in the presence of rMBF1 than in its absence (Fig. 4A). Essentially the same results were obtained when the mixture contained BmFTZ-F1 in place of FTZ622 (data not shown). These results suggest that rMBF1 interacts with FTZ-F1 (or BmFTZ-F1) and promotes the binding of FTZ-F1 (or BmFTZ-F1) to DNA. To get direct evidence for the interactions between rMBF1 and FTZ-F1, we performed GST pull-down assays (Fig. 4B). FTZ622 was recovered from the beads incubated with GST–MBF1 but not from the beads incubated with buffer or GST (lanes 2–4). These results clearly show that rMBF1 makes a direct contact with the DNA-binding domain of FTZ-F1. Deletion analysis of MBF1 demonstrates that the central region of the factor (residues 35–113) is mainly responsible for the binding of FTZ-F1 (lanes 5–10). The N- and C-terminal regions (residues 1–34 and 114–146, respectively) showed interactions above the background level but these interactions were suppressed by the neighboring regions (compare lanes 5 and 6, and lanes 10 and 9, respectively).
Figure 4.
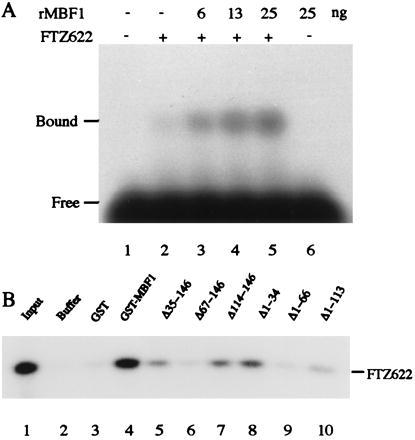
Interactions between MBF1 and FTZ-F1. (A) rMBF1 increases the DNA binding of FTZ622. The binding of FTZ622 to a 32P-labeled double-stranded oligonucleotide carrying the FTZ-F1 recognition site was analyzed by electrophoresis mobility shift assay. Each binding mixture containing 0.2 ng of FTZ622 and indicated amounts of his-tag MBF1 was incubated at 25°C for 30 min. The specific protein⋅DNA complexes were resolved by electrophoresis on a 1% agarose gel. (B) GST assay for the binding of rMBF1 to FTZ622. [32P]FTZ622 (20 ng) was incubated with buffer alone (lane 2), 200 ng of GST (lane 3), or each 200 ng of GST–MBF1 or its deletion derivative (lanes 4–10). The bound FTZ622 was collected and analyzed by 12% SDS/PAGE (Tricine buffer system). Lane 1, input [32P]FTZ622.
MBF1 Binds to FTZ-F1 Through the C-Terminal Half of the FTZ-F1 Box.
The DNA-binding domain of FTZ-F1 consists of Cys2-Cys2-type Zn fingers and a 30-amino acid basic region termed FTZ-F1 box (27). We examined the effects of amino acid substitutions and deletions in FTZ622 polypeptide on the binding of MBF1 using GST interaction assay (Fig. 5A). FTZ593 lacking the C-terminal half of the FTZ-F1 box showed severely reduced binding to GST–MBF1, whereas FTZC582 lacking the Zn finger motif region retained a significant binding activity. Amino acid substitutions in the second finger (FTZ565S) and the N-terminal half of the FTZ-F1 box (FTZ581Q, 584Q, 585I, 587A2, 589Q, and 591Q) did not affect the binding to GST–MBF1. By contrast, FTZ597Q5 carrying five amino acid substitutions in the C-terminal half of the FTZ-F1 box showed a significantly reduced level of binding. To confirm that the C-terminal half of the FTZ-F1 box is important for the binding of MBF1, we performed electrophoresis mobility shift assays including a FTZ622 derivative and the FTZ-F1-binding site probe in the presence or absence of his-tag MBF1 (Fig. 5B). MBF1-mediated increase of the DNA binding was almost abolished in FTZ593 (lanes 3 and 4) and in FTZ597Q5 (lanes 13 and 14). By contrast, the stimulatory effect of MBF1 was clearly seen in other amino acid substitution mutants in the N-terminal half of the FTZ-F1 box (lanes 5–12). These results show that MBF1 binds to FTZ-F1 through the C-terminal half of the FTZ-F1 box.
Anchorage of MBF2 to the FTZ-F1-Binding Site Through FTZ-F1 and MBF1 Is Essential for Selective Activation of ftz Transcription.
When natural MBF1 was added to a HeLa cell nuclear extract in the presence of rMBF2 and FTZ622, it supported selective activation of ftz transcription in a FTZ-F1-binding site-dependent manner (ref. 22; see also Fig. 6, lane 2). rMBF1 was also functional for the selective activation of ftz transcription (Fig. 6, compare lanes 1 and 3). Essentially the same results were obtained when we used BmFTZ-F1 in place of FTZ622 (data not shown). The selective activation of ftz transcription requires the simultaneous presence of FTZ622, rMBF1, and rMBF2. Thus, omission of rMBF2 abolished the activation (Fig. 6, lane 4). Omission of either FTZ622 or rMBF1 resulted in moderate level of transcription of both ftz and Ad2MLP (Fig. 6, lanes 5 and 6) similar to that observed when rMBF2 alone was added to the nuclear extract (Fig. 6, lane 7).
Figure 6.
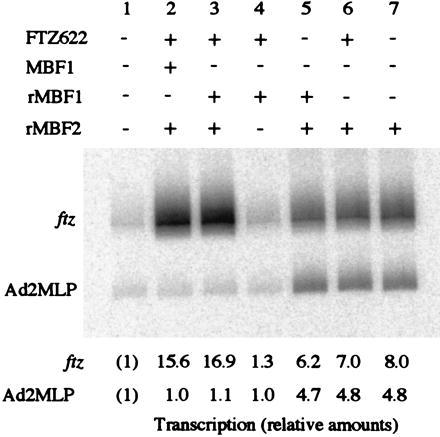
rMBF1 supports selective activation of ftz transcription. The ftz gene and Ad2MLP were transcribed in a HeLa cell nuclear extract. Where indicated, the reaction mixture contained 50 ng of either the natural MBF1 or his-tag MBF1. The relative levels of transcription are shown at the bottom.
We used two FTZ622 mutants and a mutant of MBF2 in this transcription system to test the importance of protein-DNA and protein-protein interactions. FTZ587A2 can bind to MBF1 (Fig. 5A) but does not bind to DNA (27). FTZ593 retains a significant DNA-binding activity (27) but is defective in the MBF1 binding (Fig. 5A). MBF2Δ70-92 lacking the C-terminal region is defective in the MBF1 binding, yet capable of binding to TFIIA (22). None of these mutants supported the selective activation of ftz transcription in the presence of rMBF1 (Fig. 7, lanes 3–5). Instead, transcription of both the ftz gene and Ad2MLP was enhanced to the levels observed with rMBF2 alone (Fig. 7, lane 6). These results suggest that tethering of the positive cofactor MBF2 to a FTZ-F1-binding site through FTZ-F1 and MBF1 is crucial for the selective activation in a FTZ-F1 site-dependent manner.
Figure 7.
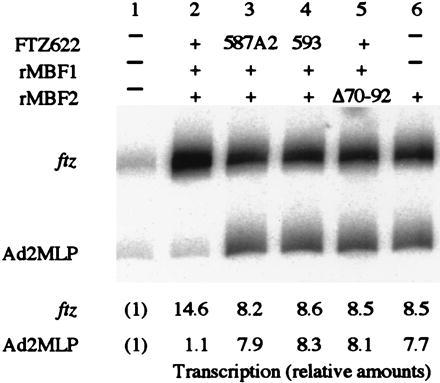
Selective activation of ftz transcription requires DNA–FTZ622, FTZ622–MBF1, and MBF1–MBF2 interactions. The ftz gene and Ad2MLP were transcribed in a HeLa cell nuclear extract. Where indicated, the transcription mixture contained 50 ng of the histidine-tagged recombinant factors.
DISCUSSION
This report describes the isolation of a cDNA encoding MBF1 and the properties of recombinant MBF1 expressed from this cDNA. Comparison of the deduced amino acid sequence of MBF1 with the sequences in the databases revealed an evolutional conservation of the factor. This indicates the presence of a common regulatory mechanism in eukaryotes, in which MBF1 or its counterpart in each organism exerts its function as a bridging molecule that connects certain sequence-specific regulator(s) and TBP, and increases the DNA binding of regulator(s). Indeed, we found that yeast MBF1 interconnects between GCN4 and TBP, and mediates transcriptional activation by GCN4 (K.-i.T., S. Harashima, H.U., and S.H., unpublished observation). Interestingly, the basic region of certain basic leucine zipper (bZIP) proteins shares homology with the C-terminal region of FTZ-F1 box to which MBF1 binds (27). Moreover, the defect of yeast mbf1 disruptant was rescued by expression of a cDNA encoding silkworm or human MBF1 (K.-i.T. and S.H., unpublished observation). MBF1 was originally termed as mediator of BmFTZ-F1 type 1 (21). Having established that the factor is conserved among eukaryotes, we renamed it as multiprotein bridging factor type 1.
Deletion analyses of B. mori MBF1 demonstrated that its central region (residues 35–113) is essential for the binding of FTZ-F1, MBF2, and TBP. The C-terminal half of this region (residues 63–113) is markedly conserved among various species and the highly conserved region extends further until the residue 132 (Fig. 2). Recent NMR study revealed the presence of four amphipathic helices in this highly conserved region (residues 63–132), that are bundled into a compact form (J. Ozaki, K.-i.T., T. Ikegami, H.U., S.H., and M. Shirakawa, unpublished observation). This raises a possibility that the compact domain constitutes binding surfaces for the protein–protein interactions. The N-terminal half of the essential region (residues 35–62) exhibits less prominent conservation. Because this region assumes a flexible structure, it might play some functional role rather than an architectural one.
The DNA-binding domain of FTZ-F1 consists of two functional subdomains. The Zn finger motif region and the N-terminal half of FTZ-F1 box constitute the first subdomain that governs the recognition of the binding sequence. The second one is the C-terminal half of FTZ-F1 box that is responsible for the high affinity binding to the recognition site (27). The present study demonstrates that B. mori MBF1 makes a direct contact with FTZ-F1 through the second subdomain and increases the DNA binding of FTZ-F1. What is the mechanism underlying the increased DNA binding? It is possible that MBF1 also makes a contact with DNA within the MBF1⋅FTZ-F1⋅DNA ternary complex and stabilizes the DNA binding since the factor is a basic protein. However, we consider this unlikely for the following reasons. First, upon addition of MBF1, no qualitative changes were observed in the DNase I footprinting patterns by BmFTZ-F1 (21). Second, only accumulation but not supershift of the FTZ-F1⋅DNA complex was detected upon addition of MBF1 to the electrophoresis mobility shift assay mixtures (ref. 21; see also Fig. 4A). Therefore, we prefer an alternative possibility that the contact of MBF1 with the second subdomain induces some conformational change (e.g., stabilization of the α-helical structure) in this region or masks the inhibitory surface to allow the increased FTZ-F1 binding to DNA. It has been reported that the human T cell leukemia virus transactivator Tax binds to the basic region of certain bZIP proteins and thus increases the DNA binding (30, 31). Although MBF1 and Tax do not share an apparent sequence similarity, they may achieve their function through a similar mechanism.
We show here that DNA–FTZ-F1, FTZ-F1–MBF1, and MBF1–MBF2 interactions are essential for the selective activation of ftz transcription. This finding strongly supports our model for tanscriptional activation through MBF1 and MBF2 in which FTZ-F1 and MBF1 recruit MBF2 to a promoter carrying the FTZ-F1-binding site and MBF2 activates transcription through its interaction with TFIIA to allow the selective activation in a FTZ-F1-binding site-dependent manner (22). D. melanogaster DHR39 is a nuclear receptor that recognizes the same sequence as FTZ-F1 does (32). In transient transfection experiments, FTZ-F1 contributed as a positive regulator whereas DHR39 did not activate the reporter gene expression (32, 33). Although the amino acid sequence of the first subdomain that governs the recognition of the binding sequence is markedly conserved between these two receptors, the sequence of the second subdomain is quite different (32, 33). This raises an intriguing possibility that DHR39 cannot activate transcription due to its lack of the MBF1-binding ability. Recent articles reported FTZ-F1–Ftz interactions required for ftz gene expression (34, 35). Whether FTZ-F1–coactivator interactions we found and FTZ-F1–Ftz interactions comprise independent pathways for the activation of ftz or they create a synergistic effect is an interesting issue to be determined.
The heterologous interaction between B. mori MBF1 and D. melanogaster FTZ-F1 is not surprising because amino acid sequence of the DNA-binding domain of FTZ-F1 is almost identical to that of BmFTZ-F1 (96% identity, 99% similarity; see ref. 20). Similarly, the marked conservation of MBF1 and TBP core sequences in eukaryotes most likely allowed the heterologous interaction between silkworm MBF1 and human TBP. Now, we are trying to isolate a cDNA encoding D. melanogaster MBF1 to study the mode of protein–protein interactions in a homologous system.
Acknowledgments
We thank R. G. Roeder for providing a human TBP cDNA clone, G.-C. Sun for the cDNA library, Marek Jindra for critical reading of the manuscript, and Y. Takada for preparation of the manuscript. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan. F.-Q.L. was supported by a fellowship from the Naito Foundation.
ABBREVIATIONS
- TBP
TATA box-binding protein
- ftz, fushi tarazu
GST, glutathione S-transferase
- Ad2MLP
adenovirus 2 major late promoter
- MBF1 and MBF2
multiprotein bridging factors 1 and 2, respectively
- r
recombinant
- rh
recombinant human
- his-tag
histidine-tagged
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AB001078).
References
- 1.Johnson P, McKnight S. Annu Rev Biochem. 1989;58:799–839. doi: 10.1146/annurev.bi.58.070189.004055. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P J, Tjian R. Science. 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 3.Lewin B. Cell. 1990;61:1161–1164. doi: 10.1016/0092-8674(90)90675-5. [DOI] [PubMed] [Google Scholar]
- 4.Roeder R G. Trends Biochem Sci. 1991;16:402–408. doi: 10.1016/0968-0004(91)90164-q. [DOI] [PubMed] [Google Scholar]
- 5.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lim S-C, Heyman R A, Rose D W, Glass C K, Rosenfeld M G. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 6.Chakravarti D, LaMorte V J, Nelson M C, Nakajima T, Schulman I G, Juguilon H, Montiminy M, Evance R M. Nature (London) 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 7.Schubart D B, Rolink A, Kosco-Vilbois M H, Botteri F, Mathias P. Nature (London) 1996;383:538–542. doi: 10.1038/383538a0. [DOI] [PubMed] [Google Scholar]
- 8.Kim U, Qin X-F, Gong S, Stevens S, Luo Y, Nussenzweig M, Roeder R G. Nature (London) 1996;383:542–547. doi: 10.1038/383542a0. [DOI] [PubMed] [Google Scholar]
- 9.Brownell J E, Zhou J, Ranalli T, Kobayashi R, Edmondson S Y, Allis C D. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 10.Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence P A, Johnston P, Macdonald P, Struhl G. Nature (London) 1987;328:440–442. doi: 10.1038/328440a0. [DOI] [PubMed] [Google Scholar]
- 12.Wakimoto B T, Kaufman T C. Dev Biol. 1981;81:51–64. doi: 10.1016/0012-1606(81)90347-x. [DOI] [PubMed] [Google Scholar]
- 13.Topol J, Dearolf C R, Prakash K, Parker C S. Genes Dev. 1991;5:855–867. doi: 10.1101/gad.5.5.855. [DOI] [PubMed] [Google Scholar]
- 14.Tsai C, Gergen P. Development (Cambridge, UK) 1995;121:453–462. doi: 10.1242/dev.121.2.453. [DOI] [PubMed] [Google Scholar]
- 15.Ueda H, Sonoda S, Brown J L, Scott M P, Wu C. Genes Dev. 1990;4:624–635. doi: 10.1101/gad.4.4.624. [DOI] [PubMed] [Google Scholar]
- 16.Lavorgna G, Ueda H, Clos H, Wu C. Science. 1991;252:848–851. doi: 10.1126/science.1709303. [DOI] [PubMed] [Google Scholar]
- 17.Woodard C T, Baehrecke E H, Thummel C S. Cell. 1994;79:607–615. doi: 10.1016/0092-8674(94)90546-0. [DOI] [PubMed] [Google Scholar]
- 18.Murata T, Kageyama Y, Hirose S, Ueda H. Mol Cell Biol. 1996;16:6509–6515. doi: 10.1128/mcb.16.11.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueda H, Hirose S. Nucleic Acids Res. 1990;18:7229–7234. doi: 10.1093/nar/18.24.7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun G-C, Hirose S, Ueda H. Dev Biol. 1994;162:426–437. doi: 10.1006/dbio.1994.1099. [DOI] [PubMed] [Google Scholar]
- 21.Li F-Q, Ueda H, Hirose S. Mol Cell Biol. 1994;14:3013–3021. doi: 10.1128/mcb.14.5.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F-Q, Takemaru K, Goto M, Ueda H, Handa H, Hirose S. Genes Cells. 1997;2:143–153. doi: 10.1046/j.1365-2443.1997.1090306.x. [DOI] [PubMed] [Google Scholar]
- 23.Fischer S G. Methods Enzymol. 1983;100:424–430. doi: 10.1016/0076-6879(83)00071-3. [DOI] [PubMed] [Google Scholar]
- 24.Ohta T, Kobayashi M, Hirose S. J Biol Chem. 1995;270:15571–15575. doi: 10.1074/jbc.270.26.15571. [DOI] [PubMed] [Google Scholar]
- 25.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B-L, Langer J A, Schwartz B, Pestka S. Proc Natl Acad Sci USA. 1989;86:558–562. doi: 10.1073/pnas.86.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueda H, Sun G-G, Murata T, Hirose S. Mol Cell Biol. 1992;12:5667–5672. doi: 10.1128/mcb.12.12.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Schägger H, von Jagow G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 30.Perini G, Wagner S, Green M R. Nature (London) 1995;376:602–605. doi: 10.1038/376602a0. [DOI] [PubMed] [Google Scholar]
- 31.Baranger A M, Palmer C P, Hamm M K, Giebler M R, Brauweiler A, Nyborg J K, Schepatz A. Nature (London) 1995;376:606–608. doi: 10.1038/376606a0. [DOI] [PubMed] [Google Scholar]
- 32.Ohno C, Ueda H, Petkovich M. Mol Cell Biol. 1994;14:3166–3175. doi: 10.1128/mcb.14.5.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayer S, Walker N, Mosammaparast M, Nelson J P, Shilo B-Z, Benyajati C. Nucleic Acids Res. 1993;7:1619–1627. doi: 10.1093/nar/21.7.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guichet A, Copeland J W R, Erdelyi M, Hlousek D, Zavorsky P, Ho J, Brown S, Percival-Smith A, Krause H M, Ephrussi A. Nature (London) 1997;385:548–552. doi: 10.1038/385548a0. [DOI] [PubMed] [Google Scholar]
- 35.Yu Y, Li W, Su K, Yussa M, Han W, Perrimon N, Pick L. Nature (London) 1997;385:552–555. doi: 10.1038/385552a0. [DOI] [PubMed] [Google Scholar]