

Abstract
The protein Sex-lethal (SXL) controls pre-mRNA splicing of two genes involved in Drosophila sex determination: transformer (tra) and the Sxl gene itself. Previous in vitro results indicated that SXL antagonizes the general splicing factor U2AF65 to regulate splicing of tra. In this report, we have used transgenic flies expressing chimeric proteins between SXL and the effector domain of U2AF65 to study the mechanisms of splicing regulation by SXL in vivo. Conferring U2AF activity to SXL relieves its inhibitory activity on tra splicing but not on Sxl splicing. Therefore, antagonizing U2AF65 can explain tra splicing regulation both in vitro and in vivo, but this mechanism cannot explain splicing regulation of Sxl pre-mRNA. These results are a direct proof that Sxl, the master regulatory gene in sex determination, has multiple and separable activities in the regulation of pre-mRNA splicing.
Keywords: transformer, U2AF–Sxl chimeric transgenes
Sex determination in Drosophila is controlled by the gene Sex-lethal (Sxl) (1). This gene encodes an RNA binding protein with high affinity for poly(U) sequences that induces female-specific patterns of alternative splicing in two different pre-mRNAs: the pre-mRNAs for transformer (tra) (2–4) and Sxl (5) (Fig. 1a).
Figure 1.

(a) Schematic representation of the exons and introns of tra and Sxl pre-mRNAs that are relevant for the regulated patterns of splicing controlled by the protein SXL. In the absence of SXL (male flies) the non-sex-specific (nss) 3′ splice site of tra is used, and the male-specific (ms) exon of Sxl is included. The presence of SXL in female flies induces the partial use of the female-specific (fs) site in tra and skipping of the ms exon in Sxl. (b) Representation of the structural domains of Sxl, U2AF65, USx, and RS–Sx proteins. Sxl is composed of two RNP consensus motifs (RNPa and RNPb) and an NH2-terminal glycine/asparagine-rich (GN) region. U2AF65 is composed of three RNP consensus motifs that form its RNA binding domain and an NH2-terminal region—the effector domain—composed of two subdomains: an arginine/serine-rich (RS) motif and a region involved in protein–protein interactions (PI).
In tra pre-mRNA, SXL represses the use of a non-sex-specific default 3′ splice site, thereby allowing the use of an alternative female-specific site that is not used in the absence of SXL (Fig. 1a) (6, 7). SXL binds to the polypyrimidine tract associated with the non-sex-specific site, and in vitro data indicate that there it antagonizes the general splicing factor U2AF (8). U2AF is thereby diverted to the female-specific site, for which it has lower affinity. Use of the female-specific site allows production of functional TRA protein. TRA, in addition to the product of the gene transformer 2 (tra-2) (9, 10), controls by alternative splicing the expression of the gene double-sex (dsx), which is responsible for somatic sexual differentiation (11, 12).
SXL also induces skipping of the male-specific exon 3 from its own pre-mRNA (Fig. 1a) (5, 13). The presence of this exon places a stop codon in the Sxl ORF, which gives rise to a truncated and probably nonfunctional protein. Skipping of exon 3 generates Sxl-encoding transcripts, thereby establishing a positive autoregulatory loop necessary to maintain female differentiation. High-affinity binding sites for SXL have been identified in both of the introns flanking exon 3 (14–17).
SXL is composed of two domains: an NH2-terminal region rich in glycine and asparagine residues (GN domain) (18), which has been implicated in binding cooperativity, and two ribonucleoprotein consensus (RNP-CS) motifs that are responsible for specific RNA recognition (5, 19, 20) (Fig. 1b). U2AF65 is a general splicing factor with two functional domains (21): a COOH-terminal RNA binding domain composed of three RNP-CS motifs, which is responsible for recognition of the polypyrimidine tracts associated with 3′ splice sites (19); and an NH2-terminal effector domain that promotes the association of U2 small nuclear RNP with the branch point and that is composed of an arginine/serine-rich (RS) motif and a region (PI) that establishes protein–protein interactions with other splicing factors (8, 22) (Fig. 1b).
Previous in vitro results suggested that SXL regulates tra splicing by antagonizing U2AF binding to the non-sex-specific 3′ splice site. In this report, we have used a chimeric protein in which the RNA binding domains of SXL were fused to the effector domain of U2AF65 to test whether antagonism for U2AF can explain splicing regulation of tra and Sxl in vivo. Our results confirm the model proposed for tra regulation in vitro and indicate that a different molecular mechanism operates to regulate Sxl splicing.
MATERIALS AND METHODS
Flies Stocks and Cultures.
Flies were cultured on standard food. SxlfP7BO represents a deletion for the entire Sxl transcription unit (23). Sxlf1 is a null mutation of Sxl (24). Dp(1;3)sn13a1, Sxl+ represents a transposition of material from the X chromosome, carrying the wild-type Sxl+ locus, into autosome III. TM3, Sb Ser is a balancer chromosome carrying the dominant marker mutations Sb (Stubble) and Ser (Serrate). For further description of mutations and chromosomes see Lindsley and Zimm (25).
Construction of the Transgenes.
The USx transgene was prepared by cloning a blunt-ended SacI–KpnI fragment from plasmid d7a into the pHT4 transformation vector (26) digested with Asp418 and also blunt-ended with the Klenow fragment of DNA polymerase I. Plasmid d7a was generated by cloning a 684-bp EaeI fragment of SXL cDNA into the HindIII site of pU2AF DRNP2 (21), both DNAs blunt-ended with the Klenow fragment. The resulting plasmid encodes a fusion protein between amino acids 1 and 233 of U2AF65 and amino acids 95 and 323 of SXL.
The RS–Sx transgene was prepared by cloning a PCR product of plasmid a23 (see below) flanked with KpnI sites into the KpnI site of pHT4. The PCR product contains sequences able to encode a fusion protein between the RS domain of U2AF65 (amino acids 1–55, with the initiator methionine placed in the same translational initiation context as SXL) and the complete amino acid sequence of SXL. Plasmid a23 was obtained by cloning a blunt-ended BamHI fragment containing SXL coding sequences from plasmid pGEM3-Sxl (8) into the SmaI site of pGEX-3X-U2AF65 (21).
The Sxl-ΔGN transgene was prepared by inserting a PCR product corresponding to SXL coding sequences from amino acid 95 to the stop codon into the KpnI site of pHT4 blunt-ended with the Klenow fragment. Met-95 was placed in a context appropriate for translation in Drosophila with the following sequence: CAAAATG(Met-95).
P-Element-Mediated Germ-Line Transformation.
The USx, RS–Sx, and Sxl-ΔGN transgenes were cloned into the P-element transformation vector pHT4 (26), under the control of the hsp70 promotor; and germ-line transformants were generated by standard procedures (27). Four lines for USx, two lines for RS–Sx and four lines for Sxl-ΔGN were obtained. All of the lines corresponding to each transgene behaved similarly. Then, the experiments reported in this article were performed with one and the same line for each transgene.
Reverse Transcription–PCR (RT-PCR) Analyses.
Adult flies with the indicated genotypes were maintained at 37°C (heat shock, hs) for 90 min, and then RNA was isolated either immediately after heat-shock (Sxl–lacZ RNA splicing) or after 48 h of recovery at 25°C (tra RNA splicing). RNA preparation from frozen adults was performed as described (28, 29). Ten micrograms of total RNA was reverse-transcribed by avian myeloblastosis virus reverse transcriptase from Promega. PCR amplifications were done by the procedure of Frohman et al. (30). The primers used for the RT-PCR analysis of Sxl–lacZ RNA splicing were P1 (5′-CGCATCGTAACCGTGCATCTGC-3′), P2 (5′-CGCCATTCAGGCTGCGCAACTG-3′, and P3 (5′-GTGGTTATCCCCCATATGGC-3′). The P1 and P2 primers correspond to sequences of the lacZ fragment in the reporter construct Sxl–lacZ. Reverse transcription was carried out with P1 primer, the first PCR used P1 and P3 primers, and the second PCR used P2 and P3 primers. The primers used for the RT-PCR analysis of tra RNA splicing were P1 (5′-CTTCGGCTGCGATTGCGGCTC-3′), P2 (5′-GATCTGGAGCGAGTGCGTCTG-3′), and P3 (5′-GGTCACACTGAGGAAAGTGC-3′). Reverse transcription was carried out with P1 primer, the first PCR used P1 and P3 primers, and the second PCR used P2 and P3 primers. In the first PCR, there was one cycle of 95°C for 3 min, 62°C for 2 min, and 72°C for 40 min, and then 25 cycles, each of 95°C for 45 s, 62°C for 2 min, and 72°C for 1.5 min. After the first PCR, 10% of the amplified product was reamplified by one additional round of 25 cycles (15 cycles in the case of tra RNA) each of 95°C for 45 s, 62°C for 2 min, and 72° for 1.5 min. In the second PCR, products were amplified with the same 5′ primer as for the first PCR but with a different 3′ primer that was internal to the first 3′ primer, so that it is possible to get doublets about the expected size (13, 31). The PCR products were fractionated on 1.2% agarose gels and subsequently transferred to nylon filters. Blots were prehybridized at 68°C in 5× standard saline citrate (SSC), 1% SDS, 2% of nonfat dry milk, and denatured salmon sperm DNA (0.1 mg/ml). Hybridization was carried out at 68°C for 15 h in prehybridization solution containing 32P-labeled Sxl cDNA probe (see Fig. 4) or 32P-labeled tra cDNA (see Fig. 2 and Fig. 3A). Filters were washed twice in 2× SSC/0.1% SDS at room temperature and twice in 0.1× SSC/0.1% SDS at 65°C.
Figure 4.
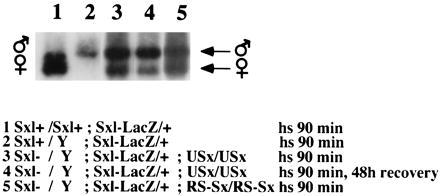
Effect of USx and RS–Sx transgenes on splicing of the Sxl–lacZ reporter gene. Transgenic flies with the indicated genotypes were maintained at 37°C for 90 min to allow expression of the transgenes, then RNA was isolated, and RT-PCR was performed to quantify the ratio between the alternatively spliced products. The positions of the RT-PCR products corresponding to the male- and female-specific spliced transcripts are indicated.
Figure 2.

Effect of USx and RS–Sx transgenes on the splicing of tra pre-mRNA. Transgenic flies with the indicated genotypes were maintained at 37°C for 90 min to allow expression of the transgenes, and RNA was isolated after 48 h at 25°C. RT-PCR was then performed to quantify the ratio between the alternatively spliced products. The positions of the RT-PCR products corresponding to the non-sex-specific and female-specific spliced transcripts are indicated. SxlcF1 refers to a female Sxl cDNA transgene (13).
Figure 3.

(a) Effect of the Sxl-ΔGN transgene on tra pre-mRNA splicing in vivo. The analysis was performed as in Fig. 2. (b) Binding of recombinant SXL and SXL-ΔGN to tra non-sex-specific 3′ splice site. Mobility-shift assays with the indicated purified recombinant SXL derivatives at a protein concentration of 5 × 10−8 M were performed. The position corresponding to the migration of unbound RNA is indicated. (c) Regulation of tra alternative 3′ splice site choice by SXL and SXL-ΔGN-in vitro. In vitro splicing reactions in the presence of the indicated purified recombinant proteins was performed and analyzed by primer extension. The products of each primer-extension reaction, corresponding to each of the spliced RNAs, are indicated.
RNA Binding Assays.
A 33-nucleotide RNA corresponding to positions 165–188 of tra pre-mRNA was synthesized as described by Milligan et al. (32). Briefly, 10 fmol of this RNA (30,000 cpm) was incubated with purified recombinant SXL protein and derivatives at a protein concentration of 5 × 10−8 M, in 20 mM Hepes, pH 8.0/0.5 mM EDTA/0.1 M KCl/20% glycerol/1 mM dithiothreitol, and in the presence of 3 μg of tRNA as unspecific competitor. The RNA–protein complexes were fractionated on a 4% native polyacrylamide gel by electrophoresis at 15 V/cm, at 4°C for 90 min. The gel was then dried and exposed to photographic film for 12 h without intensifying screen.
In Vitro Splicing Assays.
In vitro splicing reactions (30 μl) were performed with 10 fmol of M-tra pre-mRNA (8), 2.4 mM MgCl2, 67 mM KCl, 22 mM creatine phosphate, RNasin (0.9 unit/μl), 13 mM Hepes (pH 8.0), 0.13 mM EDTA, 13% glycerol, and 29% nuclear extract (∼3 μg/μl, final protein concentration), in the presence of the indicated purified recombinant SXL protein or derivatives at 0.3 μg/μl, at 30°C for 90 min. The reactions were stopped by digestion with proteinase K, phenol/chloroform extraction, and precipitation with ethanol.
To analyze the spliced products, the RNA isolated from the splicing assays was resuspended in 24 μl of water and divided in two aliquots. Each aliquot was hybridized to 120 fmol of 32P-labeled oligodeoxynucleotide complementary to the last 5 nucleotides of tra exon 1 and to the first 13 nucleotides of either tra non-sex-specific exon or the female-specific exon. These splice junction primers can only hybridize with either the non-sex-specific or the female-specific spliced products, respectively (8). The hybridization reactions were performed in 40 mM Pipes, pH 6.4/1 mM EDTA/400 mM NaCl/0.2% SDS, at 37° C for 12 h and stopped by addition of 180 μl of 0.5 M ammonium acetate and precipitation with isopropanol. The pellet was washed with ethanol 80% and resuspended in 9.5 μl of RT mixture containing 50 mM Tris⋅HCl (pH 8.3), 74 mM KCl, 6 mM MgCl2, all four dNTPs (each at 1 mM), 10 mM dithiothreitol, RNasin (0.5 unit/μl), and avian myeloblastosis virus reverse transcriptase (Promega; 0.5 unit/μl). The primer-extension reactions were incubated at 42°C for 60 min, stopped by addition of 15 μl of formamide loading dye, boiled for 5 min, loaded on a 10% polyacrylamide denaturing gel, and electrophoresed at 15 V/cm for 90 min. The gel was exposed to photographic film for 12 h with one intensifying screen and the products of primer extension corresponding to each of the the spliced products were displayed together for ease of comparison, as described (8).
Recombinant Proteins.
All the recombinant proteins used were expressed in and purified from Escherichia coli as fusions with glutathione S-transferase (GST) (33), as described (8). GST–Sxl was expressed from the plasmid pGEX-2T-Sxl (8). GST–Sxl (DGN) was expressed from plasmid pGEX-2T-Sxl (DGN), which was obtained by cloning a Klenow-treated 684-bp EaeI fragment from Sxl cDNA, corresponding to the sequences encoding the two RNP motifs of the SXL protein (amino acids 95–323), into the SmaI site of pGEX-2T. GST–Sxl (RNPb−), which contains residues 1–206 of SXL fused to GST and, therefore, lacks the second RNP motif of SXL, was expressed from plasmid pGEX-Sxl (RNPb−). This plasmid was prepared by cloning a BamHI–ClaI fragment from pGEM3-Sxl (8), in which the ClaI site was blunt-ended with the Klenow fragment, into pGEX-2T digested with BamHI and SmaI.
RESULTS AND DISCUSSION
By using an in vitro alternative splicing assay for tra pre-mRNA, it has been reported (8) that when the RNA binding domains of SXL were fused to the effector domain of U2AF65 (USx, Fig. 1b), the chimeric protein retained SXL RNA binding properties but stimulated, rather than inhibited, the use of the non-sex-specific site. This result indicated that SXL antagonizes U2AF65 function to regulate tra splicing. To test whether this mechanism also operates in vivo and whether a similar mechanism operates in the control of Sxl alternative splicing, we generated transgenic flies transformed with the USx chimeric gene under the control of a heat shock promoter.
We first studied the effect of USx expression on tra pre-mRNA splicing. To separate the effect of the transgene from the effect of endogenous SXL protein that might be induced by the transgene acting on endogenous Sxl transcripts, we used males deficient for the Sxl gene. Fig. 2 shows that, as reported (13, 31), expression of a Sxl cDNA (SxlcF1) under the heat shock promoter induces the production of female-specific transcripts in males deficient for the endogenous Sxl gene (lane 1). Expression of USx under the same experimental conditions, however, was unable to induce female-specific splicing in tra pre-mRNA (lane 2). In contrast, when tra pre-mRNA splicing was analyzed in USx males containing an endogenous Sxl+ gene, activation of female-specific tra splicing was observed (lane 3). This result could be explained if expression of USx was by itself unable to activate female-specific tra pre-mRNA splicing but was able to activate the female-specific splicing of the endogenous Sxl gene, therefore stimulating the production of endogenous SXL protein and consequently activating female-specific tra splicing (see below).
As a further test for specificity, we obtained transgenic flies expressing a chimeric protein (RS–Sx) in which only the arginine/serine-rich region of U2AF65 was fused to the NH2 terminus of SXL (Fig. 1b). This fusion protein does not have U2AF activity because it lacks an essential region for the interaction of U2AF65 with a splicing factor of the DEAD box family of RNA-dependent ATPases with homology to RNA helicases. This fusion protein fails to complement U2AF-depleted nuclear extracts and behaves as SXL for splicing regulation of tra in vitro (J. Fleckner, M. Zhang, J.V., and M.R.G., unpublished results). Contrary to USx, RS–Sx induced the female-specific splicing of tra pre-mRNA (Fig. 2, lane 4). These results indicate that RS–Sx has Sxl+ function on tra regulation and argue that USx is unable to regulate tra splicing because it contains a complete functional U2AF65 effector domain.
The chimeric USx protein lacks the NH2-terminal 94 amino acids of SXL. This region is rich in glycine and asparagine residues and has been implicated in cooperative binding of SXL to RNA (18). To rule out that the inability of USx to regulate tra splicing was due to this difference with wild-type SXL, we prepared transgenic flies expressing a SXL protein truncated at its NH2-terminal domain (Sxl-ΔGN). Fig. 3a shows that this NH2-terminal domain is not required to regulate tra splicing in vivo. Female-specific splicing was induced on transgenic male flies by expression of the Sxl-ΔGN transgenes (Fig. 3a, lane 3). Fig. 3b shows that this domain is not required for binding of SXL to the non-sex-specific site of tra, and Fig. 3c shows that it is not required to mediate tra alternative splicing in vitro. In contrast, a SXL mutant lacking one of the RNA binding domains abolished both activities. We conclude that the NH2-terminal domain of SXL is dispensable for the regulation of tra splicing and, therefore, that the absence of regulatory activity in USx can be attributed to the presence of U2AF65 effector domain.
Next, we analyzed the direct effect of expression of the USx and RS–Sx transgenes in Sxl pre-mRNA splicing. To separate the effect of the transgene from the effect of endogenous SXL protein that might be induced by the transgene acting on endogenous Sxl transcripts, we used males deficient for the Sxl gene. These males also contained a Sxl–lacZ reporter gene that contains all the cis-regulatory sequences necessary for sex-specific splicing but does not encode any functional SXL protein (13). Expression of Sxl cDNA has been shown to induce, at least partially, the use of female-specific spliced transcripts of this reporter (13). Fig. 4 shows that expression of USx (lane 3), or RS–Sx (lane 5), also induces female-specific splicing of Sxl–lacZ RNA. Heat shock treatment alone did not affect the sex-specific pattern of Sxl alternative splicing, because control males and females have, respectively, the corresponding male and female patterns of splicing in Sxl–lacZ RNAs (lanes 1 and 2). We conclude that expression of the chimeric USx, or RS–Sx, protein induces female-specific splicing of Sxl pre-mRNA and, therefore, that USx and RS–Sx resemble wild-type SXL for this type of splicing regulation. The extent of female-specific splicing induction by the transgenes is similar to that previously reported for expression of a wild-type Sxl cDNA (13). Although we cannot exclude minor quantitative differences among the activities of USx, RS–Sx, and SXL, viability results presented below indicate that the transgenes have enough Sxl activity to regulate Sxl splicing.
In addition, the results of this experiment also indicates that the failure of USx to induce the female-specific splicing of tra pre-mRNA is not due to a possible degradation of the USx protein during the 48 h of recovery after heat shock. Fig. 4, lane 4, shows that when the splicing pattern of Sxl–lacZ RNA was analyzed in males Sxl−/Y; Sxl–lacZ/+ homozygous for USx, subject to the the same experimental conditions (90 min of heat shock and 48 h of recovery at 25°C), female-specific splicing of the Sxl–lacZ reporter gene could be detected. This result indicates that USx showed Sxl+ function for Sxl pre-mRNA splicing, but Sxl− for tra splicing. These results argue that providing U2AF activity to the splicing regulator SXL abolishes its regulatory function on tra splicing but not on Sxl splicing.
To further confirm these results in a more physiological and biologically meaningful context, we studied the viability of USx and RS–Sx transgenic males after heat shock. Because SXL also controls the process of dosage compensation (hypertranscription of the single male X chromosome), gain of Sxl function results in male-specific lethality (1). If USx and RS–Sx behave as Sxl+, their expression should cause lethality in males but have no effect on female viability. This expectation proved to be true. Female flies, either heterozygous or homozygous for USx or RS–Sx are fully viable. Males, on the contrary, showed reduced viability, which is more pronounced in homozygosis than in heterozygosis (data not shown). To confirm that this male-specific lethality depends on Sxl function, two complementary analyses were performed. (i) Duplications of endogenous Sxl+ alleles increased the lethality of males heterozygous for USx or RS–Sx (Fig. 5A). (ii) The lethality of the males homozygous for USx, or RS-Sx, was reversed in Sxl-deficient flies (Fig. 5A). Therefore, expression of USx, or RS-Sx, causes Sxl-dependent male-specific lethality. This effect can be explained as indicated in Fig. 5B. Usx (or RS–Sx) are transiently expressed during heat shock, and then USx (or RS–Sx) proteins accumulate. If the transgenic protein shows Sxl+ function, it will induce female-specific splicing of the endogenous Sxl pre-mRNA. Consequently, endogenous Sxl protein is produced, establishing a positive feedback loop on its own pre-mRNA that maintains both the induction of female-specific splicing and the continuous production of endogenous Sxl protein. This protein will prevent hypertranscription of the single male X chromosome, causing the lethality of the transgenic males but have no effect on female viability. For males heterozygous for the transgene, the probability of the endogenous Sxl feedback positive loop to become established would be higher if the males carry two copies of Sxl+ alleles. However, if the transgenic males are deficient for endogenous Sxl+ alleles, no endogenous Sxl protein is produced by the transient expression of USx (or RS–Sx). Consequently, the males will be viable. The results of our viability studies, therefore, confirm our molecular analysis indicating that the chimeric USx and RS–Sx proteins provide sufficient Sxl function to regulate Sxl splicing in vivo.
Figure 5.

(A) Effect of the USx and RS–Sx transgenes on male viability. The transgenic flies were raised at 29°C throughout development and during the second larval instar they were heat-shocked for 90 min at 37°C. Viability of males heterozygous for the transgenes and carrying a duplication of Sxl+ is shown. The crosses were USx/USx or RS–Sx/RS–Sx females and y cm SxlfP7BO; Dp(1;3)sn13a1, Sxl+/TM3, Sb Ser males. In both crosses, the control was females carrying the duplication of Sxl. The number of control flies was 170 and 276 for USx and RS–Sx transgenes, respectively. Viability of males homozygous for the transgenes and carrying a deficiency of Sxl+ is shown. The crosses were cm Sxlf1 ct6/+; USx/TM3, Sb females and BSY; USx/USx males, or cm Sxlf1 ct6/+; RS–Sx/TM3, Sb females and BSY; RS–Sx/RS–Sx males. In both crosses, the control was females homozygous for the transgene and with two Sxl+ copies. The number of control flies was 122 for both USx and RS–Sx transgenes. In A, males Sxl− stand for males carrying the null Sxlf1 mutation. (B) Schematic representation of the effect of USx and RS–Sx transgenes on the viability of males. For the endogenous Sxl primary transcript, only the second and fourth exons (open boxes) and the male-specific third exon (solid box) were drawn. For explanations see text.
CONCLUDING REMARKS
Previous studies have established that regulation of tra splicing by SXL involves a blockage mechanism (6, 7), which was further characterized in vitro by the identification of U2AF as the antagonized target molecule (8). A blockage mechanism has also been proposed for the regulation of Sxl pre-mRNA splicing (34). However, distinct features of this pre-mRNA, including the number and location of the cis-acting signals required for proper regulation (14–18), suggested that distinct mechanisms could operate to regulate tra and Sxl splicing. Our results directly test and confirm this hypothesis by revealing that, in vivo, blocking U2AF binding can explain the regulatory activity of SXL on tra but not on Sxl pre-mRNA.
This different role of the SXL protein in Sxl and tra splicing may be related to the presence of multiple SXL binding sites in Sxl but not in tra pre-mRNA (14–18). The glycine/asparagine-rich NH2-terminal region (GN domain) of SXL has been implicated in RNA binding cooperativity and has been shown to be important for Sxl pre-mRNA splicing regulation in cultured cells (18). Our results show that this domain is dispensable for tra splicing regulation both in vivo and in vitro. Our data also show that the USx protein, which lacks this domain, displays Sxl+ function in Sxl pre-mRNA splicing. It is possible that the region of U2AF65 present at the NH2 terminus of USx provides an equivalent function to that of the GN domain in SXL. In any case, these observations further support the notion that SXL controls the splicing of different pre-mRNAs through diverse mechanisms that involve distinct protein domains, a common theme in transcriptional regulators that can now be extended to the regulation of postranscriptional processing.
Acknowledgments
We thank D. Mateos and R. de Andrés for their technical assistance. L.O.F.P. was the recipient of Instituto de Cooperacion Iberoamericana Ministerio de Educación y Ciencia. J.V. was supported in part by fellowships from European Molecular Biology Organization and Spanish Ministerio de Educación y Ciencia. This work was supported by Grant PB92–0006 from Direccion General de Investigacion Cientifica y Técnica to L.S., European Molecular Biology Organization funds to J.V., and a National Institutes of Health grant to M.R.G.
ABBREVIATIONS
- RNP
ribonucleoprotein
- GST
glutathione S-transferase
- RT-PCR
reverse transcription–PCR
References
- 1.Cline T W. Genetics. 1978;90:683–698. doi: 10.1093/genetics/90.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boggs R T, Gregor P, Idriss S, Belote J M, McKeown M. Cell. 1987;50:739–747. doi: 10.1016/0092-8674(87)90332-1. [DOI] [PubMed] [Google Scholar]
- 3.Nagoshi R N, McKeown M, Burtis K, Belote J M, Baker B S. Cell. 1988;53:229–236. doi: 10.1016/0092-8674(88)90384-4. [DOI] [PubMed] [Google Scholar]
- 4.Belote J M, McKeown M, Boggs R T, Ohkawa R, Sosnowski B A. Dev Genet. 1989;10:143–154. doi: 10.1002/dvg.1020100304. [DOI] [PubMed] [Google Scholar]
- 5.Bell L R, Maine E M, Schedl P, Cline T W. Cell. 1988;55:1037–1046. doi: 10.1016/0092-8674(88)90248-6. [DOI] [PubMed] [Google Scholar]
- 6.Sosnowski B A, Belote J M, McKeown M. Cell. 1989;58:449–459. doi: 10.1016/0092-8674(89)90426-1. [DOI] [PubMed] [Google Scholar]
- 7.Inoue K, Hoshijima K, Sakamoto H, Shimura Y. Nature (London) 1990;344:461–463. doi: 10.1038/344461a0. [DOI] [PubMed] [Google Scholar]
- 8.Valcárcel J, Singh R, Zamore P D, Green M R. Nature (London) 1993;362:171–175. doi: 10.1038/362171a0. [DOI] [PubMed] [Google Scholar]
- 9.Goralski T J, Edström J-E, Baker B S. Cell. 1989;56:1011–1018. doi: 10.1016/0092-8674(89)90634-x. [DOI] [PubMed] [Google Scholar]
- 10.Amrein H, Maniatis T, Nöthiger R. EMBO J. 1990;9:3619–3629. doi: 10.1002/j.1460-2075.1990.tb07573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burtis K C, Baker B S. Cell. 1989;56:997–1010. doi: 10.1016/0092-8674(89)90633-8. [DOI] [PubMed] [Google Scholar]
- 12.Hoshijima K, Inoue K, Higuchi I, Sakamoto H, Shimura Y. Science. 1991;252:833–836. doi: 10.1126/science.1902987. [DOI] [PubMed] [Google Scholar]
- 13.Bell L R, Horabin J I, Schedl P, Cline T W. Cell. 1991;65:229–239. doi: 10.1016/0092-8674(91)90157-t. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto H, Inoue K, Higuchi I, Ono Y, Shimura Y. Nucleic Acids Res. 1992;20:5533–5540. doi: 10.1093/nar/20.21.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horabin J I, Schedl P. Mol Cell Biol. 1993;13:7734–7746. doi: 10.1128/mcb.13.12.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bopp D, Calhoun G, Horabin J I, Samuels M, Schedl P. Development (Cambridge, UK) 1996;122:971–982. doi: 10.1242/dev.122.3.971. [DOI] [PubMed] [Google Scholar]
- 17.Penalva L O F, Sakamoto H, NavarroSabate A, Sakashita E, Granadino B A, Segarra C, Sanchez L. Genetics. 1996;144:1653–1664. doi: 10.1093/genetics/144.4.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Bell L R. Genes Dev. 1994;8:2072–2085. doi: 10.1101/gad.8.17.2072. [DOI] [PubMed] [Google Scholar]
- 19.Singh R, Valcárcel J, Green M R. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 20.Kanaar R, Lee A L, Rudner D Z, Wemmer D E, Rio D C. EMBO J. 1995;14:4530–4539. doi: 10.1002/j.1460-2075.1995.tb00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamore P D, Patton J G, Green M R. Nature (London) 1992;355:609–614. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Zamore P D, Carmo-Fonseca M, Lamond A I, Green M R. Proc Natl Acad Sci USA. 1992;89:8769–8773. doi: 10.1073/pnas.89.18.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salz H K, Cline T W, Schedl P. Genetics. 1987;117:221–231. doi: 10.1093/genetics/117.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cline T W. Genetics. 1980;96:903–926. doi: 10.1093/genetics/96.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsley D L, Zimm G. The Genome of Drosophila melanogaster. San Diego: Academic; 1992. [Google Scholar]
- 26.Schneuwly S, Klemenz R, Gehring W J. Nature (London) 1987;325:816–818. doi: 10.1038/325816a0. [DOI] [PubMed] [Google Scholar]
- 27.Spradling A C. In: P-Element-Mediated Transformation. Roberts D B, editor. Oxford, U.K.: IRL; 1986. pp. 175–198. [Google Scholar]
- 28.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 29.Campuzano S, Balcells L L, Villares R, Carramolino L, García-Alonso L, Modolell J. Cell. 1986;44:303–312. doi: 10.1016/0092-8674(86)90764-6. [DOI] [PubMed] [Google Scholar]
- 30.Frohman M A, Dush M K, Marin G R. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granadino B, Penalva L O F, Sanchez L. Mol Gen Genet. 1996;253:26–31. doi: 10.1007/s004380050292. [DOI] [PubMed] [Google Scholar]
- 32.Milligan J F, Groebe D R, Witherell G W, Uhlenbeck O C. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 34.Horabin J I, Schedl P. Mol Cell Biol. 1993;13:1408–1414. doi: 10.1128/mcb.13.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]