

Abstract
Niemann–Pick disease type C (NP-C) is an autosomal recessive lipidosis linked to chromosome 18q11–12, characterized by lysosomal accumulation of unesterified cholesterol and delayed induction of cholesterol-mediated homeostatic responses. This cellular phenotype is identifiable cytologically by filipin staining and biochemically by measurement of low-density lipoprotein-derived cholesterol esterification. The mutant Chinese hamster ovary cell line (CT60), which displays the NP-C cellular phenotype, was used as the recipient for a complementation assay after somatic cell fusions with normal and NP-C murine cells suggested that this Chinese hamster ovary cell line carries an alteration(s) in the hamster homolog(s) of NP-C. To narrow rapidly the candidate interval for NP-C, three overlapping yeast artificial chromosomes (YACs) spanning the 1 centimorgan human NP-C interval were introduced stably into CT60 cells and analyzed for correction of the cellular phenotype. Only YAC 911D5 complemented the NP-C phenotype, as evidenced by cytological and biochemical analyses, whereas no complementation was obtained from the other two YACs within the interval or from a YAC derived from chromosome 7. Fluorescent in situ hybridization indicated that YAC 911D5 was integrated at a single site per CT60 genome. These data substantially narrow the NP-C critical interval and should greatly simplify the identification of the gene responsible in mouse and man. This is the first demonstration of YAC complementation as a valuable adjunct strategy for positional cloning of a human gene.
Keywords: Chinese hamster ovary cells, gene transfer, cholesterol
Niemann–Pick disease type C (NP-C) is an autosomal recessive neurodegenerative disorder with heterogeneous manifestations. The classic clinical profile includes variable hepatosplenomegaly, vertical supra-nuclear ophthalmoplegia, progressive ataxia, dystonia, and dementia (1). The metabolic lesion is characterized by lysosomal sequestration of low-density lipoprotein (LDL)-derived cholesterol leading to delayed cholesterol-mediated homeostatic responses (2, 3). Lysosomal cholesterol accumulation can be depicted cytochemically by the cholesterol-specific fluorescent dye, filipin. Normally, endocytosed LDL-derived cholesterol is mobilized from lysosomes to the endoplasmic reticulum for esterification. As a result, there is little free cholesterol accumulation in lysosomes detectable by filipin staining in normal cells. In contrast, in NP-C cells the lysosomal accumulation of the endocytosed LDL-derived free cholesterol results in a specific perinuclear filipin-staining pattern. Biochemically, the NP-C phenotype can most conveniently be monitored by LDL-induced cholesterol ester synthesis. Cholesterol ester synthesis is markedly stimulated by LDL in normal cells, but not in NP-C cells.
The underlying genetic defect of this fatal neurological disease is unknown. Efforts thus far to isolate a gene responsible for NP-C using positional cloning have established an NP-C critical interval of 1 centimorgan on human chromosome 18, flanked by the markers D18S44 and NPC-B42 (GDB D18S1388) (refs. 4–6 and E.D.C., unpublished work). In addition, there are two independent murine mutants with an autosomal recessive lysosomal cholesterol storage defect (7–10). The pathological features of these murine mutants are similar to human NP-C (11, 12). Linkage analysis using one of these mutant strains (an inbred C56BL/KsJ sphingomyelinosis strain) placed the NP-C locus near the glucocorticoid receptor 1 gene on mouse chromosome 18 (13, 14). Restoration of the normal intracellular distribution and esterification of exogenous cholesterol was achieved when human chromosome 18, a chromosome partly syntenic to mouse chromosome 18 (15), was transferred into an immortalized cell line derived from this strain (SPM-3T3 cells) (16). Genetic cross breeding studies suggested that the gene responsible for NP-C in the C56BL/KsJ sphingomyelinosis mice was the same gene mutated in the other strain (NP-C BALB/c) (17), whereas cell fusion studies with a simian virus 40-transformed ovarian granulosa cell line (18) derived from the NP-C BALB/c mice suggested that these mice were genotypically allelic to human NP-C linked to chromosome 18 (data not shown). These data were consistent with the linkage of human NP-C to chromosome 18q11–12 (4, 5) and strongly suggested that murine and human NP-C are caused by mutations in the same gene.
In addition to the human and mouse NP-C phenotypes, a Chinese hamster ovary (CHO) cell line, which exhibits the NP-C phenotype (CT60), has also been described (19). This cell line was generated from normal CHO cells using chemical mutagenesis, and was selected based on its cholesterol metabolism profile. The CT60 cell line displays sequestration of unesterified cholesterol in the acidic compartment of the lysosomal/endosomal fraction and markedly reduced activation of cholesterol ester synthesis by LDL. Thus, it appears that CT60 has a remarkable phenotypic resemblance to human and mouse NP-C, and that the basic defect is in translocation of lysosomal LDL-derived cholesterol to the endoplasmic reticulum for esterification. Although these cells remained to be confirmed genotypically, the data supported CT60 as a potential CHO counterpart of NP-C.
One of the major advantages of using CHO cell mutants for complementation studies is the ease with which CHO cells take up and express exogenous DNA (20). Here we show data that suggest that the mutant CHO cell line, CT60, is genetically comparable to mouse and therefore human NP-C. This cell line was used to evaluate three yeast artificial chromosomes (YACs) spanning the NP-C genetic interval for their ability to complement the NP-C phenotype. The data indicate that: (i) the CT60 mutation can be rescued by a normal murine genome, (ii) YAC 911D5 complements the NP-C phenotype in the CT60 cells, and (iii) the region covered by YAC 911D5, specifically proximal to marker NPC-B42, constitutes the most critical region for isolation of the gene responsible for NP-C.
MATERIALS AND METHODS
Cells and Culture Media.
All cell lines described in this paper were grown as monolayers in tissue culture flasks or dishes. The CHO cell mutant CT60 and its variant CT60 neoR (resistant to neomycin) HATS (hypoxanthine, aminopterin, and thymidine sensitive) were provided by T. Y. Chang (Dartmouth College of Medicine, Hanover, NH) and grown in CT60 medium: Ham’s F-12 medium (F-12) (Biofluids, Rockville, MD) supplemented with 10% fetal bovine serum (FBS, HyClone), 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. The mouse ovarian granulosa cell lines, ELN and ELC, obtained from normal and NP-C BALB/c mice, respectively, by simian virus 40 transformation (18) were grown in a 1:1 mixture of F-12 and Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% FBS and the above reagents. Yeast spheroplast fusion clones derived from CT60 cells were selected and maintained in CT60 medium as described above, plus 400 μg/ml G-418 (GIBCO/BRL). CT60–ELN/ELC cell fusion clones were selected and propagated in the same medium plus 1× HAT (10−4M hypoxanthine, 4 × 10−7 M aminopterin, 1.6 × 10−5M thymidine). Normal human fibroblasts (derived from normal volunteers of the Developmental and Metabolic Neurology Branch, National Institute of Neurologic Disorders and Stroke, National Institutes of Health, under guidelines approved by National Institutes of Health clinical research committees) and NP-C human fibroblasts (3123; NIGMS Human Genetic Mutant Cell Repository, Coriell Institute for Medical Research, Camden, NJ) were propagated in DMEM supplemented with 10% FBS.
Cell Fusions.
Cell fusions were performed using polyethylene glycol according to Davison et al. (21), with the following modifications: CT60 neoR HATS and ELN or ELC cells were seeded as a mixture (1:1 ratio) at 1.4 × 106 cells per 100-mm dish and grown in CT60 medium at 37°C overnight. Cells were washed with PBS, treated with the dropwise addition of 4 ml of F-12 medium containing 40% polyethylene glycol (PEG 1500, Boehringer Mannheim) and 5% dimethyl sulfoxide, and incubated at room temperature for 2 min. Plates were washed twice with CT60 medium and incubated in the same media for 24 hr. Cells from each fusion were trypsinized and seeded onto three 100-mm plates, cultured for 2 more days, and then switched to double selection media consisting of F-12/10% FBS with 400 μg/ml G-418 and 1 × HAT. Clones were isolated and expanded prior to analysis.
Yeast Spheroplast Fusion.
All YACs used for spheroplast fusions were modified with a gene conferring neor by homologous recombination using the pRAN4 vector as described (22). Yeast spheroplasts were prepared and fused to recipient cells and clones were derived as described by Mogayzel et al. (20) and Huxley et al. (23).
Filipin Staining.
Clones were stained with filipin and viewed by fluorescence microscopy according to Kruth and Vaughan (24) and Pentchev et al. (25), with the following modifications. Cells were plated in 2-well chamber slides (Nunc) in F-12 media supplemented with 5% lipoprotein-depleted serum (PerImmune, Rockville, MD) for 2–3 days before human LDL (PerImmune) was added (50 mg/ml). After overnight incubation in the presence of LDL, cells were washed three times with PBS and fixed with 3% paraformaldehyde in PBS for 30 min. Cells were washed in PBS (three times for 5 min), quenched with glycine (1.5 mg/ml in PBS; 10 min), and 2 ml of fresh filipin (Sigma) solution (0.05 mg/ml in PBS) was added to each slide (30 min). Slides were rinsed with PBS, gaskets were removed, a drop of phenylenediamine/glycerol was added, and slides were mounted with coverslips.
Fluorescence signals were detected using a Zeiss Axiovert 405M microscope equipped with a filter for observation at the appropriate wavelengths: 365 nm excitation and 395 nm emission. Photographs were obtained using 4-sec exposures at a magnification of ×320.
Lipid Analysis.
When LDL is endocytosed and hydrolyzed into free cholesterol in lysosomes, cholesterol ester synthesis is activated by the increase in the free cholesterol pool. This reaction may be monitored in vitro by the incorporation of exogenously derived [3H]oleic acid into cholesterol [3H]oleate.
The amount of radioactive tracer [3H]oleic acid (DuPont/New England Nuclear; specific activity of 200,000 dpm/nmol) incorporation into cholesterol ester and triglyceride was measured by thin layer chromatography as described by Pentchev et al. (25, 26), with the following modifications. Cells were seeded in 6-well plates and incubated with [3H]oleic acid (100 mM) and human LDL (50 mg/ml). After 18 hr incubation, lipids and proteins were extracted. Nonradioactive cholesterol oleate and triolein were added to the lipid extract before thin layer chromatography was performed. Proteins were assayed by the Lowry method (27). The amount of cholesterol oleate was normalized to the amount of protein (in milligrams) and compared in the presence or absence of LDL (Δ cholesterol [3H]oleate synthesis). The Δ was compared with controls using a nonpaired, two-tailed t test.
DNA Preparation for STS Content Characterization.
Cell hybridization and yeast spheroplast fusion-derived clones were frozen at 3 × 106 cells per ml in microcentrifuge tubes. For DNA lysates, each tube was thawed, cells were pelleted, washed once with PBS, and resuspended in 50 μl of lysis buffer [1× Taq buffer (Perkin–Elmer)/0.05 mg/ml proteinase K/20 mM DTT/0.5 μg/ml sarkosyl]. The suspension was incubated overnight at 37°C, boiled for 15 min, then pelleted at 13,000 rpm for 5 min. An aliquot of the supernatant was used for PCR using primers D18Mit64 and D18Mit146 (Massachusetts Institute of Technology database) and an annealing temperature of 55°C.
Northern Blot Analysis.
RNA was extracted from cell monolayers using Trizol (GIBCO/BRL), following the product instructions. For each RNA sample, 10 μg were loaded on a 1.2% agarose/formaldehyde gel. Northern blotting and hybridization were performed as described (20). Probes were labeled by random oligo extension (Ready-To-Go kit, Pharmacia), following the kit instructions. After a 10-min denaturation, repeated sequences were blocked by incubating the probe with unlabeled human placental and Cot-1 DNA at 65°C for 30 min.
RESULTS
Formation of Heterokaryons Between CT60 Cells and Normal or NP-C Mouse Ovarian Cells.
To assess whether the CHO mutant CT60 is genotypically allelic to mouse NP-C, cell fusion studies were performed. A CT60 variant that is resistant to neomycin (neoR) and sensitive to HAT (CT60 neoR HATS; T. Y. Chang, personal communication) was fused to simian virus 40 transformed mouse ovarian granulosa cell lines, ELN or ELC, derived from normal or NP-C BALB/c mice, respectively. We hypothesized that if the mutation causing the NP-C-like phenotype in CT60 cells was allelic to the gene responsible for murine NP-C, heterokaryons of CT60 neoR HATS and mouse cells would show phenotypic correction with ELN, but not with ELC cells (Table 1). After 2 weeks of neo/HAT double selection, clones were isolated and expanded. The efficiency of heterokaryon formation was low (8.5 × 10−6). As expected, the morphology of the heterokaryons differed from both of the parental lines used for cell fusion. In addition, heterokaryon propagation was less successful compared with the parental lines, presumably due to the instability of the aneuploid cells. However, six clones each were successfully propagated from fusions performed with CT60 neoR HATS and either ELN or ELC cells and characterized for retention of the gene responsible for murine NP-C on mouse chromosome 18 by PCR using two polymorphic sequence-tagged site (STS) markers unique to the mouse NP-C interval [D18Mit64 proximal to and D18Mit146 distal to the gene responsible for murine NP-C (14)]. These two markers were present in all six CT60 neoR HATS–ELN fusion clones and all six CT60 neoR HATS–ELC fusion clones, but not in the parental cell line CT60 neoR HATS, suggesting that the heterokaryons retained at least one copy of the gene responsible for murine NP-C on chromosome 18. Representative clones are shown in Fig. 1, including those evaluated in subsequent studies.
Table 1.
Summary of filipin staining and cholesterol esterfication analyses of ELN, ELC, CT60 neoR HATS, and CT60 neoR HATS–ELN/ELC fusion derived clones
| ELN | ELC | CT60: ELN | CT60: ELC | CT60 | |
|---|---|---|---|---|---|
| Filipin | No | Yes | No | Yes | Yes |
| (lysosomal | |||||
| accumulation) | |||||
| Cholesterol | |||||
| ester synthesis | Yes | No | Yes | No | No |
| (LDL | |||||
| stimulation) | |||||
| Genotype | Normal | NP-C | Normal | NP-C | NP-C |
Figure 1.
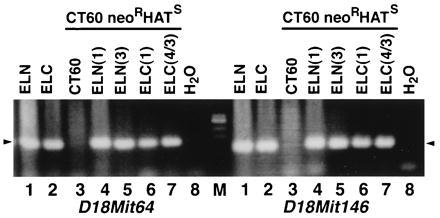
PCR with the mouse-specific polymorphic markers D18Mit64 and D18Mit146 to confirm the presence of the mouse NP-C segment in fused cell lines. The lane order is the same in the left and right halves. Lanes: 1, ELN; 2, ELC; 3, CT60 neoR HATS; 4–7, CT60 neoR HATS fused to lanes 4 and 5 (ELN) and lanes 6 and 7 (ELC); 8, H2O. Middle lane (size marker, M): λ/HindIII fragments plus øx 174 RF DNA/HaeIII fragments. Arrowheads indicate the expected size PCR products (D18Mit64, 172 bp; D18Mit146, 138 bp). Clone identification numbers are indicated in parentheses.
Complementation of CT60 Cells by Normal Mouse Ovarian Cells.
ELN- and ELC-derived CT60 neoR HATS heterokaryon clones were evaluated for normalization of the NP-C phenotype by filipin staining and cholesterol esterification analysis (Fig. 2 and Table 1). Each of the six CT60 neoR HATS–ELN clones containing the mouse NP-C interval demonstrated correction of the NP-C phenotype as indicated by filipin staining. The overall staining intensity was much reduced in the CT60 neoR HATS–ELN fusion clones, in contrast to the uniform perinuclear staining observed in NP-C cells (ELC), CT60 neoR HATS, and the CT60 neoR HATS–ELC fusion clones. An example of each type of these fusion clones is shown in Fig. 2A. In addition, patches of cells without lysosomal staining (resembling the absence of staining in the normal ELN cells) were observed in the CT60 neoR HATS–ELN fusion clones. Thus, complementation appeared to vary among the cells in the clones, presumably because of abnormal segregation of the chromosomes in the heterokaryons. Correction of the NP-C phenotype was not observed in any of the six CT60 neoR HATS–ELC fusion clones by filipin staining.
Figure 2.
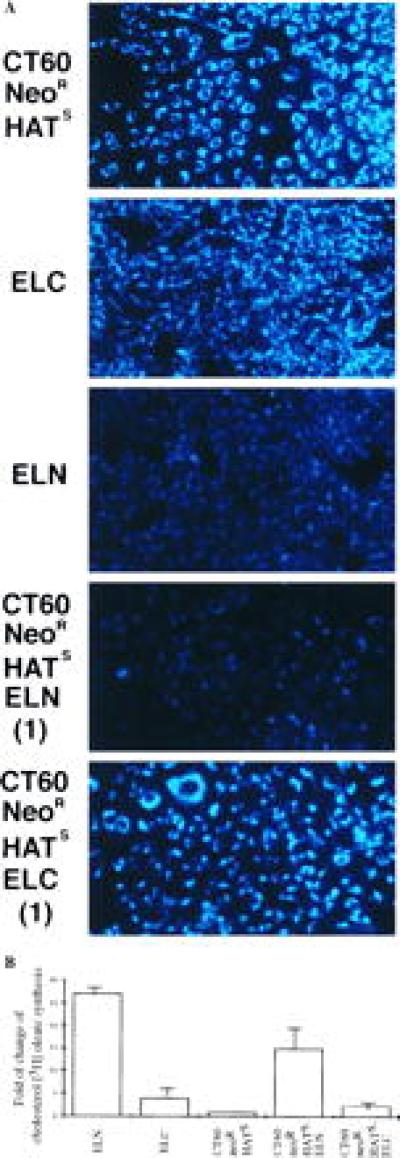
(A) Filipin staining of unesterified cholesterol in response to LDL uptake, in CT60 neoR HATS, ELN, ELC, and two of the cell fusion-derived clones. The intense staining in CT60 neoR HATS and ELC cells and the CT60 neoR HATS–ELC (1) fusion clone has the perinuclear pattern of lysosomal distribution seen in NP-C (ELC), but not in normal cells (ELN). Clone identification numbers are indicated in parentheses. (B) Stimulation of [3H]oleic acid incorporation into esterified cholesterol oleate in ELN, ELC, CT60 neoR HATS, and CT60 neoR HATS–ELN/ELC fusion-derived cells. The mean fold of change in [3H]cholesterol oleate synthesis is plotted for the unfused cell lines, each assayed in triplicate. For each heterokaryon type, the mean fold of change was calculated from three clones, each assayed in triplicate. SD are: ELN:1.15, ELC:1.64, CT60 neoR HATS:0.33, CT60 neoR HATS–ELN:4.50, and CT60 neoR HATS–ELC:1.01.
Cholesterol esterification analyses were performed on parental cell lines and fusion clones as a secondary method of screening for correction of the NP-C phenotype (Table 1 and Fig. 2B). Following overnight uptake of LDL by normal cells (ELN), cholesterol ester synthesis was markedly increased. In contrast, stimulation of cholesterol ester synthesis was much less in CT60 neoR HATS and in the NP-C cells (ELC). In the CT60 neoR HATS–ELN fusion clones, cholesterol ester synthesis was significantly increased compared with CT60 neoR HATS–ELC fusion clones (P ≈ 10−7), demonstrating complementation of the CT60 NP-C defect by the normal, but not the NP-C genome.
Transfer of YACs from the NP-C Interval to CT60 Cells by Yeast Spheroplast Fusion.
The human gene responsible for NP-C has been localized to a genomic region on chromosome 18, flanked by the genetic markers D18S44 centromerically and NPC-B42 telomerically (refs. 4–6 and E.D.C., unpublished work). The physical map assembled across the interval includes three overlapping YACs, 877F12, 844E3, and 911D5, which span the interval completely (Fig. 3, ref. 6, and E.D.C., unpublished work). These three YACs were modified with a neor gene by homologous recombination and then introduced into the NP-C CHO cell line, CT60, by spheroplast fusion. Similarly, another YAC that contains the cystic fibrosis transmembrane conductance regulator gene (CFTR) from chromosome 7, modified with a neor gene (yCFTR325-Neo), was used for fusion as an unlinked YAC control (20).
Figure 3.
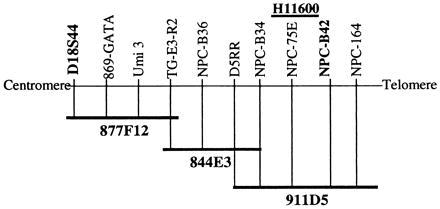
Schematic of the YAC coverage of the NP-C interval and the position of the expressed sequence tag (EST), H11600. The boundary markers of the physical interval are shown in boldfaced type. Other markers across the region are indicated, but relative distances are not to scale.
Spheroplast fusion resulted in neor clones from each of the YACs 877F12, 844E3, and 911D5 (22, 29, and 37 clones, respectively). Neor clones were also obtained from fusions with the control YAC, yCFTR325-Neo. Clonal cell lines were expanded and initially characterized by STS content mapping using STS primers from the NP-C locus. The expected STS contents were found in 8, 10, and 34 clones derived from 877F12, 844E3, and 911D5, respectively. The detailed analyses described below were carried out in two clones from 877F12, two clones from 844E3, and five clones from 911D5.
Complementation of the NP-C Phenotype in CT60 Cells by a Single Human YAC from the NP-C Interval.
As in the heterokaryon study, complementation in the YAC fusion clones was analyzed cytologically and biochemically. Filipin staining indicated that all clones evaluated from 911D5 fusions no longer accumulated perinuclear lysosomal cholesterol. An example is shown in Fig. 4A (clone 911D5A1). In contrast, parental CT60 cells, as well as clones derived from 844E3 (Fig. 4A), 877F12, and CFTR (data not shown) displayed extensive perinuclear staining. These findings indicated that the NP-C phenotype was corrected specifically only when YAC 911D5 was present in these cells.
Figure 4.
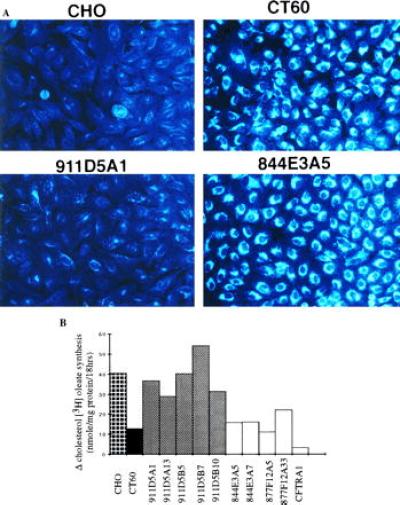
(A) Filipin staining of unesterified cholesterol in response to LDL uptake in CHO (normal), CT60 cells (NP-C), and YAC spheroplast fusion-derived clones from YAC 911D5 (911D5A1), and 844E3 (844E3A5). (B) Stimulation of [3H]oleic acid incorporation into esterified cholesterol oleate in normal CHO, CT60, and yeast spheroplast fusion clones. The change (Δ) in cholesterol [3H]oleate synthesis in response to LDL is plotted against the cell lines or fusion clones, each assayed in triplicate.
In support of the cytological data, when cellular cholesterol ester synthesis was analyzed, LDL-stimulated esterification was significantly increased compared with CT60 in all clones derived from 911D5, but not in either clone derived from 844E3 or clone A5 derived from 877F12 (Table 2, Fig. 4B). Clone 877F12A33 displayed modest stimulation of cholesterol esterification compared with CT60, but remained significantly different from control CHO cells (P < 0.05). While this data could be taken to suggest partial complementation by 877F12, the filipin staining result clearly demonstrated that 877F12 did not complement the NP-C associated lysosomal accumulation of cholesterol. Together, the phenotypic analyses suggested that 911D5 harbors a human gene capable of correcting the NP-C phenotype, and that the critical interval for identification of the gene responsible for NP-C has been narrowed from the 1-centimorgan interval defined by markers D18S44 and NPC-B42 to the region contained within YAC 911D5, specifically the 300–400 kb proximal to NPC-B42.
Table 2.
Statistical analysis of the change in cholesterol ester synthesis (Δ cholesterol [3H]oleate) in response to LDL in YAC fusion clones
| Cell type | n | Mean Δ, nmol/mg protein | P value (vs. CT60) | P value (vs. CHO) |
|---|---|---|---|---|
| CHO | 9 | 40.38 | 0.0001 | |
| CT60 | 6 | 12.78 | 0.0001 | |
| 911D5 A1 | 6 | 36.60 | 0.0001 | 0.49 |
| A13 | 6 | 28.79 | 0.002 | 0.04 |
| B5 | 3 | 40.15 | 0.00002 | 0.97 |
| B7 | 3 | 54.01 | 0.000003 | 0.07 |
| B10 | 3 | 31.26 | 0.0007 | 0.21 |
| 844E3 A5 | 6 | 15.73 | 0.22 | 0.0002 |
| A7 | 3 | 15.98 | 0.25 | 0.004 |
| 877F12A5 | 6 | 11.10 | 0.60 | 0.0001 |
| A33 | 3 | 22.12 | 0.008 | 0.02 |
| CFTR A1 | 3 | 3.30 | 0.007 | 0.0002 |
Gene Expression from YAC 911D5 in the Complemented Clones.
The complemented cell lines derived from 911D5 were also tested for gene expression using a strategy based upon ESTs mapped to YAC 911D5 (E.D.C., et al., unpublished work). Fig. 5 shows a Northern blot of YAC fusion clones hybridized with a probe derived from one of these ESTs, H11600 (GenBank, March 1997; shown in Fig. 3). This probe detected a message of approximately 5.0 kb in normal and NP-C human fibroblasts, and in clones 911D5A1 and A13, but not in CT60 cells and clones 877F12A5, 844E3A5, and CFTRA1. The H11600 probe also detected another message of approximately 2.6 kb in 911D5A1 and 911D5A13 and very faintly, in the human cell lines (Fig. 5). This message was subsequently shown to be the consequence of expression of another gene (GenBank accession no. R42429), which overlaps with the 3′ untranslated region of H11600, but is transcribed in the opposite direction (14).
Figure 5.
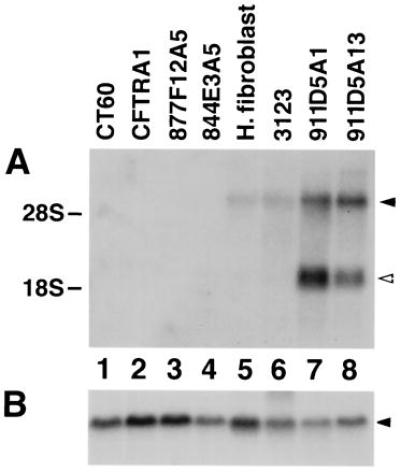
Northern blot analysis of YAC spheroplast fusion-derived clones. Each lane contains 10 μg of total RNA. (A) Blot hybridized with a probe derived from an EST in the NP-C interval (H11600; see Fig. 3), which detects transcripts of approximately 5.0 and 2.6 kb, as indicated by the closed and open arrows respectively. Lanes: 1, CT60; 2, CFTRA1 (clone derived from YAC yCFTR325-Neo); 3, 877F12A5 (clone derived from YAC 877F12); 4, 844E3A5 (clone derived from YAC 844E3); 5, normal human fibroblast; 6, 3123 (NP-C fibroblast); 7 and 8, clones derived from YAC 911D5 (lane 7, 911D5A1; lane 8, 911D5A13). (B) Same blot hybridized with a human β actin probe.
Fluorescent in situ hybridization (FISH) analyses were performed using labeled YAC DNA or yeast genomic DNA as probes. Only a single site of integration was visualized in metaphases of all analyzed clones derived from 911D5. The integration site was different among the clones, with the exception of 911D5A1 and A13 (data not shown), suggesting that these two clones could be siblings. No hybridization was observed in untransfected CT60 cells, indicating that the hybridization was specific to the transferred YAC, 911D5, which hybridized specifically to the pericentric region of chromosome 18q in human cells, consistent with the physical location of 911D5 in the NP-C interval (data not shown). A PCR assay was performed using primers from the NP-C interval to assess the copy number of YAC 911D5 in the CT60 genome by comparing the products with those amplified using known amounts of human genomic DNA or CT60 DNA mixed with YAC DNA at known copy number. The data indicated that there were on the order of 1–10 copies of YAC DNA per CHO cell genome (data not shown). Specifically, the clones 911D5A51 and A13 harbor only on the order of 1 copy of YAC 911D5, while clones 911D5B5, 911D5B7, and 911D5B10 contain on the order of 2–10 copies of the same YAC.
DISCUSSION
We have shown that a single YAC, spanning less than one-third of the NP-C minimal genetic interval, complements the NP-C phenotype in CT60 cells. The fact that the other two YACs from the NP-C interval or the control CFTR YAC did not complement suggests that the procedures of yeast spheroplast fusion along with the introduction of yeast genomic and irrelevant human DNA do not cause any substantial alteration of the NP-C phenotype. These data establish the utility of YAC complementation as a valuable adjunct strategy for positional cloning of human genes, when a phenotypic alteration linked to a gene can be monitored.
We found that the feasibility of YAC transfer is vitally dependent on the choice of cell lines. While it is logical to transfer human YACs from the NP-C interval to confirmed human or mouse NP-C cells, no neor clones as a consequence of stable YAC integration were ever obtained using untransformed and transformed human fibroblasts and transformed mouse ovarian cells as fusion recipients. A similar lack of success in deriving stably integrated YACs in human and mouse cell lines has been reported (28), although the reason for the failure in these cell lines is unclear. It has been proposed that the difficulties encountered (at least with human fibroblasts) are related to the limited efficiency of stable integration of human genomic DNA into the genome of human cells (29–33).
In contrast, CHO cells appear to be a good recipient for YAC spheroplast fusion. This study suggests that the mutant CHO cell line, CT60, has an alteration(s) in the hamster homolog(s) of the gene responsible for NP-C. Two lines of evidence support the idea that the hamster CT60 genotype is recessive. First, complementation was achieved in cell fusions between CT60 neoR HATS cells and normal mouse ovarian cells, and not in the parallel fusions between CT60 neoR HATS and NP-C mouse ovarian cells. Second, the CT60 mutation was corrected by a YAC containing genetic material from the human NP-C genetic interval.
The complementation of the NP-C phenotype by human genomic DNA contained within a YAC raises the question of how and why this phenomenon occurs in CT60 clones. Northern blot analyses indicated that a gene contained within YAC 911D5 is expressed in two of the complemented cell lines. FISH and PCR suggested that a single copy of 911D5 was incorporated at a single site in the genome of these cell lines. It is unclear why expression of the human gene harbored by the YAC integrated in these clones is greater than the expression of the same gene in the human control cells, although a similar result in CHO cells was recently described (20). In that study, CFTR was expressed in two YAC fusion clones in which each of two different CFTR YACs were integrated at a single site and as a single copy. The level of CFTR expression in both clones was higher than that expected from the knowledge of the level of endogenous CFTR expression in most human cells. Analogous to the CFTR YAC study and this work in which hamster cells were used, Perou et al. (28) reported complementation of the beige mutation in murine cells. However, complementation was observed only in clones containing a YAC that had been amplified and existed extrachromosomally, and not in clones containing the YAC incorporated into the mouse genome. While these studies indicate that YAC transfer can be used to express exogenous genes in mammalian cells, the mechanisms that govern the level of gene expression after successful stable transfer are far from clear. Whether gene expression from YAC 911D5 is modified by the positional effects of integration remains to be elucidated. Nevertheless, these clones are potentially very useful for subsequent steps in cloning the NP-C and other genes on YAC 911D5 by representational difference analysis, cDNA microarray, and other approaches that target the difference in gene expression patterns in cell lines. In addition, the proof that the hamster CT60 cells are capable of expressing a human gene responsible for NP-C suggests that the human promoter is contained and functional within the YAC. Thus, CT60 cells should be valuable as recipients for future NP-C complementation analyses and the subdivision of YAC 911D5 should be useful for future studies of regulation of the gene responsible for NP-C.
Acknowledgments
We wish to thank Ta-Yuan Chang (Dartmouth College of Medicine, Hanover, NH) for the gift of the CT60 CHO cell line, Jerome Strauss (Philadelphia) for performing murine cell transformations with simian virus 40, Karla Henning for advice and assistance with YAC manipulations, Vince Coviello and Dietrich Stephan (Laboratory of Cancer Genetics, National Human Genome Research Institute) for assistance, Elizabeth Novotny for FISH expertise, and Min Chen for providing preliminary data regarding YAC transfer to CHO cells. We would like to acknowledge the National Niemann–Pick Disease Foundation and the Ara Parseghian Medical Research Foundation for their generous support of this work. Grant support was also obtained from the Arkansas Governor’s Developmental Disabilities Planning Counsel.
ABBREVIATIONS
- NP-C
Niemann–Pick disease type C
- CHO
Chinese hamster ovary
- YAC
yeast artificial chromosome
- LDL
low-density lipoprotein
- neoR
resistant to neomycin
- HATS
hypoxanthine, aminopterin, and thymidine sensitive
- STS
sequence-tagged-site
- CFTR
cystic fibrosis transmembrane conductance regulator gene
- EST
expressed sequence tags
- FISH
fluorescent in situ hybridization
References
- 1.Pentchev P G, Vanier M T, Suzuki K, Patterson M C. In: The Metabolic and Molecular Base of Inherited Disease. Scriver C R, editor. New York: McGraw–Hill; 1995. pp. 2625–2639. [Google Scholar]
- 2.Pentchev P G, Brady R O, Blanchette-Mackie E J, Vanier M T, Carstea E D, Parker C C, Goldin E, Roff C F. Biochim Biophys Acta. 1994;1225:235–243. doi: 10.1016/0925-4439(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 3.Pentchev P G, Comly M E, Kruth H S, Tokoro T, Butler J, Sokol J, Filling-Katz M, Quirk J M, Marshall D C, Patel S, Vanier M T, Brady R O. FASEB J. 1987;1:40–45. doi: 10.1096/fasebj.1.1.3609608. [DOI] [PubMed] [Google Scholar]
- 4.Carstea E D, Polymeropoulos M H, Parker C C, Detera-Wadleigh S D, O’Neil R R, Patterson M C, Goldin E. Proc Natl Acad Sci USA. 1993;90:2002–2004. doi: 10.1073/pnas.90.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carstea E D, Parker C C, Fandino L B, Vanier M T, Overhauser J, Weissenbach J, Pentchev P G. Am J Hum Genet Suppl. 1994;55:A182. (abstr.). [Google Scholar]
- 6.Carstea, E. D., Morris, J. A., Coleman, K. G., Loftus, S. K., Zhang, D., et al. (1997) Science, in press.
- 7.Morris M D, Bhuvaneswaran C, Shio H, Fowler S. Am J Pathol. 1982;108:140–149. [PMC free article] [PubMed] [Google Scholar]
- 8.Miyawaki S, Mitsuoka S, Sakiyama T, Kitagawa T. J Hered. 1982;73:257–263. doi: 10.1093/oxfordjournals.jhered.a109635. [DOI] [PubMed] [Google Scholar]
- 9.Sakiyama T, Tsuda M, Kitagawa T, Fujuta R, Miyawaki S. J Inherited Metab Dis. 1982;5:239–240. doi: 10.1007/BF02179154. [DOI] [PubMed] [Google Scholar]
- 10.Pentchev P G, Boothe A D, Kruth H S, Weintroub H, Stivers J, Brady R O. J Biol Chem. 1984;259:5784–5791. [PubMed] [Google Scholar]
- 11.Higashi Y, Pentchev P G, Murayama S, Suzuki K. In: Neuropathology in Brain Research. Ikuta F, editor. New York: Elsevier; 1991. pp. 85–102. [Google Scholar]
- 12.Ohno K, Nanba E, Miyawaki S, Sakiyama T, Kitagawa T, Takeshita K. Cell Struct Funct. 1992;17:229–235. doi: 10.1247/csf.17.229. [DOI] [PubMed] [Google Scholar]
- 13.Sakai Y, Miyawaki S, Shimizu A, Ohno K, Watanabe T. Biochem Genet. 1991;29:103–113. doi: 10.1007/BF00578243. [DOI] [PubMed] [Google Scholar]
- 14.Loftus, S. K., Morris, J., Carstea, E., Gu, J., Cummings, C., Brown, A., Ellison, J., Ohno, K., Rosenfeld, M. A., Pentchev, P., Tagle, D. A. & Pavan, W. J. (1997) Science, in press. [DOI] [PubMed]
- 15.McKusick V A, editor. Mendelian Inheritance in Man. 9th Ed. Baltimore: Johns Hopkins Univ. Press; 1990. p. clxvii. [Google Scholar]
- 16.Kurimasa A, Ohno K, Oshimura M. Human Genetics. Heidelberg: Springer; 1993. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto T, Iwasawa K, Tokoro T, Eto Y, Maekawa K. No To Hattatsu. 1994;26:318. [PubMed] [Google Scholar]
- 18.Amsterdam A, Hanukoglu I, Suh B S, Keren-Tal D, Plehn-Dujowich D, Sprengel R, Rennert H, Strauss J F., III J Steroid Biochem Mol Biol. 1992;43:875–884. doi: 10.1016/0960-0760(92)90315-A. [DOI] [PubMed] [Google Scholar]
- 19.Cadigan K M, Spillane D M, Chang T Y. J Cell Biol. 1990;110:295–308. doi: 10.1083/jcb.110.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mogayzel P J, Henning K A, Bittner M L, Novotny E A, Schwiebert E M, Guggino W B, Jiang Y, Rosenfeld M A. Hum Mol Genet. 1997;6:59–68. doi: 10.1093/hmg/6.1.59. [DOI] [PubMed] [Google Scholar]
- 21.Davison R L, O’Malley K A, Wheeler T B. Somatic Cell Genet. 1976;3:271–280. doi: 10.1007/BF01538965. [DOI] [PubMed] [Google Scholar]
- 22.Srivastava A, Schlessinger D. Gene. 1991;103:53–59. doi: 10.1016/0378-1119(91)90390-w. [DOI] [PubMed] [Google Scholar]
- 23.Huxley C, Hagino Y, Schlessinger D, Olson M. Genomics. 1991;9:742–750. doi: 10.1016/0888-7543(91)90369-p. [DOI] [PubMed] [Google Scholar]
- 24.Kruth H S, Vaughan M. J Lipid Res. 1980;21:123–130. [PubMed] [Google Scholar]
- 25.Pentchev G P, Comly M E, Kruth H S, Vanier M T, Wenger D A, Patel S, Brady R O. Proc Natl Acad Sci USA. 1985;82:8247–8251. doi: 10.1073/pnas.82.23.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pentchev G P, Kruth H S, Comly M E, Butler J D, Vanier M T, Wenger D A, Patel S. J Biol Chem. 1986;35:16775–16780. [PubMed] [Google Scholar]
- 27.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Perou C M, Justice M J, Pryor R J, Kaplan J. Proc Natl Acad Sci USA. 1996;93:5905–5910. doi: 10.1073/pnas.93.12.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colbere-Garapin F, Ryhiner M L, Stephany I, Kourilsky P, Garapin A C. Gene. 1986;50:279–288. doi: 10.1016/0378-1119(86)90332-x. [DOI] [PubMed] [Google Scholar]
- 30.Hoeijmakers H J H, Odijk H, Westerveld A. Exp Cell Res. 1987;169:111–119. doi: 10.1016/0014-4827(87)90230-8. [DOI] [PubMed] [Google Scholar]
- 31.Mayne L V, Jones T, Dean S W, Harcourt S A, Lowe J E, Priestley A, Steingrimsdotter H, Sykes H, Green M H L, Lehmann A R. Gene. 1988;66:65–76. doi: 10.1016/0378-1119(88)90225-9. [DOI] [PubMed] [Google Scholar]
- 32.Lohrer H, Blum M, Herrlich P. Mol Gen Genet. 1988;212:474–480. doi: 10.1007/BF00330852. [DOI] [PubMed] [Google Scholar]
- 33.Lambert C, Schultz R A, Smith M, Wagner-McPherson C, McDaniel L D, Donlon T, Stanbridge E J, Friedberg E C. Proc Natl Acad Sci USA. 1991;88:5907–5911. doi: 10.1073/pnas.88.13.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]