

Abstract
Phosphatidylinositol 3-kinases (PI 3-kinases) have been implicated in membrane trafficking in the secretory and endocytic pathways of yeast and mammalian cells, but the molecular mechanisms by which these lipid kinases operate are not known. Here we identify a protein of 170 kDa that is rapidly released from cell membranes in response to wortmannin, a potent inhibitor of mammalian PI 3-kinases. The amino acid sequence of peptides from p170 reveal its identity to early endosomal antigen (EEA) 1, an endosomal antigen with homology to several yeast proteins genetically implicated in membrane trafficking. Immunofluorescence analysis of 3T3-L1 adipocytes with antisera against p170/EEA1 reveal a punctate peripheral pattern that becomes diffuse in response to wortmannin. In vitro, p170/EEA1 binds specifically to liposomes containing PIns(3)P, suggesting that the effect of wortmannin on cells is due to inhibition of PIns(3)P production. Thus, p170/EEA1 may define a family of proteins that mediate the regulatory effects of 3′-phosphoinositides on membrane trafficking in yeast and mammalian cells.
Phosphatidylinositol 3-kinases (PI 3-kinases) are a family of enzymes that, in mammalian cells, catalyze the synthesis of PIns (phosphatidylinositol)-(3)P, PIns(3,4)P2, and PIns(3,4,5)P3 (1). In yeast, the only known PI 3-kinase, vps34p, catalyzes the synthesis of PIns(3)P (2). This activity is essential for the sorting of newly synthesized proteins (2), as well as for endocytosis (3), into the vacuole. A conserved role for PI 3-kinase in membrane trafficking in mammalian cells has been suggested by the finding that trafficking and degradation of platelet-derived growth factor receptors requires the presence of PI 3-kinase binding sites on the receptor cytoplasmic domain (4). In addition, numerous membrane trafficking processes that take place in the endocytic pathway, such as platelet-derived growth factor receptor down-regulation (5), glucose transporter recycling (6, 7), transferrin-receptor recycling (8), and transcytosis (9) are profoundly affected by treatment of cells with wortmannin, a potent inhibitor of the activities of several mammalian PI 3-kinase isoforms (10, 11). Some of the effects of wortmannin on membrane trafficking in the endocytic pathway can be mimicked by procedures that directly interfere with PI 3-kinase function, such as the micro-injection of anti-PI 3-kinase antibodies or dominant-negative constructs (7). Thus, at low concentrations, wortmannin is likely to be exerting its effects through inhibition of PI 3-kinase catalytic activity.
The mechanism of PI 3-kinase action in endocytic trafficking is unclear. However, wortmannin induces a pronounced change in the morphology of endosomes (12), reminiscent of the effect of brefeldin A, an agent known to block the activity of ARF1 and inhibit the assembly of cytosolic coat proteins onto membranes (13). Based on these observations, we reasoned that 3′ phosphoinositides might serve as docking sites or activators of docking sites for peripheral proteins that reversibly interact with membranes. These peripheral membrane proteins could, in turn, regulate membrane trafficking, protein sorting, or both. Thus, we undertook experiments to identify downstream effectors of PI 3-kinase by searching for peripheral membrane proteins that interact with membranes in a wortmannin-sensitive manner. In this manuscript we describe the identification and characterization of one such protein, and demonstrate that, in vitro, it interacts with 3′ phosphoinositides.
METHODS
Identification of p170.
3T3-L1 fibroblasts (American Type Culture Collection) were grown in 150-mm dishes and used after 7 days of differentiation, when lipid droplets were observed in 95–100% of the cells. Cells were treated without or with wortmannin (50 nM, Sigma) for 15 min. To prepare a crude membrane fraction, cells were washed twice with ice-cold PBS, once with an ice-cold buffer (cytosol buffer) containing sucrose (0.2 M), Hepes at pH 7.0 (25 mM), potassium acetate (125 mM), magnesium acetate (2.5 mM), DTT (1 mM), benzamidine (1 mM), 1, 10 phenanthroline (1 mM), phenylmethylsulfonyl fluoride (0.2 mM), tosylarginine methyl ester (10 μg/ml), leupeptin (5 μg/ml), ATP (1 mM), creatine phosphate (5 mM), and creatine kinase (10 μg/ml) (Boehringer Mannheim). The buffer was aspirated, and monolayers were scraped into the residual buffer remaining on the dish. Typically 1 ml of broken cell suspension was collected from a 150-mm dish. Cells were homogenized with seven passes through a 27-gauge needle, and nuclei and unbroken cells removed by centrifugation for 5 min at 500 × g. The supernatant was then centrifuged at 300,000 × g for 15 min in a TL-100 tabletop centrifuge, and the pellet resuspended in 150 μl of cytosol buffer using a tuberculin syringe. Potassium chloride was then added from a 10 × stock to a final concentration of 0.5 M. After 15 min on ice, membranes were separated by ultracentrifugation as described above, and the supernatant loaded on a 4.6-ml continuous gradient (10–30% wt/wt) of sucrose in cytosol buffer. Gradients were centrifuged for 18 hr at 35,000 rpm in a Beckman SW 50.1 rotor, and fractionated from the top into 10 fractions, which were resolved by electrophoresis in 5–15% SDS/PAGE. Gels were silver-stained (Bio-Rad).
Sequencing of p170.
p170 derived from 20 150-mm plates of differentiated 3T3-L1 cells was obtained as described above, concentrated, separated by 5–15% SDS/PAGE, stained with Coomassie blue, destained extensively, and excised. After in-gel S-carboxyamidomethylation, the band was subjected to in-gel endoproteinase Lys C (Wako) digestion, without addition of 0.02% Tween (14). Candidate ions for sequencing were determined by microcapillary HPLC coupled to a triple mass spectrometer (Finnigan TSQ7000 with electrospray ionization source (San Jose, CA) as described by Hunt et al. (15). The ion intensities observed corresponded to a load of 100–300 fmol by comparison with the average ion abundance of a standard protein (BSA) LysC digest. Direct peptide sequence information was obtained by collisionally induced dissociation on an equivalent injection of the digest mixture. The resulting MS/MS spectra were manually interpreted. The database-searching algorithm sequest (16) was also used to facilitate interpretation of MS/MS spectra.
Immunofluorescence.
3T3-L1 cells on glass coverslips were serum-deprived for 4 hr and incubated for 10 min without or with wortmannin as indicated. Cells were washed twice with ice-cold PBS and fixed on ice for 15 min with 4% formaldehyde. Cells were permeabilized by immersion in methanol at −20°C for 7 min. Early endosomal antigen (EEA) 1 was detected with human autoimmune antiserum to EEA1, obtained from a patient identified at the Monash Clinical Immunology Laboratory (17, 18). The antiserum was used at a 1:10,000 dilution, and was detected with goat antibodies to human IgG coupled to fluorescein (Zymed). GLUT4 was detected with a rabbit antiserum to GLUT4 and goat antibodies to rabbit IgG coupled to rhodamine.
Binding of p170/EEA1 to Liposomes.
Liposomes were prepared by mixing phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, PIns, and PIns(3)P at the proportions indicated in each experiment, drying the mixture under nitrogen, and resuspending to a final concentration of 1 mg of total phospholipid/ml in a buffer composed of Hepes at pH 7.2 (50 mM), NaCl (100 mM), and EDTA (0.5 mM). Resuspended lipids were sonicated in a bath sonicator until a homogeneous suspension was formed (5 min). Liposomes were collected by centrifugation at 16,000 × g for 10 min and resuspended in cytosol buffer at 2 mg/ml of total lipid. A cytosolic fraction was prepared from 3T3-L1 adipocytes as described above, but the cytosol buffer was supplemented with sodium vanadate (1 mM) and sodium fluoride (20 mM). Aliquots of cytosol (100 μl, approximately 5 mg/ml) were placed in 1.5 ml Eppendorf tubes, and where indicated treated for 5 min with wortmannin (50 nM). Tubes were placed at room temperature, and 50 μl of the indicated liposome mixture was added. Tubes were vortexed once, and after 15 min centrifuged at 16,000 × g for 10 min. The cytosolic supernatant (S) was removed, and the liposome pellet (P) resuspended in 130 μl of cytosol buffer. Samples were analyzed for the content of EEA1 by 5–15% SDS/PAGE and immunoblotting.
Production of Phosphoinositides.
PIns(3)P was produced enzymatically with the mouse homologue of the yeast and human VPS34p (ref. 19; J.V. and M. P. Czech, unpublished work), expressed as a glutathione S-transferase (GST) fusion protein in Sf9 insect cells. Expression and purification of the protein were as previously described (20). Mixed liposomes of phosphatidylinositol/phosphatidylserine (1:1) were incubated with GST-mVPS34 in 20 mM Tris⋅HCl, pH 7.5/100 mM NaCl/3.5 mM MnCl2/0.5 mM EGTA/20 μM ATP for 2 hr at room temperature. Lipids were extracted in chloroform/methanol (1:1) and washed twice with methanol/1 M HCl (1:1). Product yield was determined by including trace amounts of [γ-32P]ATP and comparing starting and chloroform-soluble counts. The only radiolabeled chloroform-soluble product detected was PIns(3)P, as assessed by thin-layer chromatography (20).
RESULTS AND DISCUSSION
To search for peripheral membrane proteins that could be downstream targets of PI 3-kinases, we used 3T3-L1 adipocytes. In these cells PI 3-kinase activity appears to regulate GLUT4 trafficking between endosomal membranes and the cell surface (6, 7). Thus, these cells are excellent candidates to identify downstream targets of PI 3-kinase involved in membrane trafficking. 3T3-L1 adipocytes were treated without or with wortmannin, homogenized, and a crude membrane fraction prepared from postnuclear supernatants. Peripheral membrane proteins were extracted with 0.5 M KCl, separated by velocity centrifugation on 10–30% sucrose gradients and analyzed by SDS/PAGE and silver staining. A protein of approximately 170 kDa (p170) was eluted from control cell membranes, but was greatly diminished in membranes from wortmannin-treated cells (Fig. 1A, fraction 2, arrow).
Figure 1.

(A) Peripheral membrane proteins from 3T3-L1 adipocyte membranes. 3T3-L1 adipocytes were treated without (−) or with (+) wortmannin (50 nM) for 15 min. Crude membrane fractions were prepared and peripheral membrane proteins released with 0.5 M KCl. Proteins were separated by velocity centrifugation on 10–30% sucrose gradients, and 10 fractions were collected. Fractions were analyzed by 5–15% SDS/PAGE and silver staining. The first six fractions are shown. The arrow indicates the position of a 170-kDa band, present in the second fraction, that is greatly reduced in extracts from wortmannin-treated cells. Molecular size markers are indicated to the right in kilodaltons. (B) Sequence of peptides derived from p170. Approximately 5 pmol of p170, obtained from untreated cells (right lane), were excised and subjected to lysyl endoproteinase digestion, separation, and sequencing by microcapillary HPLC-electrospray ionization tandem MS. The sequences of three peptides that aligned with the sequence of EEA1 are shown. (C) Domain structure of EEA1 (18).
To determine the identity of p170, larger amounts of protein were obtained, separated by SDS/PAGE, stained with Coomassie blue (Fig. 1B), excised, and digested in-gel. Released peptides were sequenced by tandem MS. The sequences of three peptides were identical to sequences within a human protein previously identified as an antigen in subacute systemic lupus erythematosus, EEA1 (Fig. 1B). The EEA1 molecule has been previously characterized (17, 18) as a ubiquitously expressed 162-kDa protein containing extensive predicted coiled-coil regions, as well as an IQ calmodulin-binding domain, and two regions, at the N and C termini, with homology to Zn2+ finger motifs (Fig. 1C).
To confirm the identity of p170 with EEA1, membrane and cytosolic fractions from 3T3-L1 adipocytes were analyzed by immunoblotting with human anti-EEA1 antiserum (Fig. 2). This antiserum recognizes a single band of 170 kDa in both membrane and cytosolic fractions (Fig. 2 Upper). Treatment of 3T3-L1 cells with increasing concentrations of wortmannin causes a redistribution of this protein from the membrane to the cytosolic fraction, with a half-maximal inhibitory effect observed at 10 nM. At this concentration wortmannin inhibits PI 3-kinases, but no other documented catalytic activities. The effect of wortmannin is rapid, with maximal redistribution observed after 5 min of exposure to the toxin (not illustrated).
Figure 2.
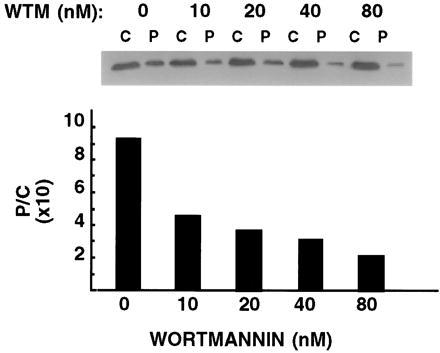
Concentration-dependence of wortmannin-induced redistribution of EEA1. 3T3-L1 adipocytes were treated with increasing concentrations of wortmannin (WTM) for 15 min, and fractionated into a crude membrane (P) and cytosolic (C) fraction. Proportional amounts of the two fractions were separated by SDS/PAGE and analyzed by immunoblotting with human anti-EEA1 antiserum and 125I-labeled protein A. A single protein band at 170 kDa was detected (Inset) in both cytosol (C) and pelleted membrane (P) fractions. The intensity of the bands was quantified by densitometric scanning. Plotted are the ratios (P/C) of the densitometric values obtained.
To visualize the effect of wortmannin in intact cells, 3T3-L1 adipocytes were analyzed by immunofluorescence with human antiserum to EEA1. Serum-deprived cells were used to restrict PI 3-kinase activity to that which operates in the absence of specific hormonal stimulation (12). A distinctive peripheral, vesicular pattern was observed after staining with anti-EEA1 antiserum, consistent with the reported localization of EEA1 on early endosomes (Fig. 3 Upper Left). Treatment of cells with wortmannin causes a dramatic decrease in vesicular staining, suggesting that the association of EEA1 with early endosomes is regulated by basal PI 3-kinase activity (Fig. 3 Upper Right). In contrast, the subcellular localization of GLUT4, a transmembrane protein that is not restricted to early endosomes (21), is not markedly altered by wortmannin treatment under these conditions (Fig. 3 Lower). Thus, wortmannin causes a pronounced dissociation of p170/EEA1 from peripheral endosomes in intact cells, under conditions in which the general organization of endosomal membranes is not markedly disrupted. The relationship between EEA1 localization and the regulation of GLUT4 trafficking by insulin and wortmannin is currently under further investigation in our laboratory.
Figure 3.

Localization of EEA1 in 3T3-L1 adipocytes. Cells were serum-deprived for 3 hr, treated for 15 min without (Left) or with (Right) wortmannin (20 nM), fixed and double-stained with antibodies to EEA1 (Upper) and GLUT4 (Lower). Images were collected using a cooled charged-coupled device camera. Equivalent exposures are shown.
The concentration dependency and time course of EEA1 dissociation in response to wortmannin parallels the decrease in endogenous PIns(3)P and PIns(3,4,5)P3 previously observed to occur in response to the toxin (12). These data suggest that one or both of these PI 3-kinase products may regulate the association of EEA1 to endosomes. Further support for this hypothesis is provided by the finding that incubation of cytosol from 3T3-L1 cells with liposomes containing PIns results in a dramatic adsorption of EEA1 to such liposomes (Fig. 4 Upper). This adsorption is specific, as it is not observed when liposomes contain only phosphatidylserine or phosphatidylethanolamine. The adsorption of EEA1 to liposomes composed of PIns is completely blocked by pre-treatment of the cytosol with wortmannin (Fig. 4, lanes marked +). The effect of wortmannin is selective, as the majority of the cytosolic polypeptides that adsorb to liposomes are unaffected by wortmannin treatment (Fig. 4 Lower). These results suggest that phosphorylation of PIns in liposomes by PI 3-kinases present in the cytosol causes the subsequent adsorption of EEA1.
Figure 4.

Adsorption of EEA1 to liposomes containing PIns. Cytosolic extracts (S) from 3T3-L1 cells were treated without (−) or with (+) wortmannin (WTM) at 50 nM for 5 min, and then incubated without (−) or with liposomes composed of phosphatidylinositol (PI) phosphatidylserine (PS), and phosphatidylethanolamine (PE). Liposomes were centrifuged and pellets (P) analyzed by immunoblotting with antibodies to EEA1 (Upper) and by silver staining (Lower).
The hypothesis that EEA1 binds to phosphorylated PIns is supported by an experiment in which PIns(3)P, generated with mammalian Vps34 produced in Sf9 cells, is incorporated into the liposomes. Incubation of cytosol with liposomes containing increasing amounts of PIns(3)P results in an increasing adsorption of EEA1 to the liposomes, which is now insensitive to wortmannin (Fig. 5). These results strongly suggest that the effect of wortmannin to block the binding of EEA1 to endosomal membranes can be attributed to inhibition of PI 3-kinase activity and PIns(3)P production.
Figure 5.

Adsorption of EEA1 to liposomes containing PIns(3)P. (A) Cytosolic extracts from 3T3-L1 cells were treated without (−) or with (+) wortmannin (WTM) at 50 nM for 5 min, and then incubated with liposomes composed of phosphatidylserine (PSer), PIns, and PIns(3)P, at the proportions depicted above. Liposomes were centrifuged and cytosolic supernatants (S) and liposome pellets (P) analyzed by PAGE and immunoblotting with antibodies to EEA1. (B) The intensity of the EEA1 bands in the liposome pellets was quantified by densitometry and plotted against the content of PIns(3)P.
Many reported effects of wortmannin in intact cells, such as the impairment of transferrin receptor recycling, platelet-derived growth factor receptor down-regulation, glucose transporter recycling, and the alteration in endosome morphology, are all consistent with a requirement for PI 3-kinase at early stages of the endocytic pathway (5–12). Our data, indicating that an early event in PI 3-kinase inhibition is the loss of EEA1 binding to endosomes, suggests an important role for this protein in meditating the effects of PI 3-kinase on membrane trafficking.
The specific function of EEA1 is presently not known. However, it is interesting that the Zn2+ finger motif at the C-terminus of EEA1 is highly homologous to that found in three yeast proteins, Vac1, Vps27, and Fab1, which have been genetically implicated in membrane trafficking. Vac1 is required for vacuolar protein sorting and vacuolar inheritance (22), both processes that also require vps34, the yeast form of PI 3-kinase (23). Vps27p has been implicated in the control of traffic through an endocytic prevacuolar compartment (24), a site at which vps34 may also function (3). Thus, the possibility exists that the particular Zn2+ finger motif found in EEA1, Vps27p, and Vac1p may be involved in an interaction with PI 3-kinase products. In mammalian cells, two proteins have been found to contain a Zn2+ finger motif homologous to that in EEA1. One, the mouse Hrs protein (25), is a substrate for tyrosine phosphorylation in response to growth factors, and recently has been implicated in the control of vesicle trafficking protein complexes (26). If the Hrs protein, like EEA1, binds to 3′ phosphoinositides, it becomes an attractive candidate for a mediator of growth-factor-induced vesicle trafficking events such as insulin-stimulated glucose transporter translocation and platelet-derived growth factor-induced receptor down-regulation.
In summary, our finding that EEA1 is a downstream target of PI 3-kinase suggests that proteins with homology to EEA1 may mediate other regulatory effects of 3′-phosphoinositides in eukaryotic cells. Clearly, more work is required to delineate the structural features of EEA1 required for interaction with 3′ phosphoinositides and the role of this interaction in both basal and regulated membrane traffic through the early endosome.
Acknowledgments
We thank R. Robinson, J. Neveu, T. Addona, and E. Spooner of the Harvard Microchemistry facility for expertise in the HPLC, mass spectrometry, and peptide sequencing. We are also grateful to Drs. Pietro DeCamilli and Mike Czech for reading the manuscript and for helpful suggestions. Immunofluorescence analysis was done with instrumentation and support from the Biomedical Imaging Group, University of Massachusetts Medical School. We thank Mike Czech for generously providing mammalian vps34 for this project. This work was supported in part by National Institutes of Health Grant DK 52228.
ABBREVIATIONS
- PI 3-kinase
phosphatidylinositol 3-kinase
- EEA
early endosomal antigen
- PIns
phosphatidylinositol
References
- 1.Auger K, Serunian L, Soltoff S, Libby P, Cantley L. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 2.Stack J H, Emr S D. J Biol Chem. 1994;269:31552–31562. [PubMed] [Google Scholar]
- 3.Munn A L, Riezman H. J Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joly M, Kazlauskas A, Fay F, Corvera S. Science. 1994;263:684–687. doi: 10.1126/science.8303278. [DOI] [PubMed] [Google Scholar]
- 5.Joly M, Kazlauskas A, Corvera S. J Biol Chem. 1995;270:13225–13230. doi: 10.1074/jbc.270.22.13225. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Clarke J F, Ester C J, Young P W, Kasuga M, Holman G D. Biochem J. 1996;313:125–131. doi: 10.1042/bj3130125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haruta T, Morris A J, Rose D W, Nelson J G, Mueckler M, Olefsky J M. J Biol Chem. 1995;270:27991–27994. doi: 10.1074/jbc.270.47.27991. [DOI] [PubMed] [Google Scholar]
- 8.Martys J L, Wjasow C, Gangi D M, Kielian M, McGraw T E, Backer J M. J Biol Chem. 1996;271:10953–10962. doi: 10.1074/jbc.271.18.10953. [DOI] [PubMed] [Google Scholar]
- 9.Hansen S H, Olsson A, Casanova J E. J Biol Chem. 1995;270:28425–28432. doi: 10.1074/jbc.270.47.28425. [DOI] [PubMed] [Google Scholar]
- 10.Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. J Biol Chem. 1994;269:3563–3567. [PubMed] [Google Scholar]
- 11.Thelen M, Wymann M P, Langen H. Proc Natl Acad Sci USA. 1994;91:4960–4964. doi: 10.1073/pnas.91.11.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shpetner H S, Joly M, Hartely D, Corvera S. J Cell Biol. 1996;132:595–605. doi: 10.1083/jcb.132.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klausner R D, Donaldson J G, Lippincott-Schwartz J. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellman U, Wernstedt C, Gonez J, Heldin C-H. Anal Biochem. 1995;191:332–336. doi: 10.1006/abio.1995.1070. [DOI] [PubMed] [Google Scholar]
- 15.Hunt D F, Henderson R A, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox A L, Appella E, Engelhard V H. Science. 1992;255:1261–1263. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- 16.Eng J K, McCormick A L, Yates J R., III J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 17.Mu F T, Callaghan J M, Steele-Mortimer O, Stenmark H, Parton R G, Campbell P L, McCluskey J, Yeo J P, Tock E P, Toh B-H. J Biol Chem. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 18.Stenmark H, Assland R, Toh B-H, D’Arrigo A. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- 19.Volinia S, Dhand R, Vanhaesbroeck B, MacDougall L K, Stein R, Zvelibil M J, Domin J, Panaretou C, Waterfield M D. EMBO J. 1995;14:3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Virbasius J V, Guilherme A, Czech M P. J Biol Chem. 1996;271:13304–13307. doi: 10.1074/jbc.271.23.13304. [DOI] [PubMed] [Google Scholar]
- 21.Slot J W, Geuze H J, Gigengack S, James D E, Lienhard G E. Proc Natl Acad Sci USA. 1991;88:7815–7819. doi: 10.1073/pnas.88.17.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisman L S, Wickner W. J Biol Chem. 1992;267:618–623. [PubMed] [Google Scholar]
- 23.Herman P K, Emr S D. Mol Cell Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piper R C, Cooper A A, Yang H, Stevens T H. J Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komada M, Kitamura N. Mol Cell Biol. 1995;15:6213–6221. doi: 10.1128/mcb.15.11.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bean A J, Seifert R, Chen Y A, Sacks R, Scheller R H. Nature (London) 1997;385:826–829. doi: 10.1038/385826a0. [DOI] [PubMed] [Google Scholar]