

Abstract
It was previously assumed that the import of cytoplasmically synthesized precursor proteins into mitochondria occurs through a single structure spanning both outer and inner membranes at contact sites. Based on recent findings, however, the two membranes appear to contain independent translocation elements that reversibly cooperate during protein import. This feature makes it difficult to generate a means of isolating a fully integrated and functional translocation complex. To study these independent translocases in vitro and in vivo, we have constructed a chimeric protein consisting of an N-terminal authentic mitochondrial precursor (delta1-pyrroline-5-carboxylate dehydrogenase) linked, through glutathione S-transferase, to IgG binding domains derived from staphylococcal protein A. This construct becomes trapped en route to the matrix, spanning both outer and inner membranes in such a way that the entire signal-less delta1-pyrroline-5-carboxylate dehydrogenase moiety reaches the matrix, while only the folded protein A domain remains outside. During in vivo import of this precursor, outer and inner membranes of yeast mitochondria become progressively “zippered” together, forming long stretches of close contact. Using this novel intermediate, the outer and inner mitochondrial membrane channels, which normally interact only transiently, can be tightly joined (both in vitro and in vivo), forming a stable association. This suggests a method for isolating the functional translocation complex as a single entity.
While mitochondria manufacture some polypeptides, most (>95%) mitochondrial proteins are encoded by nuclear DNA. These are synthesized on cytoplasmic ribosomes and transported into, or across, one or both mitochondrial membranes to reach their destination in the outer membrane, intermembrane space, inner membrane, or matrix. One approach towards understanding this process has been to dissect import stages using translocation intermediates (for reviews see refs. 1 and 2). For example, a precursor protein en route to the matrix could be trapped in such a way that its amino terminus has already reached the matrix. The signal sequence could then be cleaved by a matrix localized signal peptidase, while the C-terminal domain of the intermediate remains outside the mitochondrion. Such intermediates spanning both mitochondrial membranes have been achieved in several ways: (i) by performing the import reaction at low temperature (3), (ii) by lowering ATP levels to deprive the translocation reaction of needed energy (1, 2), (iii) by prebinding polyclonal antibodies (4) or by covalently attaching a highly cross-linked protein moiety [e.g., bovine pancreatic trypsin inhibitor (BPTI)] to the precursor’s C terminus to “plug up” the translocation machinery (5), and (iv) by using ligands (e.g., methotrexate, biotin) to stabilize the tertiary structure of the preprotein [e.g., dihydrofolate reductase (DHFR), avidin], preventing it from unfolding (6–9), thereby blocking its complete import. These in vitro intermediates have helped define the energy requirement of protein import (3, 10). They have revealed the folding state of precursor proteins as they are imported (6–8) and they have proven useful in identifying components of the mitochondrial import machinery through cross-linking studies (11–14). Nevertheless, a complete and functional inner/outer membrane translocation complex has, thus far, not been isolated by means of the currently available intermediates.
Although the previously described translocation intermediates (spanning both membranes) do not spontaneously detach from the import channels, there is little information regarding the stability with which they hold the two independent channels together. An ideal intermediate should have an effective “plug” on both sides of the two-membrane complex. The need for such a structure is based on two recent findings. First, using a series of chimeric precursors consisting of various lengths of the N terminus derived from an authentic mitochondrial precursor protein fused to DHFR, it has been shown that the translocation is reversible (i.e., the preprotein can “fall out”) unless a critical length of the N terminus has reached the matrix (15), so that the emerging presequence can interact with the matrix-localized Hsp70/TIM44 (TIM44 is a component of the inner membrane channel) complex, trapping the protein inside (16–18).
Second, in vitro evidence suggests that the outer and inner mitochondrial membranes contain separate translocation-competent elements that transiently cooperate during the import process (19–21). Indeed, Horst et al. (22) have shown that antibodies against TOM40 (an outer membrane component) coprecipitate mitochondrial Hsp70/TIM44 only in the presence of a trapped intermediate (with BPTI as the C-terminal block), which spans both membranes. Conversely, antibodies to either mitochondrial Hsp70 or TIM44 coprecipitated TOM40 only when the intermediate was present. However, none of the other TOM and TIM proteins considered to be components of the import channels (2) were coprecipitated with this intermediate.
We reasoned that without an efficient block on the matrix side, it is unlikely that translocation intermediates will remain firmly and simultaneously associated with the outer and inner membrane import elements during the subsequent manipulations required for characterization. The stability of an intermediate spanning both membranes might be significantly increased if most or all of the mature portion of an authentic precursor were imported all the way into the matrix. The authentic protein might then have an opportunity to properly fold (and to oligomerize and/or associate with its functional counterparts) in its normal matrix environment, thereby creating a more efficient block on the matrix side. Such a translocation intermediate, with efficient blocks on both cis and trans sides, should be very stable, since it is “frozen” within the import machinery. Inserting a long enough spacer between the mitochondrial preprotein and the C-terminal blocking domain would be a requirement for achieving such a construct. Endo et al. (9) emphasized this requirement when describing their construct pCOXIV-DHFR-spacer-avidin. An intermediate could be generated only after this chimeric precursor was allowed to form a tetramer in the presence of biotin. But such a tetrameric protein can use only a small portion of available import sites, resulting in low efficiency.
Isolated mitochondria have a limited number of import sites (5, 8). However, based on observations that (i) the outer membrane translocation components are randomly distributed and (ii) a close apposition between outer and inner membranes may exist in vivo over a much larger area than seen in vitro, it has been proposed that protein import sites may occur over the entire mitochondrial boundary in vivo (20, 21). Import efficiency in vivo is thus expected to be much higher than in vitro. Translocation intermediates generated in vivo should therefore provide a superior molecular probe. However, previous studies on in vivo ligand-induced translocation arrest have yielded conflicting results (23, 24).
We have constructed a novel chimeric precursor [delta1-pyrroline-5-carboxylate dehydrogenase precursor (pPut)-glutathione S-transferase (GST)-protein A (pPGPrA)], containing an authentic mitochondrial precursor [pPut (25, 26)] at the N terminus linked, through GST (27), to a polypeptide containing the IgG binding domains of staphylococcal protein A (28). This hybrid precursor becomes trapped en route to the matrix both in vitro and in vivo. The intermediate thus generated spans both the outer and inner membrane protein import channels in yeast mitochondria in such a way that the entire signal-less Put moiety resides inside the matrix, while the folded protein A domain remains exposed outside the organelle. In vitro, the intermediate is localized at or near morphologically distinct contact sites, where the outer and inner membranes are closely apposed. In contrast, during in vivo import of pPGPrA, the outer and inner membranes are progressively “zippered” together forming long stretches of close contact. Together, biochemical and ultrastructural data strongly suggest that the normally transient interactions between the outer and inner membrane channels are stabilized by the translocation intermediate described here, making it a powerful tool for isolating the outer and inner membrane channel complexes as a single stoichiometric entity.
MATERIALS AND METHODS
Constructs.
A segment of the staphylococcal protein A gene was amplified by the PCR using the pRIT5 plasmid (Pharmacia) as a template. The segment encodes residues −10–271 of the protein A polypeptide and contains five potential IgG binding domains, but excludes the entire membrane-spanning domain and most of the signal sequence of protein A (28). The PCR product was inserted into the ClaI–BamHI-digested pBluescript (Stratagene)/GST (which codes for GST with a C-terminal thrombin-specific cleavage site, LVPR↓GS; D.P., unpublished work). The KpnI–BamHI fragment of the resulting plasmid, pBluescript/GST-protein A, was then subcloned into the same sites of pNYHM170 [pPut in pET3a (Novagen; ref. 26)] to obtain the plasmid pET3a/pPut-GST-protein A. The encoded chimeric protein (with a thrombin-specific cleavage site introduced between GST and protein A) is under the control of the T7 promoter. This construct will be called pPGPrA. To construct pET3a/pPut-protein A (called pPPrA, i.e., without the GST spacer between pPut and protein A), the KpnI–BamHI fragment of the protein A PCR product was directly subcloned into the same sites of pNYHM170 (see above).
To express pPGPrA as an endogenous precursor in yeast, the KpnI–BamHI fragment of pBluescript/GST-protein A was subcloned into pYES2/pPut [where pPut is under the control of the GAL1 promoter in pYES2 (Invitrogen); N.S. and D.P., unpublished work]. The resulting plasmid pYES2/pPGPrA was used to transform (29) a protease deficient strain of Saccharomyces cerevisiae (ABYS 86, MATα pra1–1 prb1–1 prc1–1 cps1–3 ura3Δ5 leu2–3, 112; obtained from R. Erdmann, University of Bochum, Germany). Transformants (Ura+) were selected by growth on selective synthetic minimal media (29).
Import of pPGPrA and pPPrA into Isolated Mitochondria.
The plasmids (pET3a/pPGPrA and pET3a/pPPrA) were linearized downstream from the insert with BamHI and transcribed by T7 RNA polymerase. The transcripts were translated in rabbit reticulocyte lysate in the presence of [35S]methionine, and a post-ribosomal supernatant (PRS) containing the newly synthesized protein (pPGPrA or pPPrA) was obtained as described (30). Mitochondria were isolated (25) from S. cerevisiae strain D273–10B (ATCC 24657). Import reactions consisting of mitochondria (100 μg protein) and a 35S-labeled hybrid precursor were carried out essentially as described (30). Following import at 30°C for 30 min, the mixture was diluted with HS buffer (20 mM Hepes/KOH, pH 7.5/0.6 M sorbitol) containing 0.1 mg/ml BSA and the mitochondria were sedimented at 4°C in a microfuge (15,000 × g) for 2 min. Each pellet was resuspended in 50 μl of HS buffer containing 0.1 mg/ml BSA and either left untreated or treated with protease(s) for 30 min on ice in the presence or absence of Triton X-100. Reaction mixtures were then diluted with HS buffer containing 0.1 mg/ml BSA, 5 mg/ml soybean trypsin inhibitor, 100 units/ml Trasylol, and 1 mM phenylmethylsulfonyl fluoride. Mitochondria were reisolated and washed with 10% trichloroacetic acid. Samples for lanes 8 and 9 (see Fig. 1) were directly precipitated with 10% trichloroacetic acid. Samples were analyzed by SDS/7.5% PAGE and autoradiography. Precursor and corresponding signal-less mature forms are indicated.
Figure 1.
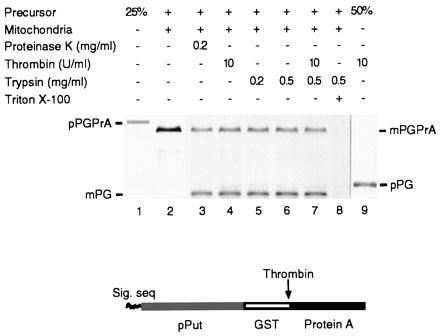
Generation of a translocation intermediate spanning both outer and inner mitochondrial membranes in vitro. (Lower) Schematic representation of the pPGPrA construct. (Upper) A PRS of a reticulocyte lysate translation mixture containing newly synthesized [35S]methionine-labeled pPGPrA (lane 1) was incubated with isolated yeast mitochondria. Following import, mitochondrial pellets were resuspended in buffer and either left untreated (lane 2) or treated for 30 min on ice with proteinase K (lane 3), thrombin (lane 4), two different concentrations of trypsin (lanes 5 and 6), a mixture of thrombin and trypsin (lane 7), or trypsin plus 0.5% Triton X-100 (lane 8). Lane 9, PRS alone treated with thrombin. Lanes 1 and 9 show 25 and 50%, respectively, of the amount of PRS used in the import assay. Samples were analyzed by SDS/PAGE followed by autoradiography.
Immunoelectron Microscopy.
Translocation intermediates were generated in vitro as follows. Mitochondria (250 μg protein), which had been incubated with pPGPrA under import conditions (see Materials and Methods) were reisolated, resuspended in HS buffer, and then allowed to settle onto a poly-l-lysine-coated polystyrene dish for 1 hr at 4°C. The remainder of the procedure was exactly as previously described (30), except the primary antibody (against mitochondrial proteins) was replaced by nonspecific rabbit IgG. Translocation intermediates were generated in vivo as follows. Yeast cells carrying the plasmid pYES2/pPGPrA were grown at 30°C in selective synthetic minimal media (29) containing 0.1% (instead of 2%) dextrose and 2% lactate. Expression of pPGPrA was induced by 1 mM galactose for either 30 min or 4 hr. Mitochondria were purified and processed for immunogold electron microscopy as above.
RESULTS
We designed a chimeric precursor, pPGPrA, consisting of three parts: an authentic mitochondrial precursor (pPut) at the N terminus, followed by GST (26 kDa), followed by a portion of staphylococcal protein A containing five potential IgG binding domains at the C terminus. In addition, a specific thrombin cleavage site was inserted between the GST and protein A moieties. The advantages of this construct are several: (i) The import characteristics of pPut are well defined (25, 26). (ii) GST serves as a long spacer between pPut and the C-terminal block, which should allow the entire pPut sequence to reach the matrix. (iii) As a C-terminal block, we chose protein A over a ligand-induced block (e.g., binding of methotrexate to DHFR) or a cross-linked moiety (such as BPTI) because of their inherent limitations. Ligand-induced block requires bound ligand and the block is easily released by simple dilution (8). Excess ligand in solution is necessary to maintain the block, which precludes subsequent affinity chromatography. Likewise, BPTI must have three intramolecular disulfide bridges to serve as a C-terminal block, a prerequisite that restricts its in vivo use since these disulfide bonds are unlikely to form in the reducing environment of the cytosol. By contrast, if protein A is allowed to fold properly during the translocation reaction, it is expected to block the completion of import (or slow down the import kinetics) even without added IgG. The high affinity of protein A for nonspecific rabbit IgG can subsequently be exploited to localize the translocation intermediate, either on intact mitochondria or in subfractions. It also enables one to purify, by chromatography of detergent-solubilized material on IgG-Sepharose, components of the translocation machinery that remain associated with the intermediate. A protein A tag for generating translocation intermediates has been successfully used to isolate components of the chloroplast import machinery (28, 31). (iv) Finally, specific cleavage by thrombin should provide an easy way to map the topology of the intermediate in the import channel. It also provides a means of generating reversible intermediates. More importantly, simple thrombin digestion will release channel proteins after affinity chromatography of detergent solubilized material, obviating stringent conditions (e.g., low or high pH, SDS, etc.).
Import of 35S-labeled pPGPrA was assayed with isolated yeast mitochondria (30). Following incubation, mitochondria, where required, were treated with the proteases indicated in Fig. 1 Upper, and analyzed by SDS/PAGE and autoradiography. More than 70% of the pPGPrA (molecular mass ≈117 kDa) used in the import assay (lane 1 shows only 25% load) was converted to mature protein (mPGPrA; lane 2, molecular mass ≈114 kDa) through cleavage of the signal sequence by a signal peptidase in the matrix. Lanes 3–7 show that ≈75% of the total mature protein (as judged from band intensities) was clipped by external protease (proteinase K, thrombin, or trypsin) to yield an 83-kDa fragment. The remaining 25% was immune to proteases, presumably because it had become sequestered within the matrix. All three protease treatments left the 83-kDa segment intact. This segment corresponds to the mPut-GST region, with the protein A segment (≈31 kDa) clipped off. This indicates that the protein A segment was, 75% of the time, fully exposed and unable to be transported further. After membrane disruption with Triton X-100, the protein was fully degraded by trypsin (lane 8). Lane 9 shows the result of cleavage of the precursor protein by thrombin in the absence of mitochondria: two fragments [pPut-GST (pPG) and protein A] were generated. The resulting large fragment has a molecular mass of about 86 kDa, as predicted for pPG with intact signal sequence. While both pPG and protein A bands are visible when analyzed by SDS/12% PAGE (not shown), only the former can be detected on 7.5% gels (lane 9). Lower percentage gels were used to distinguish precursor and mature forms of PGPrA.
The protein A portion of the trapped construct was localized by tagging with colloidal gold-labeled IgG (Fig. 2). The gold particles cluster only at contact sites where the outer and inner membranes are closely apposed, confirming that the protein A moiety remains outside the organelle. The position of the remaining segments is deduced from the following observations. Cleavage of the signal sequence indicates that at least the N terminus of the construct, which contains the signal peptidase processing site, is inside. In addition, GST must be entirely on the trans side of the outer membrane because it is immune to attack by external proteases. Since the GST segment (≈26 kDa) is much longer than the minimum polypeptide length required to span contact sites [about 50 amino acid residues; 18 nm in a fully extended conformation (32)], most of the mPut segment is likely to be within the matrix. This suggests that the trapped construct has the dumbbell shape shown in Fig. 3.
Figure 2.
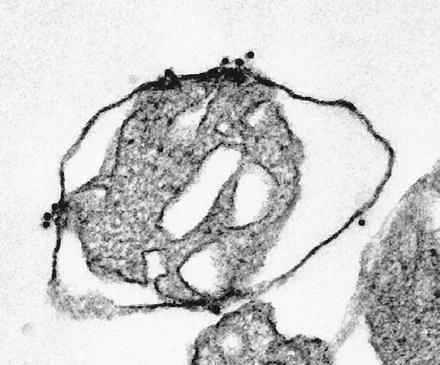
Immunoelectron microscopic localization of translocation intermediates generated in vitro. Mitochondria with translocation intermediates (as in Fig. 1, lane 2) were immobilized, fixed, and incubated sequentially with rabbit IgG, and goat anti-rabbit IgG conjugated to colloidal gold (10 nm). Small clusters of gold particles indicate the location of the protein A moiety in trapped intermediates. Protein A remains accessible to IgG. (×60,000.)
Figure 3.
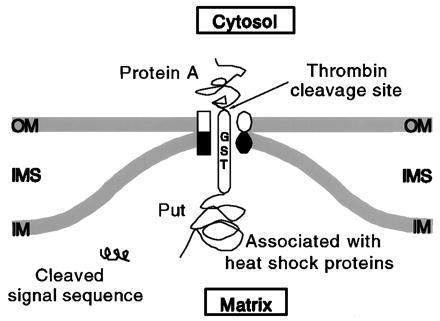
Schematic representation of the translocation intermediate. OM, outer membrane; IM, inner membrane; IMS, intermembrane space. See text for details.
Under the present import conditions, only protein A, and not GST, caused translocation arrest. Although the exact mechanism remains to be investigated, we believe that GST as a spacer provides time for protein A to fold sufficiently. In fact, when the GST fragment was deleted, the resulting chimera (pPPrA; Fig. 4, lane 1) evidently was imported entirely into the matrix (mPPrA, lanes 2 and 3), since the signal-less form remained quantitatively protected from externally added trypsin and no intermediate was generated (mPPrA, lanes 4 and 5).
Figure 4.
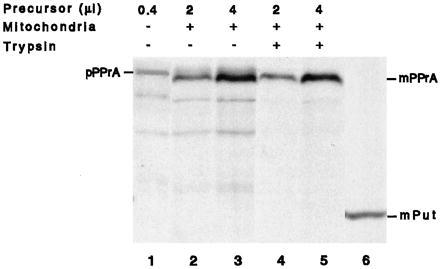
Protein A does not cause translocation arrest in the absence of GST as a spacer. Differing amounts (2 and 4 μl) of PRS of a reticulocyte lysate translation mixture containing newly synthesized [35S]methionine-labeled pPPrA were incubated with isolated yeast mitochondria. Following import, mitochondrial pellets were resuspended in buffer and either left untreated (lanes 2 and 3) or treated for 30 min on ice with trypsin (lanes 4 and 5). Samples were analyzed by SDS/PAGE followed by autoradiography. Lane 1 shows 0.4 μl of PRS in the absence of mitochondria. Lane 6 shows the migration position of mPut.
To further validate our in vitro findings, this time in intact cells, we expressed pPGPrA in yeast under the control of a GAL1 promoter; expression was monitored by immunoblots using affinity-purified anti-pPut. Following induction with galactose, mitochondria were isolated and protein A was tagged with immunogold particles. After a short (30 min) induction period, the mitochondria appeared normal, with readily visible cristae; the regions of contact between outer and inner membranes were small and an occasional gold particle was found at these sites (Fig. 5A). The appearance changed dramatically after 4 hr of induction (Fig. 5 B–E): the mitochondria became grossly distorted, often containing dense bodies in their matrix most likely representing some completely imported chimeric molecules, as suggested by the in vitro import experiments (Fig. 1). The cristae were replaced by large areas of contact (Fig. 5 B–E) where the two membranes were closely zippered together, and the outer one liberally tagged with colloidal gold. Contacts were numerous and large: most were longer than normal, almost half of them 0.5–1.2 μm (Fig. 6). Frequently, long stretches of membrane contacts containing clusters of gold particles were found predominantly over one pole, forming a “cap” (Fig. 5C), which suggests that the import channels (like most membrane proteins) freely diffuse in the entire membrane. If the import sites were confined to specific areas, one would have expected short stretches of membrane contact distributed relatively symmetrically rather than a cap of stitched membranes covering almost half the organelle.
Figure 5.
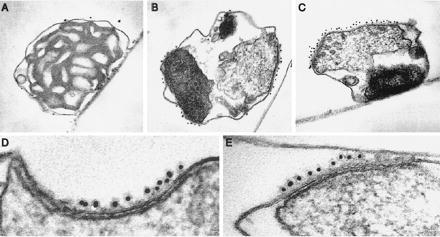
Visualization of translocation intermediates generated in vivo by immunogold electron microscopy. Mitochondria were isolated from yeast cells after induction of plasmid-borne pPGPrA with galactose for either 30 min (A) or 4 hr (B–E), immobilized, fixed, and incubated sequentially with rabbit IgG and goat anti-rabbit IgG conjugated to colloidal gold (10 nm). Clusters of gold particles are located over short contacts between outer and inner membranes and long rows of gold particles cover extended regions of zippering of the two membranes. (A and C, ×50,000; B, ×40,000; D and E, ×165,000.)
Figure 6.
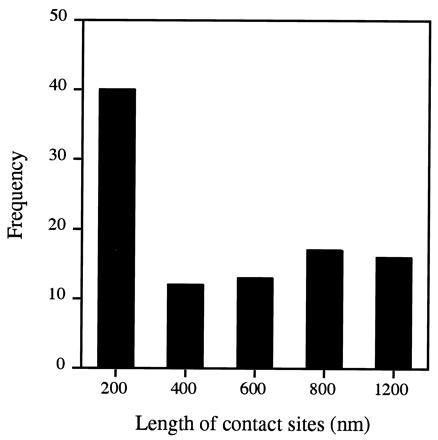
Length distribution of contact sites generated by translocation intermediates in vivo. Immunoelectron microscopy was performed on mitochondria isolated from yeast cells that had been induced with galactose for 4 hr to express pPGPrA as an endogenous precursor (see Fig. 5). For morphometric analysis (30), electron micrographs were viewed at a magnification of 80,000–125,000, and the length of contact sites decorated by gold particles was measured and grouped into five intervals. Gold particles within 30 nm or less from the membrane were considered to be membrane associated. Outer and inner membranes were considered to be in close proximity (contact sites) at regions where the distance between the centers of the inner and outer membranes was ≤10 nm. Numbers in each group were plotted against the length of contact sites.
Initiation of new import site proteins to accommodate an increased demand for import seems unlikely since mitochondrial membrane protein abundance in induced and control cells (containing the plasmid pYES2/pPGPrA) was virtually identical (not shown). Immunogold particles were found almost exclusively at intermembrane contact sites. These gold-labeled intermediates appear to represent molecules that are physically trapped during their transit through import channels. It is the failure of PGPrA to exit from the import channels that stitches the membranes together, permanently “freezing” the normally transient contact between outer and inner import channels. The progressive stitching is so strong that it grossly distorts mitochondrial structure, causing the cristae to unfold as large areas of intermembrane contact are formed.
DISCUSSION
We have constructed a simple yet powerful tool to study the mitochondrial translocation apparatus through parallel in vitro and in vivo studies. The translocation intermediate described here is trapped en route to the matrix so that the entire signal-less Put moiety is inside the matrix, while the folded protein A domain remains outside the mitochondrion (Fig. 1). This was made possible by the GST sequence, which serves as a long spacer between pPut and protein A. A thrombin-specific cleavage site between GST and protein A allowed us to determine the precise topology of the intermediate. Because the entire Put moiety reached its intended destination, it had a better opportunity to properly fold or at least remain associated with matrix heat shock proteins. Such a translocation intermediate would therefore be very stable, having blocks both cis and trans of the import machinery that “freeze” it within the import channel (Fig. 3). While we have not directly demonstrated that the trans block we observe is indeed due to folding of the mPut moiety or its association with heat shock proteins, available data from other laboratories (see Introduction), together with our observations, certainly render this interpretation likely. Both biochemical (Figs. 1 and 3) and immunoelectron microscopic (Figs. 5 and 6) data further indicate that the outer and inner membrane channels are stably held together by the PGPrA intermediate.
Our results are consistent with a model in which constituents of the protein import channels in the inner and outer mitochondrial membrane normally interact transiently (19–22). We postulate that the two channels become joined when an inner membrane import channel encounters a signal peptide protruding from the outer membrane channel. This transitory association usually ends when protein translocation through the outer membrane is complete. Our chimeric protein construct pPGPrA, however, becomes trapped in the outer/inner membrane channel complex. We suggest that the presence of one or a few such contacts containing trapped PGPrA, and the resultant apposition of the membranes, facilitate the formation of additional contacts nearby and lead to a progressive stitching together of the two membranes. Under physiological conditions, the outer and inner membrane channels presumably close in sequence upon completion of translocation across each membrane; the membranes then separate. Zippering of membranes is thus not expected under “normal” import conditions, and should only be seen when the normally transient interactions between outer and inner membrane channels are made irreversible by an artificial precursor such as our pPGPrA construct. Our data also support an earlier proposal (21) that the outer and inner import channels, rather than being confined to specific areas (such as morphologically distinct membrane adhesion or contact sites), are free to diffuse over the entire membrane.
As was the case for other reported in vitro translocation intermediates (4), we too found no in vitro zippering of outer and inner mitochondrial membranes by our construct. This could be due to less efficient protein import in vitro compared with in vivo. Alternatively, the swelling of spheroplasts that is known to occur during the standard isolation procedure could have separated the membranes (21) sufficiently to reduce the probability of a precursor-mediated interaction. Protein import into isolated mitochondria may therefore take place at or near those remaining contact sites where the two membranes are already closely apposed (33). In light of earlier findings that isolated mitochondria have only limited areas [≈5–10% of periphery (20, 21)] of contact/adhesion and small numbers of import sites [102-103 per mitochondrion (5, 8)], it is not surprising that intermediates generated in vitro are detected at only a few contact sites (Fig. 2).
For precursors en route to the matrix, exit of the signal sequence from the trans side of the outer membrane channel is likely to be coupled to its recognition by the inner membrane import machinery. Inner membrane import sites are more abundant than outer membrane sites (34, 35); the probability of a productive interaction with the signal sequence emerging from the outer membrane is therefore high. No preprotein in transit to the matrix has ever been found freely soluble in the intermembrane space (36, 37). We propose that structural proteins of the outer and inner membranes reversibly interact to stabilize precursor-induced import sites, yielding a continuous tunnel and preventing premature refolding/misfolding of the preprotein in the now virtual intermembrane space. The interaction between the structural components of the two membranes is probably weak in the absence of a preprotein in transit so that the channels easily dissociate following completion of translocation.
Several outer and inner membrane proteins are known to be involved in mitochondrial protein import (2). Although some have been studied in detail, their in vivo functions remain unknown. The novel intermediate reported here is capable of holding outer and inner membrane channels stably together both in vitro and in vivo. By doing so, the PGPrA intermediate constitutes a potentially powerful tool for isolating both outer and inner membrane translocation channels simultaneously as a single stoichiometric complex from detergent solubilized mitochondrial membranes.
Acknowledgments
We thank Günter Blobel for encouraging and supporting the initial experiments. We also thank P. de Weer, C. Deutsch, and D. Schwartz for their valuable comments on the manuscript, and C. Franzini-Armstrong and H. Shio for help with electron microscopy. This work was supported by grants from the American Heart Association and the W. W. Smith Charitable Trust.
ABBREVIATIONS
- pPut
delta1-pyrroline-5-carboxylate dehydrogenase precursor
- GST
glutathione S-transferase
- pPGPrA
pPut-GST-protein A
- pPPrA
pPut-protein A
- PRS
post-ribosomal supernatant
- BPTI
bovine pancreatic trypsin inhibitor
- DHFR
dihydrofolate reductase
References
- 1.Cyr D M, Ungermann C, Neupert W. Methods Enzymol. 1995;260:241–252. doi: 10.1016/0076-6879(95)60142-2. [DOI] [PubMed] [Google Scholar]
- 2.Kübrich M, Dietmeier K, Pfanner N. Curr Genet. 1995;27:393–403. doi: 10.1007/BF00311207. [DOI] [PubMed] [Google Scholar]
- 3.Schleyer M, Neupert W. Cell. 1985;43:339–350. doi: 10.1016/0092-8674(85)90039-x. [DOI] [PubMed] [Google Scholar]
- 4.Schwaiger M, Herzog V, Neupert W. J Cell Biol. 1987;105:235–246. doi: 10.1083/jcb.105.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vestweber D, Schatz G. J Cell Biol. 1988;107:2037–2043. doi: 10.1083/jcb.107.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eilers M, Schatz G. Nature (London) 1986;322:228–232. doi: 10.1038/322228a0. [DOI] [PubMed] [Google Scholar]
- 7.Chen W-J, Douglas M G. J Biol Chem. 1987;262:15605–15609. [PubMed] [Google Scholar]
- 8.Rassow J, Guiard B, Wienhues U, Herzog V, Hartl F-U, Neupert W. J Cell Biol. 1989;109:1421–1428. doi: 10.1083/jcb.109.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Endo T, Nakayama Y, Nakai M. J Biochem. 1995;118:753–759. doi: 10.1093/oxfordjournals.jbchem.a124976. [DOI] [PubMed] [Google Scholar]
- 10.Eilers M, Oppliger W, Schatz G. EMBO J. 1987;6:1073–1077. doi: 10.1002/j.1460-2075.1987.tb04860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vestweber D, Brunner J, Baker A, Schatz G. Nature (London) 1989;341:205–209. doi: 10.1038/341205a0. [DOI] [PubMed] [Google Scholar]
- 12.Scherer P E, Manning-Krieg U C, Jenö P, Schatz G, Horst M. Proc Natl Acad Sci USA. 1992;89:11930–11934. doi: 10.1073/pnas.89.24.11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan K R, Jensen R E. J Biol Chem. 1993;268:23743–23746. [PubMed] [Google Scholar]
- 14.Söllner T, Rassow J, Wiedmann M, Schlossmann J, Keil P, Neupert W, Pfanner N. Nature (London) 1992;355:84–87. doi: 10.1038/355084a0. [DOI] [PubMed] [Google Scholar]
- 15.Ungermann C, Neupert W, Cyr D M. Science. 1994;266:1250–1253. doi: 10.1126/science.7973708. [DOI] [PubMed] [Google Scholar]
- 16.Schneider H C, Berthold J, Bauer M F, Dietmeier K, Guiard B, Brunner M, Neupert W. Nature (London) 1994;371:768–774. doi: 10.1038/371768a0. [DOI] [PubMed] [Google Scholar]
- 17.Rassow J, Maarse A C, Krainer E, Kübrich M, Müller H, Meijer M, Craig E A, Pfanner N. J Cell Biol. 1994;127:1547–1556. doi: 10.1083/jcb.127.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kronidou N G, Oppliger W, Bolliger L, Hannavy K, Glick B S, Schatz G, Horst M. Proc Natl Acad Sci USA. 1994;91:12818–12822. doi: 10.1073/pnas.91.26.12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glick B, Wachter C, Schatz G. Trends Cell Biol. 1991;1:99–103. doi: 10.1016/0962-8924(91)90037-a. [DOI] [PubMed] [Google Scholar]
- 20.Pfanner N, Rassow J, van der Klei I J, Neupert W. Cell. 1992;68:999–1002. doi: 10.1016/0092-8674(92)90069-o. [DOI] [PubMed] [Google Scholar]
- 21.van der Klei I J, Veenhuis M, Neupert W. Microsc Res Tech. 1994;27:284–293. doi: 10.1002/jemt.1070270404. [DOI] [PubMed] [Google Scholar]
- 22.Horst M, Hilfiker-Rothenfluh S, Oppliger W, Schatz G. EMBO J. 1995;14:2293–2297. doi: 10.1002/j.1460-2075.1995.tb07223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wienhues U, Becker K, Schleyer M, Guiard B, Tropschug M, Horwich A L, Pfanner K, Neupert W. J Cell Biol. 1991;115:1601–1609. doi: 10.1083/jcb.115.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiki M, Verner K. J Biol Chem. 1993;268:1914–1920. [PubMed] [Google Scholar]
- 25.Murakami H, Pain D, Blobel G. J Cell Biol. 1988;107:2051–2057. doi: 10.1083/jcb.107.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakami H, Blobel G, Pain D. Proc Natl Acad Sci USA. 1993;90:3358–3362. doi: 10.1073/pnas.90.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith D B, Davern K M, Board P G, Tiu W U, Garcia E G, Mitchell G F. Proc Natl Acad Sci USA. 1986;83:8703–8707. doi: 10.1073/pnas.83.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnell D J, Blobel G. J Cell Biol. 1993;120:103–115. doi: 10.1083/jcb.120.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman F, Fink G R, Hicks J B. Laboratory Course Manual for Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 30.Pain D, Murakami H, Blobel G. Nature (London) 1990;347:444–449. doi: 10.1038/347444a0. [DOI] [PubMed] [Google Scholar]
- 31.Schnell D J, Kessler F, Blobel G. Science. 1994;266:1007–1012. doi: 10.1126/science.7973649. [DOI] [PubMed] [Google Scholar]
- 32.Rassow J, Hartl F-U, Guiard B, Pfanner N, Neupert W. FEBS Lett. 1990;275:190–194. doi: 10.1016/0014-5793(90)81469-5. [DOI] [PubMed] [Google Scholar]
- 33.Hackenbrock C R. Proc Natl Acad Sci USA. 1968;61:598–605. doi: 10.1073/pnas.61.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang S, Jascur T, Vestweber D, Pon L, Schatz G. J Cell Biol. 1989;109:487–493. doi: 10.1083/jcb.109.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohba M, Schatz G. EMBO J. 1987;6:2117–2122. doi: 10.1002/j.1460-2075.1987.tb02478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rassow J, Pfanner N. FEBS Lett. 1991;293:85–88. doi: 10.1016/0014-5793(91)81157-4. [DOI] [PubMed] [Google Scholar]
- 37.Hwang S T, Wachter C, Schatz G. J Biol Chem. 1991;266:21083–21089. [PubMed] [Google Scholar]