

Abstract
The yeast Sec1p protein functions in the docking of secretory transport vesicles to the plasma membrane. We previously have cloned two yeast genes encoding syntaxins, SSO1 and SSO2, as suppressors of the temperature-sensitive sec1–1 mutation. We now describe a third suppressor of sec1–1, which we call MSO1. Unlike SSO1 and SSO2, MSO1 is specific for sec1 and does not suppress mutations in any other SEC genes. MSO1 encodes a small hydrophilic protein that is enriched in a microsomal membrane fraction. Cells that lack MSO1 are viable, but they accumulate secretory vesicles in the bud, indicating that the terminal step in secretion is partially impaired. Moreover, loss of MSO1 shows synthetic lethality with mutations in SEC1, SEC2, and SEC4, and other synthetic phenotypes with mutations in several other late-acting SEC genes. We further found that Mso1p interacts with Sec1p both in vitro and in the two-hybrid system. These findings suggest that Mso1p is a component of the secretory vesicle docking complex whose function is closely associated with that of Sec1p.
Keywords: exocytosis, Saccharomyces cerevisiae, secretory pathway, vesicular transport
The compartmentalization of biochemical reactions in eukaryotic cells requires an efficient sorting of polypeptides in the secretory pathway (1, 2). In yeast, this process was first studied using a set of temperature-sensitive sec mutations, which block the transport of polypeptides at specific points in the secretory pathway (3, 4). One of the corresponding genes, SEC1, is required at the terminal stage of secretion: docking and fusion of secretory vesicles to the plasma membrane. Thus, sec1 mutant cells cease to secrete proteins at restrictive temperatures, and secretory vesicles accumulate in the cytosol (3). The cloning of SEC1 revealed that it encodes a large hydrophilic protein (5).
We previously have cloned two overexpression suppressors of the sec1–1 mutation. These two genes, SSO1 and SSO2, encode two closely related membrane proteins that together perform an essential function in yeast (6). Their mammalian homologs are known as syntaxins, and members of this protein family are conserved in all eukaryotes (7). The syntaxins are located in the plasma membrane, and in vitro studies of synaptic vesicle transport led to the proposal that the syntaxins interact with two proteins on the transport vesicle surface, synaptobrevin and synaptotagmin (8, 9). Genetic evidence in yeast suggests that a number of other proteins, including Sec1p, also participate in vesicle docking and/or fusion to the plasma membrane (6). Physical interactions have been demonstrated between several of these proteins both in yeast and in mammalian cells (10–12).
We now have cloned a third suppressor of sec1–1. This gene, MSO1, encodes a small hydrophilic protein that shows no similarity to the syntaxins or to any other protein in the databases. We find that MSO1 is a highly specific suppressor of sec1 mutations. Moreover, a disruption of MSO1 shows synthetic lethality with mutations in SEC1, SEC2, and SEC4, and synthetic phenotypes with mutations in several other SEC genes. We further found that the mso1-disrupted cells accumulate secretory vesicles in the bud, which shows that Mso1p, like Sec1p, functions in the terminal stage of secretion. Finally, we show that the Mso1p protein interacts physically with Sec1p. These findings suggest that Mso1p is a component of the secretory vesicle docking complex that is closely associated with Sec1p.
MATERIALS AND METHODS
Yeast Strains.
The yeast strains used are shown in Table 1. MSO1 was disrupted by cloning a URA3 HindIII-SmaI fragment between the EcoRI and BamHI sites (Fig. 1). Suppression of a sec1 disruption, a slp1(vam5) disruption and of Sly1p depletion was tested as previously described using strains D121, YW21–1A, and GSF4, respectively (6). Suppression of syntaxin depletion was tested in strain H458, which lacks SSO2 and has SSO1 under control of the GAL1 promoter. It was made from H440 (6) through one-step replacement of sso1-δ1::URA3 by sso1-δ1::LEU2, using direct selection for LEU2. Strain Y190 (13) was used for the two-hybrid experiments.
Table 1.
Yeast strains
| Name | Genotype | Source |
|---|---|---|
| H613 | a ura3-52 leu2-3,112 mso1-δ1::URA3 | This work |
| H614 | α ura3-52 leu2-3,112 mso1-δ1::URA3 | This work |
| H629 | α ura3-52 leu2-3,112 | This work |
| NY3 | a ura3-52 sec1-1 | P. Novick |
| NY24 | a ura3-52 sec1-11 | P. Novick |
| NY179 | a ura3-52 leu2-3,112 | P. Novick |
| NY770 | α ura3-52 leu2-3,112 sec2-41 | P. Novick |
| NY772 | a ura3-52 leu2-3,112 sec3-2 | P. Novick |
| NY774 | α ura3-52 leu2-3,112 sec4-8 | P. Novick |
| NY776 | α ura3-52 leu2-3,112 sec5-24 | P. Novick |
| NY778 | α ura3-52 leu2-3,112 sec6-4 | P. Novick |
| NY780 | α ura3-52 leu2-3,112 sec8-9 | P. Novick |
| NY782 | a ura3-52 leu2-3,112 sec9-4 | P. Novick |
| NY784 | a ura3-52 leu2-3,112 sec10-2 | P. Novick |
| NY786 | a ura3-52 leu2-3,112 sec15-1 | P. Novick |
| NY1213 | α ura3-52 leu2-3,112 sec19-1 | P. Novick |
| sf821-8A | a ura3-52 leu2-3,112 his4-580 | R. Schekman |
| trp1-289 sec7-1 | ||
| mBY12-16D | α ura3-52 leu2-3,112 his4-580 | R. Schekman |
| trp1-289 sec18-1 | ||
| BY55 | α ura3-52 sec17-1 | P. Brennwald |
All H and NY strains are congenic to NY179.
Figure 1.
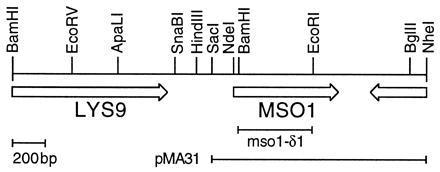
Restriction map of MSO1. ORFs are shown as arrows. Also shown is the mso1-δ1 deletion used in one-step gene disruptions, and the insert of the smallest plasmid with suppressing activity, pMA31. The insert of pMA30 extends beyond the right end of the map.
Plasmids.
Plasmid pMA30 is pHR81 (14) with a 4.3-kb genomic DNA insert that includes MSO1. Plasmid pMA31 (Fig. 1) has a SacI-NheI fragment of pMA30 cloned between the SacI and XbaI sites of pHR81. The MSO1 ORF with SmaI sites added at both ends was PCR-amplified from pMA30 and cloned into the SmaI site of pBluescript KS(−). The amplified sequence was verified on both strands. The MSO1 SmaI fragment was then cloned into the SmaI site of pGAT-4 (a kind gift from J. Peränen, Institute of Biotechnology, University of Helsinki), producing pHis6-GST-MSO1. Finally, pHis6-MSO1 was made by cutting this plasmid with SpeI and religating it, thus removing the glutathione S-transferase (GST) DNA.
For two-hybrid experiments, we used the system of Fields and Song (15). Plasmid pMA36 is pGBT9 (16) with an ApaLI-XbaI fragment of the original SEC1 plasmid (5) cloned into the SmaI site. Both sites were filled in before ligation. Plasmid pMA42 has an NdeI-BglII fragment of MSO1 cloned between the EcoRI and BamHI sites of pGBT9. The NdeI and EcoRI sites were filled in. Plasmid pMA43 has the same MSO1 NdeI-BglII fragment cloned between the EcoRI and BamHI sites of pGAD424. Plasmid pMA44 was made by filling in the EcoRI site in pMA43. Plasmids pMA46 and pMA47 were made by filling in the BlnI and AgeI sites in pMA36. Plasmid pMA49 is pGBT9 cut with BamHI, filled in, cut with SalI and ligated to a BlnI-XhoI fragment of SEC1. Plasmid pMA50, finally, has the EcoRI fragment of pMA49 cloned into the EcoRI site of pGBT9.
Preparation of Cell Lysates.
Yeast cells were resuspended in 2% SDS, 1 mM EDTA, and the following protease inhibitors: 2 mg/ml antipain, 2 mg/ml approtinin, 2 mg/ml chymostatin, 2 mg/ml leupeptin, 5 mg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride. The cells were broken by vortexing in the presence of acid-washed 45-μm glass beads. The lysates were centrifuged to remove debris and the amount of protein in the supernatants was measured (17). For the fractionation experiments, cells were grown to an OD600 of 1.0, washed in 10 mM NaN3, and then used to prepare spheroplasts (18). The spheroplasts were resuspended in 0.8 M sorbitol, 1 mM EDTA, and 10 mM ethanolamine (pH 7.2) supplemented with protease inhibitors (see above), and then broken using a ball-bearing homogenizer with a cut-off of 25 μm (19). The homogenate was centrifuged at 500 × g for 10 min to remove unbroken cells and nuclei (pellet P1). The supernatant (S1) was then centrifuged at 10,000 × g for 10 min to obtain pellet P2. The resulting S2 supernatant was centrifuged at 100,000 × g for 1 hr to obtain the microsomal pellet P3 and the S3 supernatant. Aliquots containing equal amounts of protein then were analyzed in Western blots. Membrane association was studied by incubating the P3 pellet for 30 min on ice in 10 mM Hepes buffer (pH 7.4), or in buffer containing either 1 M KCl, 2.5 M urea, or 1% Triton X-100. Membranes were removed by centrifugation, and the solubilized proteins were precipitated with chloroform/methanol (20).
Antisera.
The His6-tagged Mso1 protein encoded by pHis6-MSO1 was produced in Escherichia coli strain BL21, purified on a Ni-nitrilotriacetic acid (NTA)-agarose column in the presence of 8 M urea and used for subcutaneous immunization of rabbits. To raise antibodies against Sec1p, we used a β-galactosidase-Sec1p fusion protein expressed in E. coli from a pBluescript vector and purified from the bacterial lysate on a 6% SDS/polyacrylamide gel. The antiserum against Sso2p has been described (21). To remove nonspecific reactivity, the Mso1p antiserum was pretreated with acetone powder (22) prepared from an mso1-δ1 strain. The antiserum, in working dilution, was incubated with 1% wt/vol acetone powder at 4°C for 1 hr. The powder was removed by centrifugation, and the supernatant was used in Western blots.
In Vitro Binding.
Yeast spheroplasts were solubilized for 1 hr on ice in 40 mM MOPS (pH 6.8) containing 100 mM NaCl, 1% Tween, and 2 × protease inhibitors (see above) without EDTA. The lysate was centrifuged for 10 min at 10,000 × g and then preadsorbed for 1 hr at 4°C with Ni-NTA-agarose beads (Qiagen). The lysate was then incubated with or without His6-Mso1p for 4.5 hr at 4°C. Ni-NTA-agarose beads in solubilization buffer with 20 mM imidazole were added, and the incubation proceeded for 1 hr. The beads were collected and washed three times in solubilization buffer. Bound protein was eluted and analyzed in a Western blot.
Electron Microscopy.
Wild-type and mso1-δ1 cells were grown at 24°C in yeast extract/peptone/dextrose to an OD600 of 1.0, at which point aliquots were fixed by adding an equal volume of 6% paraformaldehyde and 4% glutaraldehyde in 0.2 M potassium phosphate buffer (pH 6.5) to the growth medium. After fixation for 1 hr at 20°C, the cells were collected by centrifugation, washed three times in 0.1 M potassium phosphate buffer (pH 6.5) and three times in water, and then treated with 1% KMnO4 for 2 hr on ice, followed by three washes in water. The samples were dehydrated and embedded in Spurr’s low viscosity media (EM Science) as described by manufacturer. Thin sections (60–80 nm) were cut, stained with lead citrate and uranyl acetate, and examined in a JEOL JEM-1200EX electron microscope.
Other Methods.
Synthetic yeast media were prepared as described (23), but with a double amount of leucine. Suppression of temperature-sensitive mutations was tested on both selective and yeast extract/peptone/dextrose plates at 30°C, 35°C, and 37°C. In the two-hybrid experiments, growth without histidine was scored in the presence of 67 mM 3-aminotriazole. Proteins for Western blots were separated on 12.5% SDS/polyacrylamide gels and blotted electrophoretically to nitrocellulose filters (24). The filters were treated with specific antisera, incubated with [35S]protein A (Amersham), and subjected to autoradiography using Kodak MR film.
RESULTS
Cloning of the MSO1 Gene.
We previously cloned two genes, SSO1 and SSO2, that suppress the temperature-sensitive sec1–1 mutation when their cDNAs are overexpressed from the ADH1 promoter (6). We reasoned that overexpression from a multicopy plasmid in which the gene’s own promoter is used might reveal other suppressor genes, providing further clues to Sec1p function. We therefore screened a genomic library in the 2 μm vector pHR81 (14) for plasmids that can suppress sec1–1. Four plasmids from this screen contained a new gene, which we call MSO1 for Multicopy suppressor of Sec One. MSO1 is located on the right arm of chromosome XIV, adjacent to LYS9 (Fig. 1). It encodes a protein of 210 amino acid residues, with a predicted molecular mass of 23,350 Da. It is hydrophilic, rich in serine residues and basic, with a pI of 10.1. The Mso1p protein sequence is not obviously related to any other protein. There are no hydrophobic stretches in the sequence that could form a membrane-spanning region, nor is there an N-terminal signal sequence.
MSO1 Is a Highly Specific Suppressor of sec1 Mutations.
To gain some insight into the mechanism of suppression, the MSO1 gene was tested for its ability to suppress different temperature-sensitive sec mutations, such as mutations in the late-acting SEC genes (sec1–1, sec1–11, sec2–41, sec3–2, sec4–8, sec5–24, sec6–4, sec8–9, sec9–4, sec10–2, and sec15–1), mutations in general components of the secretory pathway (sec17–1, sec18–1, and sec19–1), and also the sec7–1 mutation, which affects both intra-Golgi transport and budding of vesicles from the Golgi complex. We found that MSO1 suppresses both sec1–1 and sec1–11 at restrictive temperatures up to 37°C. However, it does not suppress any other sec mutations tested. MSO1 differs in this respect from SSO1 and SSO2, which can suppress mutations in many late-acting SEC genes (6).
We further tested if MSO1 can suppress a disruption of SEC1. Thus, the MSO1 plasmids were transformed into the SEC1/sec1-δ1::HIS3 diploid D121 (6). This diploid was sporulated, and tetrads were dissected. The tetrads showed 2:2 segregation for lethality, which was linked to the HIS3 marker. We conclude that overexpression of MSO1 is unable to suppress a complete loss of Sec1p. Two other yeast proteins, Sly1p and Slp1p/Vps33p/Vam5p, are related to Sec1p (25) but function at other steps in intracellular transport. We therefore tested to see if MSO1 can suppress loss of these proteins. Loss of Slp1p causes temperature sensitivity, and we found that the MSO1 plasmids are unable to suppress this phenotype. To test suppression of SLY1, which is an essential gene, we used a strain in which SLY1 is expressed from the GAL10 promoter. We found that the MSO1 plasmids are unable to support growth of this strain in the absence of galactose. Finally, we tested to see if a strain that lacks the two syntaxins Sso1p and Sso2p can survive in the presence of the MSO1 plasmid. This was done by transforming pMA30 into a yeast strain that has a single syntaxin gene expressed from the GAL1 promoter (see Materials and Methods). This strain is unable to grow in the absence of galactose, and pMA30 did not suppress this phenotype. We conclude that MSO1 is unable to suppress loss of syntaxin function.
Loss of MSO1 Causes Accumulation of Secretory Vesicles in the Bud.
To obtain more information about the function of MSO1, we disrupted the gene in both haploid and diploid cells. The resulting Mso1p-deficient strains are viable and have no obvious growth phenotypes. Thus, growth on both synthetic and rich media at different temperatures and on different carbon sources is not significantly affected. Nor is mating, sporulation, spore germination, or resistance to nitrogen starvation affected by the loss of Mso1p. The absence of a clear disruption phenotype is frequently due to the presence of duplicated genes with redundant functions. However, low-stringency Southern blots did not reveal any MSO1-related gene, and the recently completed yeast genome sequence does not contain any other gene that is clearly related to MSO1.
We checked to see if any morphological changes can be detected in the absence of Mso1p. Thus, cells from both the mso1-δ1 and wild-type strains were analyzed by electron microscopy (Fig. 2). Interestingly, a significant accumulation of 60-nm vesicles is seen in the mso1-δ1 strain. The vesicles are mostly seen in the bud region and are especially abundant in small buds. Only a few such vesicles are seen in the wild-type cells. To quantitate the effect, the total number of 60-nm vesicles in the bud region was counted in thin sections of cells from both strains. We found that 43 mso1-δ1 cells had a total of 705 vesicles in their buds. In contrast, 38 wild-type cells had only 149 vesicles in their buds. We conclude that loss of Mso1p causes a more than 4-fold accumulation of 60-nm vesicles in the bud. This suggests that the final step in secretion is partially impaired in the mso1-δ1 cells.
Figure 2.
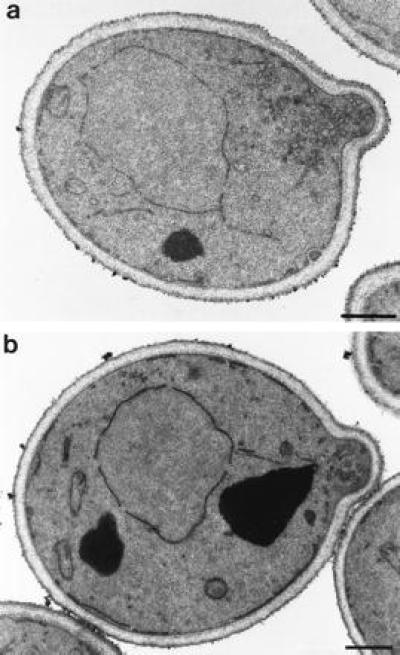
Electron micrograph of mso1 disrupted H613 cells (a) and congenic wild-type NY179 cells (b). (Bars represent 500 nm.)
Loss of MSO1 Shows Synthetic Lethality with Mutations in SEC1 and Several Other Late-Acting SEC Genes.
Because MSO1 was isolated as a suppressor of sec1–1 we tested the effect of disrupting MSO1 in a sec1–1 cell. We reasoned that loss of Mso1p might modify the phenotype of the sec1–1 mutation. Such interactions are frequently seen between genes that encode functionally related proteins. The MSO1 gene was disrupted in a wild-type strain that is congenic to the sec1–1 strain, and the two strains were mated. The resulting diploid was sporulated, and tetrads were dissected onto yeast extract/peptone/dextrose plates at a sec1–1-permissive temperature (23°C). Interestingly, we found that the mso1 disruption shows synthetic lethality with the sec1–1 mutation. Thus, all spores that contain both sec1–1 and the mso1 disruption fail to germinate, even at the permissive temperature. The synthetic lethality suggests that the function of the mutant Sec1–1 protein is partially impaired also at the permissive temperature, something that is frequently seen with temperature-sensitive mutations. It is conceivable that loss of Mso1p further reduces Sec1p function to a point where it can no longer support growth. This suggests that Mso1p and Sec1p function closely together and that Mso1p is required for full Sec1p activity.
We tested the effect of the MSO1 disruption in strains with other sec mutations. The results are summarized in Table 2. In addition to SEC1, we also found complete synthetic lethality with mutations in SEC2 and SEC4. The double-mutant spores in these crosses do not even germinate. Mutations in SEC3, SEC6, SEC8, and SEC9 have a semilethal phenotype in the absence of MSO1. Thus, some double-mutant spores germinate and form micro-colonies. Viable cells could be recovered from some of these micro-colonies, but it is conceivable that these surviving cells have acquired suppressor mutations. Mutations in SEC5, SEC10, and SEC15 are viable in the absence of MSO1, but the double-mutant strains have a clearly reduced growth at 23°C as determined from the colony size. Finally, mutations in SEC7, SEC18, and SEC19 do not show any significant interaction with the MSO1 disruption.
Table 2.
Genetic interactions of the mso1 disruption with temperature-sensitive sec mutations
| Mutation | Effect in mso1-δ1 background |
|---|---|
| sec1-1 | Lethal |
| sec1-11 | Lethal |
| sec2-41 | Lethal |
| sec3-2 | Semilethal |
| sec4-8 | Lethal |
| sec5-24 | Reduced growth |
| sec6-4 | Semilethal |
| sec7-1 | No apparent effect |
| sec8-9 | Semilethal |
| sec9-4 | Semilethal |
| sec10-2 | Reduced growth |
| sec15-1 | Reduced growth |
| sec18-1 | No apparent effect |
| sec19-1 | No apparent effect |
Semilethal means that some spores germinated and formed micro-colonies.
The Mso1 Protein Is Enriched in a Microsomal Membrane Pellet and Cofractionates with Sec1p.
A polyclonal rabbit antiserum was made against bacterially expressed histidine-tagged Mso1p. The antiserum detects some crossreacting bands in Western blots with yeast cell lysates, but pretreatment of the antiserum with aceton powder prepared from an mso1-δ1 strain significantly reduced this background. A single band representing Mso1p was identified by the fact that it is absent in the mso1-δ1 strain, but much more prominent in cells that overexpress MSO1 (Fig. 3A). The protein has an apparent molecular mass of 29 kDa. This is higher than the 23,350 Da predicted from the sequence, but close to the mobility of the bacterially expressed His6-Mso1p. It is likely that the high pI of Mso1p causes it to migrate slower during electrophoresis.
Figure 3.
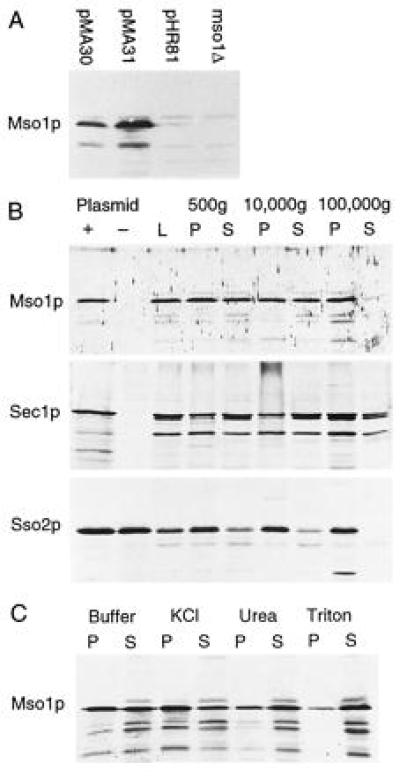
Western blots. (A) Detection of Mso1 protein in wild-type cells carrying either pMA30, pMA31, or the cloning vector pHR81. An mso1-disrupted strain is shown to the right. (B) Fractionation of yeast cell lysates by successive centrifugations at the indicated speeds. L, spheroblast lysate; P, pellet; S, supernatant. For Mso1p and Sec1p, whole cell lysates prepared in the presence and in the absence of the respective overexpression plasmid is shown (Left). There is no difference for Sso2p, because we did not need overexpression to detect this protein. (C) Membrane association of Mso1p in the 100,000 × g pellet. The pellet was treated either with Hepes buffer alone, or with buffer containing 1 M KCl, 2.5 M urea, or 1% Triton-X 100, respectively, and then separated by centrifugation at 100,000 × g.
To study the intracellular distribution of Mso1p, lysates from cells that overexpress MSO1 were fractionated by centrifugation and analyzed in Western blots. We found that Mso1p is enriched in the 100,000 × g microsomal pellet, though substantial amounts also are found in the 500 × g and 10,000 × g pellets (Fig. 3B). In contrast, only a small amount of protein is found in the 100,000 × g supernatant. This suggests that Mso1p is a membrane-associated protein. For comparison, we also studied the distribution of Sso2p and Sec1p using the same procedure (Fig. 3B). We found that Sso2p is even more tightly bound to the membranes, with no detectable protein in the 100,000 × g supernatant. This is consistent with the fact that Sso2p is an integral membrane protein. In contrast, Sec1p has a distribution that is similar to that of Mso1p, though more protein seems to be present in the final supernatant. This could mean that Sec1p is less strongly associated with the membranes than Mso1p, but the observation should be interpreted with caution because both proteins were overexpressed. We conclude that Sec1p and Mso1p to a large extent cofractionate, which is consistent with the notion that they may be present in the same complex. Finally, we note that Sec1p and Mso1p also have in common the fact that they appear to be expressed at a rather low level, as compared with Sso2p.
To investigate the nature of the membrane association, the resuspended microsomal pellet was subjected to various treatments (Fig. 3C). We found that treatment with either 10 mM Hepes buffer alone or 1 M KCl caused approximately half of the Mso1 protein to be released. In contrast, treatment with 2.5 M urea or 1% Triton X-100 released most of the protein. We conclude that while some Mso1 protein is loosely associated with the membrane, a significant fraction is tightly bound. The fact that most of the protein can be dissociated by 2.5 M urea further suggests that the binding is likely to involve polar interactions with other proteins rather than hydrophilic interactions with the membrane.
Mso1p Interacts Physically with Sec1p.
To determine if the genetic interaction between SEC1 and MSO1 reflects a direct physical interaction between the corresponding proteins, the two genes were cloned into two-hybrid vectors and tested for interaction, using both GAL1-HIS3 and GAL1-lacZ reporters. We found that the two proteins interact in this system. Thus, cells that contain both plasmids can grow in the absence of histidine, which shows that the GAL1-HIS3 reporter is expressed (Fig. 4). Expression of GAL1-lacZ also is induced in these cells. The level of β-galactosidase expression is rather low, but well above the background seen in the absence of either plasmid.
Figure 4.

Interaction of Mso1p and Sec1p in the two-hybrid system. Constructions that were tested are shown at the top. Results are shown as β-galactosidase units, with SD of three independent transformants in parentheses, and as growth in the absence of histidine. AD, activating domain; DBD, DNA binding domain.
We mapped the interacting domains within the two proteins by deletions (Fig. 4). We found that SEC1-A, in which the C-terminal part of Sec1p beyond residue 668 has been removed, can still interact with Mso1p. In contrast, SEC1-B, which retains only the 551 first residues, fails to interact with Mso1p. This suggests that the region between residues 552 and 668 is required for binding to Mso1p. However, this region is not sufficient for binding, because SEC1-D fails to interact with Mso1p. This suggests that sequences between residues 1 and 379 also are required for binding, though this region alone (SEC1-C) fails to bind Mso1p. We conclude that the binding to Mso1p requires the C-terminal region between residues 552 and 668, but also sequences N-terminal to residue 380. In this respect it resembles the interaction between the mammalian Sec1p homolog Munc-18 and syntaxin 1, which requires sequences in both the N and C termini of Munc-18 (26). A deletion also was made in MSO1, in which the sequences encoding the C-terminal 46 amino acids residues were removed. We found that this construction, MSO1-A, still interacts with Sec1p. Thus, the C-terminal part of Mso1p is not required for Sec1p binding. Finally, we note that the level of lacZ expression is 5-fold higher in cells containing SEC1-A and MSO1 than in those containing the wild-type SEC1 construct and MSO1 (Fig. 4). This suggests that the C-terminal part of the Sec1p protein may interfere with its binding to Mso1p. This could reflect a mechanism by which the Mso1p-Sec1p interaction is regulated in vivo, but other explanations are also possible.
To further verify that Mso1p interacts with Sec1p, we tested to see if histidine-tagged Mso1p can be used to isolate Sec1p from yeast cells. Thus, a lysate from the Sec1p-overexpressing strain was incubated with His6-Mso1p followed by Ni-NTA-agarose beads. Protein that bound to the beads was analyzed in a Western blot using an antiserum against Sec1p. As shown in Fig. 5, Sec1p binds to the beads in the presence, but not in the absence, of His6-Mso1p. We conclude that the in vitro association experiment confirms that Mso1p binds to Sec1p.
Figure 5.
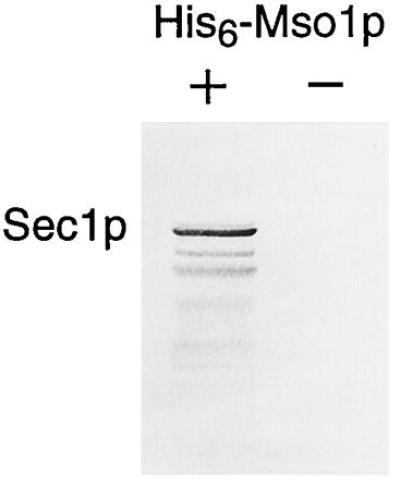
In vitro association of Sec1p and Mso1p. A lysate from the Sec1p-overproducing strain was incubated with Ni-NTA-agarose beads in the presence or in the absence of His6-Mso1p. Bound protein was analyzed in a Western blot using an antiserum against Sec1p.
DISCUSSION
Mso1: A New Protein That Is Involved in Secretion.
We previously cloned cDNAs for the SSO1 and SSO2 genes encoding yeast syntaxins by their ability to suppress the sec1–1 mutation when overexpressed (6). We now describe a third suppressor of sec1–1: the MSO1 gene. MSO1 encodes a small hydrophilic protein with no strong similarity to any previously known protein. It should be noted, however, that the sequence of Mso1p is rich in serine residues, which constitute 17% of the entire protein. It therefore shows a limited sequence similarity to several serine-rich proteins from yeast and other organisms. The absence of signal sequences in Mso1p suggests that it is a cytosolic protein. However, our results show that Mso1p is membrane associated, and that a significant fraction of the protein is released only by treatment with 2.5 M urea or 1% Triton X-100 (Fig. 2C).
Mso1p Functions in the Terminal Stage of Secretion and Is Closely Associated with Sec1p.
Several lines of evidence suggest that Mso1p functions in the terminal step of secretion, i.e., the docking of secretory vesicles to the plasma membrane, and that Mso1p is particularly closely associated with Sec1p. First, the MSO1 gene is a highly specific suppressor of SEC1. It differs in this respect from the syntaxin genes SSO1 and SSO2, which can suppress mutations in several late-acting SEC genes (6). Second, the Mso1p protein cofractionates with Sec1p (Fig. 3) and interacts physically with Sec1p (Figs. 4 and 5). Third, loss of Mso1p causes a significant accumulation of secretory transport vesicles in the bud (Fig. 2), which shows that the terminal step in secretion is partially impaired. Fourth, loss of Mso1p shows strong genetic interactions with mutations in all late-acting SEC genes that were tested (Table 2). The severity of the interaction varies from complete synthetic lethality to clearly reduced growth. It should be noted that this kind of genetic interaction is frequently seen in cases where the two encoded proteins interact directly with each other. It therefore suggests that Mso1p may interact physically with other components of the vesicle docking complex, in addition to Sec1p.
Composition of the Secretory Vesicle Docking Complex.
A large number of yeast proteins now have been identified that are involved in the docking of secretory transport vesicles at the plasma membrane. Based on the results reported here as well as previous findings, it would appear that these proteins fall into three major groups. First, there is the SNARE complex (9), which in yeast comprises the Sso1/2p, Snc1/2p, and Sec9p proteins (6, 12, 27). These proteins form a complex that is believed to play a role in vesicle docking and also in the recruitment of general factors such as Sec17p and Sec18p, that are required either for the assembly of the SNARE complex (28) or for the fusion step (29–31). A second distinct group of proteins comprises Sec3p, Sec5p, Sec6p, Sec8p, Sec10p, Sec15p, and Exo70p. These proteins are subunits of the 19.5S exocyst complex (32, 33). It is notable that the mso1 disruption shows synthetic interactions with at least six of the seven exocyst subunits (Table 2).
The third group of proteins comprises the remaining late-acting SEC gene products, i.e., Sec1p, Sec2p, Sec4p, and Mso1p. There is some evidence that these four proteins belong together. Thus, we have now shown that two of the proteins, Sec1p and Mso1p, interact physically. Moreover, the MSO1 disruption interacts most strongly with mutations in SEC1, SEC2, and SEC4, resulting in complete synthetic lethality (Table 2). This suggests that Mso1p is functionally more closely associated with Sec1p, Sec2p, and Sec4p than with the subunits of the SNARE or exocyst complexes. It is conceivable that Sec1p, Sec2p, Sec4p, and Mso1p could form a third distinct complex, though a direct interaction so far has been shown only between Sec1p and Mso1p. The fact that MSO1 suppresses only sec1 mutations, and not mutations in SEC2 or SEC4, suggests a particularly close relationship between Sec1p and Mso1p. Possible functions of Mso1p could be to regulate Sec1p function or to mediate contacts between Sec1p and other proteins involved in secretion.
Duplicated and Unique Proteins in the Secretory Pathway.
Finally, we note that no other protein that is closely related to Mso1p seems to be encoded by the yeast genome. The fact that both Sec1p and Sso1p/Sso2p have duplicated homologs that function in other transport steps previously led us to propose that the machinery for vesicle transport has been duplicated as new intracellular compartments arose during evolution (6, 25). However, these duplications seem to have been restricted to certain key components with a direct role in targeting of transport vesicles to the correct membrane. In contrast, proteins with a more general function, such as Sec18p and Sec17p, have not been duplicated and therefore are used in several different transport steps. A third group of proteins has now emerged that is involved only in the terminal step of secretion, but still lacks duplicated homologs that function in other transport steps. This group of proteins includes Mso1p and the subunits of the exocyst complex (33). The existence of this group of proteins suggests that the terminal step in secretion is particularly complex, which may reflect unique functions that are not required in other transport steps.
Acknowledgments
We thank Dan Fredriksson, Riitta Lampinen, and Mervi Lindman for skillful technical assistance, and Peter Novick for providing us with his congenic yeast strains. This work was supported by grants from the Swedish Natural Research Council (H.R.), the Swedish Cancer Society (H.R.), and the Academy of Finland (S.K.). M.A. was supported by a fellowship from the Academy of Finland and a grant from the University of Helsinki.
ABBREVIATION
- NTA
nitrilotriacetic acid
Footnotes
Data deposition: The sequence reported in this paper has been deposited with the GenBank database (accession no. U56416).
References
- 1.Jamieson J, Palade G. J Cell Biol. 1968;39:589–603. doi: 10.1083/jcb.39.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palade G. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 3.Novick P J, Schekman R. Proc Natl Acad Sci USA. 1979;76:1858–1862. doi: 10.1073/pnas.76.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novick P J, Ferro S, Schekman R. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- 5.Aalto M K, Ruohonen L, Hosono K, Keränen S. Yeast. 1991;7:643–650. doi: 10.1002/yea.320070613. , and corrigendum (1992) 8, 587–588. [DOI] [PubMed] [Google Scholar]
- 6.Aalto M K, Ronne H, Keränen S. EMBO J. 1993;12:4095–4104. doi: 10.1002/j.1460-2075.1993.tb06093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett M K, Garcia-Arraras J E, Elferink L A, Peterson K, Fleming A M, Hazuka C D, Scheller R H. Cell. 1993;74:863–873. doi: 10.1016/0092-8674(93)90466-4. [DOI] [PubMed] [Google Scholar]
- 8.Söllner T, Bennett M K, Whiteheart S W, Scheller R H, Rothman J E. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 9.Söllner T, Whiteheart S W, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman J E. Nature (London) 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 10.Hanson P I, Otto H, Barton N, Jahn R. J Biol Chem. 1995;270:16955–16961. doi: 10.1074/jbc.270.28.16955. [DOI] [PubMed] [Google Scholar]
- 11.Kee Y, Lin R C, Hsu S-C, Scheller R H. Neuron. 1995;14:991–998. doi: 10.1016/0896-6273(95)90337-2. [DOI] [PubMed] [Google Scholar]
- 12.Brennwald P, Kearns B, Champion K, Keränen S, Bankaitis V, Novick P. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 13.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 14.Nehlin J O, Carlberg M, Ronne H. Gene. 1989;85:313–319. doi: 10.1016/0378-1119(89)90423-x. [DOI] [PubMed] [Google Scholar]
- 15.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 16.Bartel P L, Chien C-T, Sternglanz R, Fields S. In: Cellular Interactions in Development: A Practical Approach. Hartley D A, editor. Oxford: Oxford Univer. Press; 1993. pp. 153–179. [Google Scholar]
- 17.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 18.Walworth N C, Novick P J. J Cell Biol. 1987;105:163–174. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balch W E, Rothman J E. Arch Biochem Biophys. 1985;240:413–425. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 20.Wessel D, Flügge U I. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 21.Jäntti J, Keränen S, Toikkanen J, Kuismanen E, Ehnholm C, Söderlund H, Olkkonen V. J Cell Sci. 1994;107:3623–3633. doi: 10.1242/jcs.107.12.3623. [DOI] [PubMed] [Google Scholar]
- 22.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 23.Sherman F, Fink G, Hicks J B. Methods in Yeast Genetics: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. [Google Scholar]
- 24.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aalto M K, Keränen S, Ronne H. Cell. 1992;68:181–182. doi: 10.1016/0092-8674(92)90462-l. [DOI] [PubMed] [Google Scholar]
- 26.Hata Y, Südhof T C. J Biol Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- 27.Protopopov V, Govindan B, Novick P, Gerst J E. Cell. 1993;74:855–861. doi: 10.1016/0092-8674(93)90465-3. [DOI] [PubMed] [Google Scholar]
- 28.Mayer A, Wickner W, Haas A. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 29.Clary D, Griff I, Rothman J E. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser C, Schekman R. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- 31.Pevsner J, Hsu S-C, Braun J E, Calakos N, Ting A E, Bennett M K, Scheller R H. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 32.TerBush D R, Novick P. J Cell Biol. 1995;130:299–312. doi: 10.1083/jcb.130.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.TerBush D R, Maurice T, Roth D, Novick P. EMBO J. 1996;15:6283–6294. [PMC free article] [PubMed] [Google Scholar]