

Abstract
Major histocompatibility complex (MHC) class II molecules displayed clustered patterns at the surfaces of T (HUT-102B2) and B (JY) lymphoma cells characterized by interreceptor distances in the micrometer range as detected by scanning force microscopy of immunogold-labeled antigens. Electron microscopy revealed that a fraction of the MHC class II molecules was also heteroclustered with MHC class I antigens at the same hierarchical level as described by the scanning force microscopy data, after specifically and sequentially labeling the antigens with 30- and 15-nm immunogold beads. On JY cells the estimated fraction of co-clustered HLA II was 0.61, whereas that of the HLA I was 0.24. Clusterization of the antigens was detected by the deviation of their spatial distribution from the Poissonian distribution representing the random case. Fluorescence resonance energy transfer measurements also confirmed partial co-clustering of the HLA class I and II molecules at another hierarchical level characterized by the 2- to 10-nm Förster distance range and providing fine details of the molecular organization of receptors. The larger-scale topological organization of the MHC class I and II antigens may reflect underlying membrane lipid domains and may fulfill significant functions in cell-to-cell contacts and signal transduction.
Keywords: major histocompatibility complex antigens, receptor patterns, atomic force microscopy, fluorescence resonance energy transfer, electron microscopy
Transmembrane receptors, antigens, and proteins have limited mobility in the plasma membrane, which can be described more precisely by a restricted diffusion model in contrast to the Singer–Nicolson fluid mosaic membrane model assuming complete diffusional freedom for membrane-bound molecules (1, 2). The limited mobility of receptor proteins may arise from extracellular, intramembrane, and intracytosolic connections. Membrane-bound molecules have a tendency to form oligomeric clusters. Accordingly, dynamic receptor co-distribution patterns have been determined by a number of biophysical and biochemical techniques—e.g., fluorescence spectroscopy, electron and scanning force microscopies, chemical crosslinking, and co-isolation methods (3–13). Such oligomeric assemblies have been described in different cellular systems at the surface of fixed and living cells in their native environments for a wide variety of receptors—e.g., among others the oligomeric structure of concanavalin A receptors or transferrin receptors; or the colocalization of interleukin 2 with HLA class I, class II, and intracellular adhesion molecule; that of the insulin receptor with major histocompatibility complex (MHC) molecules; or the supramolecular structure of the tetraspan molecules have been shown (4–10, 14–24).
Apart from multisubunit receptor structures, where subunits must be together before or upon ligand binding, seemingly unrelated proteins are co-associated in many cases and held together in a dynamic cluster, apparently by weak interactions between proteins, or between proteins and lipid moieties in the plasma membrane. Considering the surface density of these membrane components, the probability of a diffusion driven random encounter for the formation and maintenance of oligomeric homo- and/or heteroassociations without assuming cohesive forces is very low (13). Earlier theoretical and experimental considerations even suggested a genetic origin of the membrane lipid and protein mosaicism (9, 25)
The majority of the available experimental methods provides information only on the existence of receptor colocalization, without supplying further details on the degree of hetero- or homoclustering (13, 15, 26, 27). Recently, the combined application of fluorescence spectroscopy with scanning force and electron microscopies has offered a unique method to study receptor codistribution patterns at different hierarchical levels ranging from nanometer to micrometer intermolecular distances (28). Comparing quantitative electron and scanning force microscopic distributions of receptor-bound colloidal immunogold beads and considering receptor densities the degree of hetero- and homoclustering in a population of partially codistributed molecules could be estimated (28).
The present paper shows the nonrandom codistribution patterns of HLA class I and II antigen populations, representing different hierarchical levels in the plasma membrane of lymphocytes. A quantitative approach is used to characterize the number of receptors involved in partial co-clustering at the surface of human B (JY) and T (HUT-102B2) lymphoma lines.
MATERIALS AND METHODS
Cells.
The JY and HUT-102B2 cell lines originally derived from human adult B and T lymphomas, respectively, were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum and antibiotics (3).
Monoclonal Antibodies (mAbs).
Identification of MHC class I and II was carried out by the following antibodies: W6/32 (IgG2a) and KE-2 (IgG2aκ) (kind gifts of F. Brodsky, University of California, San Francisco, and M. Edidin, The Johns Hopkins University, Baltimore, respectively). Both mAbs bind to the heavy chains of HLA class I molecules. L-368 (IgG1κ) and HB28 (IgG2b) directed against β2-microglobulin (β2m) were also provided by F. Brodsky and M. Edidin, respectively. L243 mAb (IgG2a) against HLA class II (DR) molecules was provided by F. Brodsky. Antibodies were labeled with fluorescein isothiocyanate (FITC) and tetramethylrhodamine isothiocyanate (TRITC) (Molecular Probes). The dye to protein labeling ratios varied between 2 and 4 for the different aliquots of the antibodies as determined by spectrophotometry (26). The fluorescently tagged antibodies retained their biological activity in binding to cell membranes of living cells as determined in competition experiments using unlabeled antibodies. Fab fragments of the above antibodies were prepared from the mAb by papain digestion and fluorescently labeled as the whole antibodies (29).
Labeling Procedures.
Freshly harvested cells were labeled before or after gentle fixation with 0.5% paraformaldehyde for 1 hr at 4°C. After washing the cell suspension twice in phosphate-buffered saline (PBS, pH 7.4), the cell pellet was resuspended in 100 μl of PBS and incubated with 100 μg/ml of FITC- or TRITC-conjugated mAb for 40 min at 4°C.
Using FITC-conjugated antibodies as first antibodies before immunogold labeling was useful for checking antibody binding. Labeled cells were washed twice in cold PBS and fixed at this point of preparation. For scanning force microscopic (SFM) and transmission electron microscopic (TEM) measurements the cell suspension was further incubated with 10 μl of Aurogamig G-30 (Amersham; diameter, 30 ± 3 nm) or 10 μl of gold-labeled goat-anti-mouse-Ig (Sigma Aldrich; diameter, 15 μm ± 2 nm) carrying polyclonal goat anti-mouse antibodies directed against IgG Fc and the whole molecule, respectively. In some experiments we used Aurogamig G-15 (Amersham; diameter, 15 ± 1 nm) against IgG Fc. After 40 min incubation on ice, the cells were washed and stored at 4°C (28). The excess valencies of the gold beads were neutralized by adding mouse IgG in large quantity before applying the second mAb and the second gold beads. The possibility of artifactual clustering of receptors by the labeling procedure with bivalent antibodies was excluded by testing Fab fragments of every applied antibody. Furthermore, fixing the cells in 0.5% paraformaldehyde before or after the labeling did not influence the results.
Flow Cytometric Energy Transfer Measurements.
Donor and acceptor labeled mAbs carried known amounts of labeling dyes and were targeted to MHC class I and class II molecules. Measurements were carried out in a modified two laser beam Becton Dickinson FACStarPlus flow cytometer (3). Cells were illuminated at 488 and 514 nm sequentially, and the respective emission data were collected at 540 and >590 nm. Fluorescence was gated on forward angle light scatter to avoid artifacts from cell debris. Correction parameters (for spectral properties of the dyes and for instrumental factors) were calculated from data obtained with fluorescently single-labeled cells (27). Energy transfer efficiency was expressed as percentage of the excitation energy taken up by the donor (FITC) and tunneled to the acceptor (TRITC) molecules (17, 27, 30). Transfer efficiency has an inverse sixth power dependence on the donor-acceptor distance therefore the calculated efficiency is extremely sensitive to distance changes in the range of 2–10 nm (15). Two-dimensional receptor distribution maps were created using the assumption that larger energy transfer values represent a closer proximity of the investigated donor and acceptor labeled receptors, and vice versa (3, 16). Differences in actual receptor densities and dye to protein (mAb) labeling ratios were taken into account by a normalization procedure.
SFM.
A costume-made stand-alone-type atomic force microscope (Applied Optics Group, University of Twente, Twente, The Netherlands) (31) was used in combination with a Zeiss Axiovert 135 microscope in constant force or tapping modes with samples in air or under liquid, respectively. Cantilevers with Si3N4 integrated pyramidal tip with 10 and 30 nm diameters were used with an average spring constant around 0.06 N/m. Different sample surface dimensions were scanned in the different directions: 20 × 20 to 2 × 2 μm (x, y) and 4 μm (z). Scan rate was 2 lines/sec. Images of 500 × 500 pixels were collected and processed with a software including plane fitting and flattening, as well as crosssectional analysis of the sample surface for correct identification of the 30- and 15-nm diameter gold beads.
Electron Microscopy.
TEM was performed by a Jeol 100 B instrument operated at 80 kV. After proper labeling with first and second antibodies (the latter conjugated either to 15- or 30-nm diameter colloidal gold beads) the cells were fixed with 1% glutaraldehyde containing 0.1 M sodium cacodylate buffer (pH 7.2) for 1 hr on ice. The cells were subsequently washed in 0.15 M cacodylate buffer and postfixed in 1% osmium tetroxide (buffered in the same sodium cacodylate buffer) for 1 hr at 4°C. After rinsing with buffer, the cells were layered on a wet Millipore filter with 0.45-μm pore size. After overlaying with agar (1.5% in water, at 40°C) blocks were trimmed and dehydrated with ascending concentrations of ethanol (20–95%). The specimen was transferred to propylene oxide and embedded in Epon resin. One hundred-nanometer-thick slices were cut and negatively stained with 1% lead citrate and 1% uranyl acetate. Alternatively, when the surfaces of whole cells were investigated, cells were washed and sedimented onto poly-l-lysine-treated formvar/carbon coated grids. The postfixation with OsO4 was omitted. After dehydration in graded ethanol series, the cell-packed grids were air dried from ascending ether/ethanol mixtures.
Immunogold Particle Analysis.
HLA class I and II antigens of JY and HUT-102B2 cells were labeled with mAbs as defined above. Gold beads of different size, carrying anti-mouse polyclonal antibody-coating, were added after the monoclonal antibody treatments. Fig. 1 shows the main steps of the labeling procedure. In both cases the general sequence of the labeling was as follows: labeling receptor-1 (HLA class II) with first mAb (Fig. 1A); labeling with second (polyclonal) antibody carried by gold bead no. 1 (the anti-Fc antibody is not indicated; the grooves represent the binding sites for the Fc part of the first mAb) (Fig. 1B); labeling with the next mAb (directed to HLA class I) (Fig. 1C); and addition of gold bead no. 2 (Fig. 1D). Each labeling step was followed by washing with PBS. A similar type of serial washing with PBS only did not result in a detectable loss of colloidal gold beads. For some of the measurements the sequence of labeling started with the 15-nm beads. If the step in Fig. 1C (the second mAb labeling) was omitted, no binding of bead no. 2 was observed.
Figure 1.
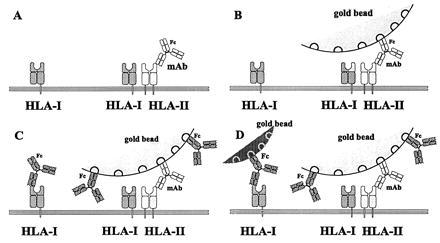
Schematic view of the steps of sequential labeling of the cell surface elements. (A) First specific labeling against antigens (HLA II) with mAb. (B) Larger gold beads attached to the Fc part of the antibody. (C) Second specific labeling of antigens (HLA I). These mAbs also cover the free binding sites of the gold beads. (D) Smaller size gold beads bind the Fc part of the second antibody.
The labeled samples were further processed for electron microscopy or scanning force measurements as described in the corresponding paragraphs. The basic assumption of the method is that those cell surface molecules that remain accessible to the second label are not within the heteroclusters of the two different antigens. The cell surface area under the “umbrella” provided by the 30-nm gold bead size is supposed to accommodate larger receptor clusters as well. Using the smaller size, 15-nm diameter beads may not absolutely be perfect because homoclusters of the second antigen might be obscured, but their easier observation justifies their use. The gold particles that were conjugated with polyclonal second antibodies could not induce clustering of the receptors, since the receptor co-clustering was observed among far distinct gold beads at a micrometer level. The distances between individual beads excluded the possibility of clustering caused by the antibodies since their distances were far beyond the size of the antibodies. This organization of cell surface receptors is called the nonrandom co-clustering at a higher hierarchical level.
Calculation of the Ratio of Homo- or Heteroclustered Receptors.
Labeling cell surface antigens of type A with the larger gold beads, and subsequently type B antigens with the smaller ones, the number of different beads can be counted in an area of SFM or TEM picture. Let us define NA and NB as the number of gold beads bound to type A and B antigens on a selected part of the cell surface, respectively. The ratio of NA and NB can be expressed as follows:
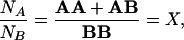 |
1 |
where AA means the number of single molecules and homo dimers of molecule type A, covered by the larger gold beads. AB refers to the number of A molecules being in the molecular vicinity of B. This fraction of the antigens of type B are supposed to be covered by the gold beads directed against antigen A. BB is the number of the smaller beads attached to single molecules or homo dimers of antigen B. This fraction of antigen B is not co-clustered with antigen A. Ratio X can easily be determined from TEM and SFM measurements counting several areas and averaging the number of the beads.
For other experiments, when the sequence of labeling was reversed—i.e., first antigen B was labeled with the larger colloidal particles and subsequently antigen A with the smaller ones, the following labeling ratio can be determined:
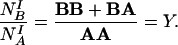 |
2 |
A normalized ratio (Z) of the total number of the two receptors can be determined from fluorescence intensity measurements.
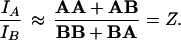 |
3 |
Using simple algebra X, Y, and the ratio of the total number of antigens (Z), the ratio of the non-(hetero)clustered ones, and the total number of the antigens can be calculated for both type A and B antigens:
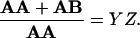 |
4 |
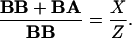 |
5 |
These equations describe the simplest case for the analysis of heteroclustering of two different kinds of receptors having comparably high (≈106) receptor numbers in the plasma membrane. Another and necessary control for the validity of the above equations is the addition of the second colloidal gold bead after the first one had been conjugated to the surface. The second gold bead should not be visible without the administration of the second mAb, since the first gold bead population is supposed to mask all available first antibodies.
In principle, the actual method of determination of the total number of the receptors should not influence the calculation. A relatively high receptor density is required to determine the relative number of the distinct (30- and 15-nm diameter) gold beads with high enough accuracy from a number of selected and representative areas of the cell membrane, because determination of their total number at the whole plasma membrane of a cell is not trivial, for technical reasons.
Quantitative Assessment of the Distribution Patterns of the Gold Markers on TEM and SFM Images.
Assuming a random distribution of the antigens on the cell surface, the corresponding spatial distribution of the gold bead labels should be Poissonian. For the purpose of analysis the images were divided into “cells” putting an equally spaced grid on them. The number of the gold beads were counted in each “cell” and the observed frequency distribution of the immunogold beads was plotted. The observed frequencies were compared with both the expected Poissonian distribution based on cell size and the total number of the gold beads, as well as to the Poissonian distribution leading to the best fit of the experimental data. Comparing the experimental frequencies to the expected and best fit Poissonian distributions one can conclude on the random or nonrandom behavior of the receptor distributions.
RESULTS AND DISCUSSION
Distribution of Immunogold Particles Directed to MHC Class I and II Antigens.
Fig. 2 shows TEM images of the surface of a JY cell, sequentially double labeled for HLA class II DR and HLA class I molecules, with 30- and 15-nm gold beads, respectively (Fig. 2A). Fig. 2B is an image of a similarly prepared sample where the sequence of the labeling was reversed. Note the difference in the ratio of the small (15 nm) and large (30 nm) beads. The total number of the HLA class I molecules exceeded that of the HLA class II by 49% as determined by fluorescence intensity measurements in a flow cytometer (Table 1). The values of X, Y, and Z could be obtained from experiments utilizing different sequence of gold bead labeling and fluorescence labeling of the antigens. The ratios characterizing the clustered number of HLA class I (A) and class II (B) molecules were calculated according to Eqs. 4 and 5. Although the surface density of the bound beads had slight sample-to-sample variation, the factors of clustered receptors were practically identical. The following values were obtained for JY cells: X ≈ 2.5; Y ≈ 0.8; Z ≈ 1.55 and the clustered ratios were:
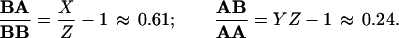 |
6 |
Figure 2.

TEM images of immunogold labeled JY cells. Labeling was done first with 30-nm gold beads against MHC class II followed by 15-nm gold particles against class I molecules (A). The same labeling procedure was applied in reversed order (B). (Bars = 500 nm.)
Table 1.
Relative antigen densities on the surfaces of the HUT-102B2 T and JY B lymphoma lines as expressed in fractions of the number density of the HLA class I antigen on the JY cells
| Antigen | Relative surface densities, %
|
|
|---|---|---|
| HUT-102B2 | JY | |
| HLA-A, -B, -C | 100 ± 10 | 100 ± 7 |
| β2m | 74 ± 10 | 69 ± 14 |
| HLA-DR | 25 ± 4 | 67 ± 5 |
| HLA-DQ | 10 ± 1 | |
Relative surface densities of antigens were determined from the mean fluorescence intensities of flow cytometric histograms, taking into account the dye-to-protein ratios of the respective mAbs and the size of the cells. Cell diameters varied between 10 and 16 μm and 20 and 25 μm for HUT-102B2 and JY cells, respectively. The W6/32 antibody raised against HLA I recognized 1.6–2.0 million binding sites per cell on JY cells, and this was taken as 100%. The cells were in logarithmic phase. Data represent mean ± SEM of at least six independent measurements.
Fluorescence Energy Transfer Results.
The fluorescence energy transfer data of Table 2 confirm the (partial) co-clustering of HLA class I and II molecules on JY (B) and HUT 102B2 (T) cells. The relatively large energy transfer value between the heavy chain of the HLA class I and the HLA class II indicates that a significant part of the two different molecule species are within the Förster distance (i.e., 2–10 nm). These data, however, do not allow us to determine the number of co-clustered receptors. Epitope density measurements, based on fluorescence intensity data, when fluorescein conjugated mAbs were attached to the epitopes in separate aliquots, gave 1:1.49 and 1:4 density ratios of the two cell surface molecules for the JY and the HUT-102B2 cells, respectively.
Table 2.
Energy transfer efficiency values between donor-acceptor pairs bound to HLA class I and HLA class II molecules
| Donor (FITC)
|
Acceptor (TRITC)
|
E ± ΔE, % (JY B) | E ± ΔE, % (HUT-102) | ||
|---|---|---|---|---|---|
| mAb | Antigen | mAb | Antigen | ||
| Part A | |||||
| L-368 | β2m | W6/32 | HLA-A, -B, -C | 13.1 ± 0.8 | 13.1 ± 0.7 |
| W6/32 | HLA-A, -B, -C | L-368 | β2m | 9.8 ± 2.3 | 9.9 ± 0.7 |
| Part B | |||||
| L243 | DR | W6/32 | HLA-A, -B, -C | 13.0 ± 0.8 | 11.9 ± 1.1 |
| L243 | DR | L-368 | β2m | 7.1 ± 1.8 | 3.8 ± 1.8 |
| W6/32 | HLA-A, -B, -C | L243 | DR | 9.2 ± 1.6 | 9.4 ± 0.4 |
| L-368 | β2m | L243 | DR | 5.5 ± 0.5 | −0.6 ± 0.7 |
| Part C | |||||
| W6/32 | HLA-A, -B, -C | W6/32 | HLA-A, -B, -C | 10.7 ± 1.4 | 12.9 ± 0.5 |
| L-368 | β2m | L-368 | β2m | 8.5 ± 0.8 | 9.6 ± 1.2 |
| L243 | DR | L243 | DR | 6.3 ± 0.8 | 5.4 ± 1.4 |
Energy transfer (E) values between FITC-labeled donor and TRITC-labeled acceptor mAbs are shown for JY and HUT-102B2 cells. Data represent mean ± SEM of at least four independent experiments. This designation means simultaneous labeling with the mAbs listed. In the case of competing mAbs, care was taken that half of the available binding sites be occupied by the TRITC-labeled antibody.
The two-dimensional map of the HLA class I and II epitopes, based on the energy transfer data of Table 2 shows that the heavy chain of the HLA class I is close to the epitope determined by the L243 mAb in the HLA class II molecules. The analysis of the homoclustering data indicated oligomeric association of both HLA class I and II molecules. Because two different types of antibodies were available for labeling HLA class I molecules, we were able to determine in accord with our earlier observations, that in their homoclusters the HLA class I molecules were oriented with an angle, relative to each other. The heavy chains were consistently closer to each other than the β2m moieties (Fig. 3). Because these conclusions were derived by averaging data which had been collected on a cell-by-cell basis, it is fair to claim that the above orientations should be prevalent among the possible molecular orientations.
Figure 3.

Two-dimensional schematic model of the proximity relationships among HLA class I and class II molecules on the surface of JY cells as predicted by the fluorescence energy transfer data of Table 2. The complexes shown indicate a possible topographical arrangement of the receptors. (Inset) Schematic vertical model of immunogold bead attachment to the HLA class II. The bead covers the co-clustered class I molecule(s) as predicted by energy transfer using site-specific labels. The quantity of the heteroclusters shown on the picture is calculated as BA in the equations.
Atomic Force Microscopy.
Fig. 4 shows the scanning force picture of part of the surface of an air-dried JY cell previously double labeled with W6/32 and L-368 mAbs binding to HLA class I antigen heavy and light (β2m) chains, respectively, and visualized with colloidal gold-conjugated secondary antibodies. Similar distribution data were obtained by tapping (oscillation) mode under buffered conditions. The nonrandom distribution of HLA class II antigens in the plasma membrane, similarly to that of HLA class I, was also confirmed by statistical analysis, as described below. The presented atomic force and electron microscopic data support our view that MHC class II antigens form homoclusters not only in the nanometer range attainable by fluorescence spectroscopy, but also at a higher hierarchical level, in the micrometer distance range. The resolution power of our earlier investigations on the co-clustering of these antigens was limited by the lateral dimensions covered by fluorescence resonance energy transfer, which falls roughly between 2 and 10 nm depending on the applied fluorescence donor–acceptor pair. This represents a rather close molecular vicinity taking into account the molecular dimensions of MHC molecules. Crystallographic data support an approximately 4 × 5 × 7 nm three-dimensional structure for MHC molecules in the plasma membrane (32). Atomic force and electron microscopy can cover a higher hierarchical level if gold beads of the size of several nm are applied and targeted to the mAb-labeled MHC receptors by second antibodies (Fig. 4). This higher hierarchical level of co-clustering simply means that a colloidal gold bead is large enough to mask a whole “receptor-island” of receptor homo- or heteroclusters built from quite a few number of molecules. Nonrandom co-clustering of the gold beads may also indicate a clustering of the clusters—i.e., the small receptor-islands in the lipid domains—which may be organized into receptor-island groups.
Figure 4.

High-resolution SFM image of the membrane surface of an air dried, immunogold labeled JY cell. The picture shows a 2 μm × 2 μm area. Sequential labeling of the HLA class II and class I molecules was done by 30- and 15-nm immunogold particles, respectively. (Inset) Direct height (z) measurement of the immunogold beads. The measured z values along the indicated trajectory (thin horizontal line, upper left quarter) clearly demonstrate the presence of the two distinct gold bead sizes on the surface of the cell. (Bar = 500 nm.)
TEM pictures are suitable targets for such investigations when measured and expected Poissonian distributions of the gold beads of the electron micrographs can be compared with determine the random or nonrandom behavior of the antigen distribution. In Fig. 5 the quantitative analysis of the distribution patterns of the immunogold markers (Inset) bound to HLA II antigens in the plasma membrane of a JY cell is displayed. The assessment of nonrandomness of the immunogold labels is based on a comparison of the experimentally observed data with both the expected and the best-fit Poissonian distribution curves. The observed positive deviation of the experimental points from both theoretical curves at higher bead numbers is a direct proof of the clustering of the underlying receptor structures at a higher hierarchical level as defined earlier. Similar results were obtained if we changed the relative size of the grids.
Figure 5.

Analysis of the nonrandom spatial distribution of immunogold labeled antigens on the surface of a JY cell. •, The measured distribution of beads among the unit cells of the grid; ○, the expected Poissonian distribution calculated using the theoretical mean. The dotted line represents the best Poissonian fit. Note the significant deviations at the right side of the curve, meaning nonstochastic spatial distributions of the clusters. (Inset) Selected area of the membrane surface of the cell used for the analysis showing the characteristic distribution of the immunogold particles. The lines indicate a possible grid layout for the analysis.
The decisive measurement of the receptor distribution, which may exclude most of the possible artifacts during sample preparation, was the atomic force detection of the similarly distributed colloidal gold beads over identical samples under buffered (i.e., under close to physiological) conditions.
A causal relationship of the observed HLA patterns to the lipid composition was assumed, as Edidin and coworkers (33–36) presented several examples by lateral diffusion measurements that comobilities and partial recoveries of fluorescence intensity can also be explained by the restricted mobility of protein molecules within the lipid domains in the plasma membranes of different cells. Recently, Jacobson et al. (37) and Feder et al. (1) presented and also reviewed data, which call for a new model of cell membrane structure. A new model is supposed to tackle the great variety of distinct receptor and other superstructures of the plasma membrane with a high number of individualized characteristics of the molecular constituents.
The biological importance of the above described receptor superstructures may be assumed in cell-to-cell communication—e.g., cytotoxic interaction and receptor crosstalk in transmembrane signal transduction and cell activation processes.
Acknowledgments
We thank Drs. T. M. Jovin and G. Vereb for helpful advice. This work was supported by the Hungarian Academy of Sciences [OTKA Grants T F 020102 (A.J.), T 023835 (L.M.), 014655 (R.G.), T 023873, and T 17592 (S.D.) from the National Research Fund, Hungary and Special Award AKP 96-316/54] and by the Ministry of Public Health and Welfare (Grant ETT 477).
ABBREVIATIONS
- MHC
major histocompatibility complex
- TEM
transmission electron microscopy
- SFM
scanning force microscopy
- FITC
fluorescein isothiocyanate
- TRITC
tetramethylrhodamine isothiocyanate
- β2m
β2-microglobulin
References
- 1.Feder T J, Brust-Mascher I, Slattery J P, Baird B, Webb W W. Biophys J. 1996;70:2767–2773. doi: 10.1016/S0006-3495(96)79846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singer S J, Nicolson G L. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 3.Bene L, Balázs M, Matkó J, Möst J, Dierich M, Szöllősi J, Damjanovich S. Eur J Immunol. 1994;24:2115–2123. doi: 10.1002/eji.1830240928. [DOI] [PubMed] [Google Scholar]
- 4.Angelisova P, Hilgert I, Horejsi V. Immunogenetics. 1994;39:249–256. doi: 10.1007/BF00188787. [DOI] [PubMed] [Google Scholar]
- 5.Clegg R M. Curr Opin Biotechnol. 1995;6:103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 6.Clegg R M. In: Fluorescence Imaging Spectroscopy and Microscopy, Chemical Analysis Series. Fengwang X, Hermann B, editors. Vol. 137. New York: Wiley; 1995. pp. 179–252. [Google Scholar]
- 7.Dale R E, Eisinger J, Blumberg W E. Biophys J. 1979;26:161–194. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale R E, Novros J, Roth S, Edidin M, Brand L. In: Fluorescent Probes. Beddard G S, West M A, editors. London: Academic; 1981. pp. 159–181. [Google Scholar]
- 9.Damjanovich S, Somogyi B, Trón L. Adv Physiol Sci. 1981;30:9–21. [Google Scholar]
- 10.Damjanovich S, Trón L, Szöllősi J, Zidovetzki R, Vaz W C L, Regaterio F, Arndt-Jovin D J, Jovin T M. Proc Natl Acad Sci USA. 1983;80:5985–5989. doi: 10.1073/pnas.80.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damjanovich S, Pieri C. Ann NY Acad Sci. 1991;621:29–39. doi: 10.1111/j.1749-6632.1991.tb16966.x. [DOI] [PubMed] [Google Scholar]
- 12.Damjanovich S, Szöllősi J, Trón L. Immunol Today. 1992;13:A12–A15. doi: 10.1016/0167-5699(92)90058-F. [DOI] [PubMed] [Google Scholar]
- 13.Damjanovich S. In: Mobility and Proximity in Biological Membranes. Damjanovich S, Edidin M, Szöllősi F, Trón L, editors. Boca Raton, FL: CRC; 1994. pp. 225–326. [Google Scholar]
- 14.Chan S S, Arndt-Jovin D J, Jovin T M. J Histochem Cytochem. 1979;27:56–64. doi: 10.1177/27.1.374620. [DOI] [PubMed] [Google Scholar]
- 15.Matkó J, Szöllősi J, Trón L, Damjanovich S. Q Rev Biophys. 1988;21:479–544. doi: 10.1017/s0033583500004637. [DOI] [PubMed] [Google Scholar]
- 16.Mátyus L, Bene L, Heilligen H, Raus J, Damjanovich S. Immunol Lett. 1995;44:203–208. doi: 10.1016/0165-2478(94)00215-d. [DOI] [PubMed] [Google Scholar]
- 17.Vereb Gy, Dale R E, Mátyus L, Bene L, Panyi Gy, Bacsó Zs, Balázs M, Matkó J, Pieri C, Ameloot M, Szöllősi J, Gáspár R, Jr, Damjanovich S. J Mol Recognit. 1995;8:237–346. doi: 10.1002/jmr.300080402. [DOI] [PubMed] [Google Scholar]
- 18.Mittler R S, Goldman S J, Spitalny G L, Burakoff S J. Proc Natl Acad Sci USA. 1989;86:8531–8535. doi: 10.1073/pnas.86.21.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabó G, Jr, Weaver J L, Pine S P, Rao P E, Aszalós A. Biophys J. 1995;68:1170–1176. doi: 10.1016/S0006-3495(95)80293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liegler T, Szöllősi J, Hyun W, Goodenow R S. Proc Natl Acad Sci USA. 1991;88:6755–6759. doi: 10.1073/pnas.88.15.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakihama T, Smolyar A, Reinherz E L. Proc Natl Acad Sci USA. 1995;92:6444–6448. doi: 10.1073/pnas.92.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakrabarti A, Matkó J, Rahaman N, Barisas G B, Edidin M. Biochemistry. 1992;31:7182–7189. doi: 10.1021/bi00146a022. [DOI] [PubMed] [Google Scholar]
- 23.Matkó J, Bushkin Y, Wei T, Edidin M. J Immunol. 1994;152:3353–3360. [PubMed] [Google Scholar]
- 24.Szöllősi J, Horejsi V, Angelisova P, Bene L, Damjanovich S. J Immunol. 1996;157:2939–2946. [PubMed] [Google Scholar]
- 25.Satir B. J Supramol Struct. 1976;5:381–389. doi: 10.1002/jss.400050310. [DOI] [PubMed] [Google Scholar]
- 26.Szöllősi J, Damjanovich S, Goldman C K, Fulwyler M J, Aszalós A A, Goldstein G, Rao P, Talle M A, Waldmann T A. Proc Natl Acad Sci USA. 1987;84:7246–7250. doi: 10.1073/pnas.84.20.7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trón L, Szöllősi J, Damjanovich S, Helliwell S H, Arndt-Jovin D J, Jovin T M. Biophys J. 1984;45:939–946. doi: 10.1016/S0006-3495(84)84240-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damjanovich S, Vereb G, Shaper A, Jenei A, Matkó J, Starink J P P, Fox G O, Arndt-Jovin D J, Jovin T M. Proc Natl Acad Sci USA. 1995;92:1122–1126. doi: 10.1073/pnas.92.4.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edidin M, Wei T. J Cell Biol. 1982;95:458–465. doi: 10.1083/jcb.95.2.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szöllősi J, Damjanovich S, Mulhern S A, Trón L. Prog Biophys Mol Biol. 1987;49:65–87. doi: 10.1016/0079-6107(87)90009-5. [DOI] [PubMed] [Google Scholar]
- 31.Van der Werf K O, Putman C A J, De Grooth B D, Segerink F B, Schipper E H, Van Hulst N, Greve J. Rev Sci Instrum. 1993;64:2892–2897. [Google Scholar]
- 32.Guo H-C, Jardetzky T S, Garrett T P J, Lane W S, Strominger J L, Wiley D C. Nature (London) 1992;360:364–366. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- 33.Edidin M, Aszalós A, Damjanovich S, Waldmann T M. J Immunol. 1988;141:1205–1210. [PubMed] [Google Scholar]
- 34.Edidin M, Kuo S C, Sheetz M P. Science. 1991;254:1379–1382. doi: 10.1126/science.1835798. [DOI] [PubMed] [Google Scholar]
- 35.Edidin M. In: Mobility and Proximity in Biological Membranes. Damjanovich S, Edidin M, Szöllősi F, Trón L, editors. Boca Raton, FL: CRC; 1994. pp. 109–135. [Google Scholar]
- 36.Edidin M, Zuniga M C, Sheetz M. Proc Natl Acad Sci USA. 1994;91:3378–3382. doi: 10.1073/pnas.91.8.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson K, Sheetz E D, Simson R. Science. 1995;268:1441–1442. doi: 10.1126/science.7770769. [DOI] [PubMed] [Google Scholar]