

Abstract
Poly(ADP-ribose) polymerase [PARP; NAD+ ADP-ribosyltransferase; NAD+: poly(adenosine-diphosphate-d-ribosyl)-acceptor ADP-d-ribosyltransferase, EC 2.4.2.30] is a zinc-finger DNA-binding protein that detects specifically DNA strand breaks generated by genotoxic agents. To determine its biological function, we have inactivated both alleles by gene targeting in mice. Treatment of PARP−/− mice either by the alkylating agent N-methyl-N-nitrosourea (MNU) or by γ-irradiation revealed an extreme sensitivity and a high genomic instability to both agents. Following whole body γ-irradiation (8 Gy) mutant mice died rapidly from acute radiation toxicity to the small intestine. Mice-derived PARP−/− cells displayed a high sensitivity to MNU exposure: a G2/M arrest in mouse embryonic fibroblasts and a rapid apoptotic response and a p53 accumulation were observed in splenocytes. Altogether these results demonstrate that PARP is a survival factor playing an essential and positive role during DNA damage recovery.
Keywords: cellular response to DNA damage, γ-rays, alkylating agents, G2 arrest, apoptosis
To protect their genome from the deleterious consequences of accumulation of unrepaired or misrepaired lesions, cells have developed an intricate DNA damage surveillance network. Through its function as a single-stranded breaks detector, poly(ADP-ribose) polymerase [PARP; NAD+ ADP-ribosyltransferase; NAD+: poly(adenosine-diphosphate-d-ribosyl)-acceptor ADP-d-ribosyltransferase, EC 2.4.2.30], a nuclear enzyme, participates to this basic process (1). PARP (113 kDa) has a modular organization (2): a N-terminal DNA-binding domain that acts as a molecular nick-sensor, encompassing two zinc-finger motifs (3) and a bipartite nuclear location signal (4), a central region bearing the auto-poly(ADP-ribosylation) sites which serves to regulate PARP–DNA interactions and a C-terminal catalytic domain involved in the nick-binding dependent poly(ADP-ribose) synthesis (5). The x-ray crystallographic structure of this domain has been recently solved revealing a surprising structural homology between the active site of PARP and that of bacterial mono-ADP-ribosylating toxins despite weak sequence homology (6).
Although the physiological role of PARP is still much debated, recent molecular and genetic approaches including expression of either a dominant-negative mutant (7–10) or antisense (11) have clearly revealed the implication of PARP in the maintenance of the genomic integrity in the base excision repair pathway (7–10, 12). To elucidate its function we disrupted the mouse PARP gene by homologous recombination and exposed the PARP-deficient mice and derived cells to various genotoxins.
MATERIALS AND METHODS
Gene Targeting in Embryonic Stem Cells and Generation of Mice.
Mouse PARP was isolated from a 129SVJ strain genomic library. The targeting vector was constructed using a 9-kb EcoRI fragment extending from intron 2 to 7 by inserting PGK-neo (phosphoglycerate kinase promoter followed by the neo gene) in the BamHI site of the 4th exon and herpes simplex virus thymidine kinase followed by the TK gene (HSV-Tk) in the XhoI site outside the sequence of the targeting vector. Following electroporation, embryonic stem cells were selected in 200 μg⋅ml−1 G418 and 2 mM of gancyclovir. A positive clone microinjected into C57BL/6 blastocysts (13) gave rise to chimaeric offspring, which in turn were mated with C57BL/6. Agouti-coat pups were genotyped by Southern blot analysis of tail DNA for germ-line transmission.
Immunoblots and Activity Blots.
Whole-cell lysates prepared from spleen were separated by 10% SDS/PAGE, transferred to a nitrocellulose membrane, and incubated either with a polyclonal antibody against full-length human PARP (1:2, 500 dilution) and stained with alkaline phosphatase-coupled antibody or with [α-32P]βNAD (nicotinamide adenoside dinucleotide) as described (14).
Chromosomal Stability Measurements.
5-Bromodeoxyuridine tablets were implanted s.c. in mice 32 h (two cell cycles of bone marrow cells) and colchicine (0.6 mg/kg) was injected 2 h before sacrifice (15). For N-methyl-N-nitrosourea (MNU) treatments peritoneal injections (80 mg/kg) were performed 9 h or 30 h (during one or two S phases), and irradiations were performed with γ-rays from a cobalt 60Co source (dose rate 0.25 Gy/min) 3 or 7 h before sacrifice. Bone marrow cells were then isolated and treated, and sister chromatid exchanges (SCEs) and chromatid breaks were scored as described (16).
Cell Culture and Flow Cytometry.
Embryonical day 13.5 mouse embryonic fibroblasts (MEFs) were grown in DMEM, supplemented with 10% fetal calf serum and antibiotics. For cell cycle, 106 passage-three MEFs were plated in 100-mm dishes and mock-treated or exposed to various doses of MNU (Sigma) at 37°C for 30 min. After 24 h of culture, cells were fixed in 80% ethanol and DNA was stained with 5 μg⋅ml−1 ethidium bromide. Cell-cycle distributions were measured with an Epics Elite (Coulter); at least 10,000 cells were analyzed per data point. Freshly isolated splenocytes from PARP+/+ and PARP−/− mice were plated at 106/ml in DMEM supplemented with 10% fetal calf serum and antibiotics.
Apoptosis and p53 Accumulation.
Splenocytes were treated with 2 mM MNU during 30 min, washed, and plated in fresh medium. Cells were harvested at times indicated, lysed with 20 mM Tris⋅HCl (pH 7.7), 2 mM EDTA, and 0.4% Triton X-100 for 15 min at 4°C, and centrifuged at 13,000 × g to eliminate cell debris and high molecular weight DNA. The supernatant was treated with phenol/chloroform (vol/vol), and the upper phase was precipitated twice by ethanol and analyzed on 1% agarose gel. p53 accumulation was detected by Western blot analysis using the Ab-7 polyclonal antibody (Calbiochem).
RESULTS
Generation of PARP−/− Mice.
PARP was inactivated by homologous recombination in embryonic stem cells from the 129/Sv mouse line by inserting PGK-neo in the fourth exon (Fig. 1A). Gene targeting was confirmed by Southern blot analysis using a 5′ probe, giving 2 positives out of 70 neomycin- and gancyclovir-resistant clones analyzed (data not shown). One heterozygote embryonic stem clone was used to generate mutant mice using standard procedures. Southern blot analysis of DNA from tail biopsies confirmed disruption of both PARP alleles in homozygous mutants (Fig. 1B). Mutant mice were completely devoid of PARP as judged by Western and activity blot analysis (14) using cells isolated from spleen (Fig. 1 C and D) and testis (data not shown).
Figure 1.

Inactivation of PARP by homologous recombination. (A) Scheme of targeting construct (Top), the PARP gene and hybridation probe (Middle), and the targeted allele (Bottom). EcoRI restriction was used to detect the targeted gene as indicated. (B) Southern blot of EcoRI-digested tail DNA from wild-type (wt) (+/+), heterozygous (+/−), and homozygous (−/−) PARP-targeted mice, using the 5′probe. The wt and mutant fragment are 9.6 and 3.3 kb, respectively. (C and D) PARP protein was not expressed and poly(ADP-ribose) activity was not detectable in PARP−/− cells isolated from spleen. B, BamHI; E, EcoRI; X, XhoI; Xb, XbaI; pGK-neo, neomycin-resistance gene driven by the pGK promoter; HSV-Tk, thymidine kinase gene driven by the herpes simplex virus promoter; PARP+/−, heterozygous PARP mutant; PARP−/−, homozygous PARP mutant.
As previously reported (17), mice lacking PARP were viable and fertile and displayed no overtly abnormal phenotype. However, we found that their average litter size (4.5 ± 2.4 pups) was smaller than those of PARP+/+ and PARP+/− mice (7.4 ± 1.9 pups). In adulthood, the weight of the homozygous mice was significantly lower than their wt littermates (PARP+/+, 23.2 ± 3.1 g; PARP−/−, 19 ± 3 g at 6 weeks). In addition, heterozygous crosses yielded 21% of PARP−/− mice (n = 237). These slightly alterated phenotypes indicate that PARP could be considered as a positive factor for everyday life. Neither skin hyperplasia nor obesity were observed in any of these PARP−/− mice in contrast to that reported for an other knock-out of PARP (17).
Sensitivity of PARP−/− Mice to DNA Damaging Agents.
The unique property of PARP to bind to DNA breaks generated by ionizing radiation or methylating agents which trigger the synthesis of poly(ADP-ribose) (18) prompted us to examine the survival of mutant mice following genotoxic stress. MNU, a monofunctional methylating agent (19), was then administrated i.p. to 6-week-old mice at various single doses. Nine PARP−/− and 12 PARP+/+ mice were treated with a single dose of 25 mg/kg body weight. Only one PARP−/− mouse (11%) died within 8 weeks postinjection, while none of the PARP+/+ mice died during 10 weeks of observation (data not shown). In contrast, injection i.p. of a single dose of 75 mg/kg body weight in 4-week-old mice resulted in 100% (19/19) mortality in PARP−/− mice and 43% (9/21) mortality in PARP+/+ mice during 6 weeks of observation (Fig. 2A). Seventy-nine percent of the mutants mice died within the first week postinjection, suggesting that a critical function was rapidly affected.
Figure 2.

Survival of PARP+/+ and PARP−/− mice after i.p. injection of MNU at 75 mg/kg body weight at 6 weeks of age (A), and γ-radiation with 8 Gy at 6–8 weeks of age (B). The percentage of alive mice at the end of a week is plotted against age.
Thirteen PARP−/− and 10 PARP+/+ mice at 6–8 weeks of age were irradiated with 8 Gy of γ-radiation. Half of the wt mice (5/10) died 15 days postirradiation. In contrast, more than half the mice lacking PARP died 4 days postirradiation (8/13), and all mutant mice were dead by 9 days postirradiation (Fig. 2B). Thus, following 8 Gy of irradiation, the half-life of PARP−/− mice (4 days) is comparable to the half-life of the exquisitely irradiation sensitive Atm-deficient mice (20). To determine the cause of this precocious lethality, we irradiated 2 PARP+/+ and 4 PARP−/− mice at 3 months of age with 8 Gy and performed an autopsy at 3 days postirradiation. Thoracic and abdominal organs appeared macroscopically normal with the exceptions of the thymus and spleen, whose sizes were reduced to the same extent in irradiated mutants and wt mice, and of the small intestine. The latter was distended by intraluminal fluid accumulation in all irradiated mutants, but not in the controls. Histological sections through the duodenum revealed a shortening of the villi in irradiated PARP+/+ mice (v in Fig. 3 B and C); however, their epithelial lining which consists in absorptive cells and rare goblet cells (a and g in Fig. 3 E and F) was intact. The intestinal villi of irradiated PARP−/− mice were considerably shortened (v and c in Fig. 3A) and almost all their epithelial cells were undergoing necrosis, as indicated by the swelling and vacuolation of their cytoplasm (Fig. 3D). In contrast, the PARP−/− epithelial cells located within the crypts displayed numerous mitotic figures or resembled normal absorptive cells, indicating that the damaged epithelium had the potential to regenerate (c in Fig. 3A and data not shown). The epithelium of the large intestine of irradiated PARP−/− mice was normal (data not shown). Altogether, these data strongly suggest that death in PARP-deficient mice is caused by dehydration and/or endotoxicosis secondary to the acute toxicity of radiation on the epithelium of the small intestine. Therefore PARP−/− mice are acutely sensitive both to γ-radiation and alkylating agents.
Figure 3.
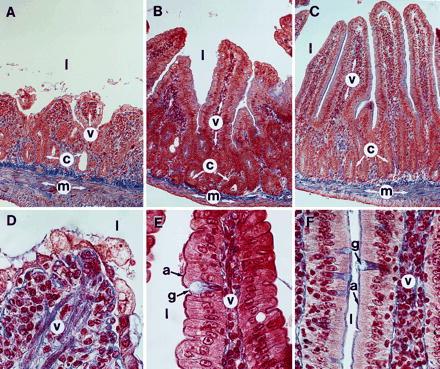
Transverse histological sections through the duodenum of an irradiated PARP−/− mouse (A and D), of an irradiated PARP+/+ mouse (B and E), and of an untreated PARP−/− mouse (C and F). (A–C) Full thickness of the duodenal wall. (D–F) Details of the epithelium near the tips of the villi. Note that the untreated PARP−/− duodenum (C and F) is histologically indistinguishable from its wt counterpart. a, Absorptive cell; c, crypt; g, goblet cell; l, lumen of the small intestine; m, muscularis; v, villi. (A–C, ×170; and D–F, ×750.)
Chromosomal Stability.
Inhibition of PARP activity in cultured cells has demonstrated the importance of PARP in an as yet unspecified step of the resolution of DNA strand breaks and this is reflected by the increase of SCEs following DNA damage (8, 21). An in vivo study of SCE and chromosome breakage was therefore undertaken in PARP null mice. SCEs were analyzed in bone marrow cells of 2-month-old mice 32 h after s.c. implant of 5-bromodeoxyuridine tablets. The rate of SCE was 4-to 5-fold higher in PARP−/− than in PARP +/+ mice (Fig. 3). When MNU (80 mg/kg) was injected during the second S phase (9 h before sacrifice), the rate of SCE in null mutants increased by more than 2-fold. When MNU was present for 30 h during two cell cycles, the rate of SCE increased by about 3-fold in null mutants, reaching very high values (on average exceeding 40 SCE per cell).
Chromosome breakage—i.e., chromatid breaks and chromatid exchanges—were studied in mice with or without 5-bromodeoxyuridine implants. No difference was found. PARP−/− mice had a slightly lower (nonsignificant) rate of chromosome breakage than PARP+/+ mice (Fig. 3). When exposed to MNU during the second S phase, a 33- to 36-fold increase was found in PARP−/− whereas this increase was much lower in PARP+/+ mice. When exposed to γ-rays, the increase of the rate of breakage was 3-fold higher in PARP−/− than in PARP+/+ mice. Both chromatid breaks, as a result of breakage without rejoining and chromatid exchanges, as a result of breakage with abnormal rejoining, were considerably increased, suggesting a serious DNA repair deficiency following exposure to ionizing radiation during G2, and above all S phase, in bone marrow cells from PARP−/− mice.
Cell Cycle Distribution and Apoptosis.
To further analyze the basis for the altered survival of animals to DNA damaging agents, primary fibroblasts were derived from mutant and wt 13.5-day embryos (MEFs). We have previously shown that blocking PARP activity by a dominant negative strategy results in G2/M accumulation and in an increased rate of cell death (8). Therefore, these two abnormalities were investigated in MEF. The progression of MEF through the cell cycle was monitored in vitro, 24 h following exposure to various doses of MNU (Fig. 4A). The relative cell cycle phase distribution in cells was similar in PARP−/− and PARP+/+ MEF in the absence of DNA damage. However, following MNU treatment, PARP−/− MEFs accumulated in G2/M in a dose-dependent manner, indicating that cells lacking PARP failed to resume progression through the cycle.
Figure 4.

Mean number of SCEs per cell in PARP−/− and PARP+/+ mice, before and after exposure to MNU during 9 or 30 h, and mean number of chromatid breaks after exposure to γ-rays 3 or 7 h before harvesting bone marrow cells.
Induction of programmed cell death was demonstrated in PARP−/− splenocytes. Following MNU treatment, PARP−/− splenocytes died apoptotically within 2–6 h and exhibited DNA fragmentation into oligonucleosome ladder (Fig. 5B) while wt splenocytes did not show DNA fragmentation during this time frame. The level of spontaneous apoptosis, probably due to the cell isolation procedure, was also higher in the PARP−/− splenocytes than in the wt (Fig. 5B). DNA flow cytometric analysis confirmed that cells lacking PARP underwent apoptosis as sub-G1 populations were observed (data not shown). The DNA damage-induced p53 accumulation was rapidly detectable in PARP−/− splenocytes but not in wt cells (Fig. 5C), indicating that PARP deficiency did not prevent, but rather led to, an elevated p53 induction presumably associated to a lack and/or delay in DNA repair.
Figure 5.

PARP deficiency affects cell cycle progression and activates the programmed cell death following MNU treatment. (A) Cell cycle progression of primary fibroblasts PARP+/+ and PARP−/− following mock or MNU treatment. (B) Time course induction of apoptosis in splenocytes lacking PARP by 2 mM MNU. (C) p53 accumulation.
DISCUSSION
The present results contrast with the conclusions reached by other authors (17) which did not find any marked defect in mice and in cells lacking PARP. Our data suggest that following genotoxic exposure, PARP has a positive function allowing the cell to receive appropriate survival signals that will enable it to repair its DNA efficiently, thus avoiding genomic rearrangement, prolonged cell cycle arrest and apoptosis. Although PARP appears to be dispensable for normal cellular activity, it should be considered as an essential survival factor for recovery from DNA damage. Cleavage of PARP and other nuclear enzymes acting at strand interruptions like DNA-PK, by interleukin 1β-converting enzyme (ICE) homologues (22, 23) during the execution phase of apoptosis, may be a fundamental feature of the apoptotic process that ensures the rapid irreversibility of cell death induced by saturating levels of DNA damage. In keeping with our previous study using a dominant-negative mutant (8), our data shed light on the importance of PARP activity [and putatively of poly(ADP-ribose) formation] in the cells’ life-or-death balance, but are not compatible with the cell suicide theory by NAD+ depletion and energy consumption (24, 25).
The extreme radiation sensitivity of the small intestine in PARP−/− mice and also in DNA-PK severe combined immunodeficient (SCID) mice (26) underline the cardinal function of DNA screening programs in rapidly proliferating tissues. The role of PARP in the base excision repair pathway is likely to be mediated through its direct or indirect association with repair proteins: XRCC1, DNA polymerase β, and DNA ligase III in a repair complex (M. Masson, and G.d.M., unpublished data). It is tempting to speculate that in the absence of PARP, these proteins are not efficiently recruited at DNA strand breaks and therefore DNA resynthesis and ligation could be compromised leading to chromosomal instability, cell cycle arrest, and cell death. As many cancer treatments kill tumor cells through apoptotic mechanisms, the increased DNA damage induced apoptosis in cells lacking PARP suggests that PARP inhibitors, based on the crystal structure of the PARP catalytic domain (6), could be useful as chemo- and radio-potentiation agents. Mice lacking PARP and derived cell lines represent attractive models to investigate in vivo PARP-mediated DNA repair by base excision, and to evaluate the potentiation of the cytotoxicity of antitumor agents by novel PARP inhibitors as well as their possible side effects.
Acknowledgments
We thank E. Flatter for animal care, and the Centre Paul Strauss (Strasbourg) for radiation experiments. This work was supported by the Centre National de la Recherche Scientifique action concertée “Radiations ionisantes,” by Association pour la Recherche Contre le Cancer, by the Ligue Nationale Contre le Cancer and by Electricité de France. C.T and F.J.O. were supported by a European Union fellowship (Human Capital Mobility) and an Association pour la Recherche Contre le Cancer fellowship, respectively.
ABBREVIATIONS
- PARP
poly(ADP-ribose) polymerase
- PGK
phosphoglycerate kinase
- MNU
N-methyl-N-nitrosourea
- SCE
sister chromatid exchange
- wt
wild type
- MEF
mouse embryonic fibroblast
References
- 1.Althaus F R, Richter C. Mol Biol Biochem Biophys. 1987;37:1–125. [PubMed] [Google Scholar]
- 2.Ménissier de Murcia J, de Murcia G. Trends Biochem. 1994;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 3.Ménissier de Murcia J, Molinete M, Gradwohl G, Simonin F, de Murcia G. J Mol Biol. 1989;210:229–233. doi: 10.1016/0022-2836(89)90302-1. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber V, Molinete M, Boeuf H, de Murcia G, Ménissier de Murcia J. EMBO J. 1992;11:3263–3269. doi: 10.1002/j.1460-2075.1992.tb05404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonin F, Ménissier de Murcia J, Poch O, Muller S, Gradwohl G, Molinete M, Penning C, Keith G, Murcia G. J Biol Chem. 1990;265:19249–19256. [PubMed] [Google Scholar]
- 6.Ruf A, Ménissier-de Murcia J, de Murcia G, Schulz G. Proc Natl Acad Sci USA. 1996;93:7481–7485. doi: 10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molinete M, Vermeulen W, Bürkle A, Ménissier de Murcia J, Küpper J H, Hoeijmakers J H J, de Murcia G. EMBO J. 1993;12:2109–2117. doi: 10.1002/j.1460-2075.1993.tb05859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreiber V, Hunting D, Trucco C, Gowans B, Grunwald D, de Murcia G, Ménissier-de Murcia J. Proc Natl Acad Sci USA. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Küpper J H, Müller M, Jacobson M K, Tatsumi-Miyajima J, Coyle D, Jacobson E L, Bürkle A. Mol Cell Biol. 1995;15:3154–3163. doi: 10.1128/mcb.15.6.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Küpper J H, Müller M, Bürkle A. Cancer Res. 1996;56:2715–2717. [PubMed] [Google Scholar]
- 11.Ding R, Smulson M. Cancer Res. 1994;54:4627–4634. [PubMed] [Google Scholar]
- 12.Satoh M S, Lindahl T. Nature (London) 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 13.Lufkin T, Dierich A, LeMeur M, Mark M, Chambon P. Cell. 1991;88:1105–1119. doi: 10.1016/0092-8674(91)90034-v. [DOI] [PubMed] [Google Scholar]
- 14.Simonin F, Briand J-P, Muller S, de Murcia G. Anal Biochem. 1991;195:226–231. doi: 10.1016/0003-2697(91)90321-j. [DOI] [PubMed] [Google Scholar]
- 15.Allen J W, Shuler C F, Mendes R W, Latt S A. Cytogenet Cell Genet. 1977;18:231–237. doi: 10.1159/000130765. [DOI] [PubMed] [Google Scholar]
- 16.Ricoul M, Dutrillaux B. Mutat Res. 1991;250:331–335. doi: 10.1016/0027-5107(91)90189-u. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z Q, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner E F. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 18.Shall S. Adv Rad Biol. 1984;11:1–69. [Google Scholar]
- 19.International Agency for Research on Cancer (1978) Some N-Nitroso Compounds, IARC Monographs on the Evaluation of the Carcinogenic Risks of Chemicals to Humans (IARC, Lyon, France), pp. 227–255.
- 20.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley J, Ried T, Tagle D, Wynshaw-Boris A. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 21.Oikawa A, Tohda H, Kanai T, Miwa M, Sugimura T. Biochem Biophys Res Commun. 1980;97:1311–1316. doi: 10.1016/s0006-291x(80)80009-x. [DOI] [PubMed] [Google Scholar]
- 22.Tewari M, Quan L T, O’Rourke K, Desnoyers S, Zeng Z, Beider D R, Poirier G G, Salvesen G S, Dixit V M. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 23.Casciola-Rosen L, Nicholson D, Chong T, Rowan K R, Thornberry N A, Miller D K, Rosen A. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger N A. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- 25.Zhang J, Dawson V L, Dawson T M, Snyder S H. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 26.Biedermann K A, Sun J, Giaccia A J, Tosto L M, Brown M. Proc Natl Acad Sci USA. 1991;88:1394–1397. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]