

Abstract
Expansion of a CTG trinucleotide repeat in the 3′ untranslated region (UTR) of DMPK, the gene encoding myotonic dystrophy protein kinase, induces the dominantly inherited neuromuscular disorder myotonic dystrophy (DM). Transcripts containing the expanded trinucleotide are abundant in differentiated cultured myoblasts, and they are spliced and polyadenylylated normally. However, mutant transcripts never reach the cytoplasm in these nonmitotic cells; instead, they form stable clusters that are tightly linked to the nuclear matrix, which can prevent effective biochemical purification of these transcripts. In DM patients, reduced DMPK protein levels, consequent to nuclear retention of mutant transcripts, are probably a cause of disease development. Formation of nuclear foci is a novel mechanism for preventing transcript export and effecting a loss of gene function.
Myotonic dystrophy (DM), an autosomal dominant neuromuscular disorder, is due to the extreme expansion of a trinucleotide (CTG) repeat in the 3′ untranslated region (UTR) of the gene encoding DM protein kinase, DMPK (1–3). Mutant DMPK alleles contain up to several thousand triplets, rather than the normal 5–35. Since the expanded repeat is not translated, and hence should not directly alter the gene’s protein kinase product, disease models based on overexpression (4) or underexpression (5) of the kinase have been proposed. Most recent studies (6–8) support a loss-of-function model for DM development; however, the cause of DMPK deficiency has been controversial.
Loss of DMPK function could result from a novel feature of mutant DMPK transcripts that was revealed by in situ hybridization. Taneja et al. (9) demonstrated that mutant DMPK transcripts form nuclear foci in DM fibroblasts and muscle; foci were not detected in control fibroblasts or muscle biopsies. It was postulated that these nuclear foci might contribute to DM pathogenesis, perhaps by disrupting transport of mRNA from DMPK and/or other genes to the cytoplasm. However, the division of fibroblasts in culture provided a potential pathway for release of nuclear RNA into the cytoplasm, preventing conclusive testing of this hypothesis. Mutant DMPK transcripts were in fact detected within the cytoplasm of DM fibroblasts; the localization and cytoskeletal association of these transcripts did not differ from those of wild-type (wt) transcripts.
The pathology of DM is most evident in differentiated muscle tissue. We have therefore examined DMPK transcription and transcript processing in control and DM cultured myoblasts, generated by MyoD retroviral infections of fibroblasts. Since differentiated myoblasts are arrested in G0 of the cell cycle, we could also test the hypothesis that the repeat expansion interferes with nuclear export of transcripts. Normal (wt) and mutant DMPK transcripts were analyzed by both Northern blotting and in situ hybridization.
Our analyses revealed that mutant DMPK transcripts were abundant in myoblasts, but could not contribute to kinase production, as the transcripts were quantitatively retained within myoblast nuclei. In contrast to fibroblasts, terminally differentiated myoblasts contained no cytoplasmic mutant transcripts detectable either by blot analysis or by in situ hybridization. Mutant transcripts formed stable, long-lived clusters that were tightly associated with the nuclear matrix. Nuclear retention was not due to disruption of splicing or polyadenylylation. The small fraction of mutant DMPK transcripts found in the cytoplasm in fibroblasts apparently bypassed normal export mechanisms and escaped during nuclear envelope breakdown. Nuclear sequestration of mutant transcripts is compatible with a loss-of-function model for DM and provides a mechanism for such a model. In addition, the abundant nuclear foci in myoblasts may contribute to disease development by affecting export of other mRNAs as well.
MATERIALS AND METHODS
Cell Lines.
DM fibroblast lines were obtained from J. David Brook (Queens Medical Centre, University of Nottingham, U.K.) and the American Type Culture Collection; control fibroblast lines were obtained from Marcy MacDonald (Massachusetts General Hospital, Harvard University, Boston) and the American Type Culture Collection. LMD25N, which produces a MyoD/NeoR amphotrophic retrovirus (10), was provided by A. Dusty Miller (Fred Hutchinson Cancer Research Center, Seattle).
Retroviral Infection.
LMD25N cells were irradiated for 45 minutes in a Gamma Cell 40, containing 2 CsCl sources with a chamber dose of 80 rad/min. Irradiated producer cells (5.5 × 106) were cocultivated with DM or control fibroblasts (1.2 × 106) in the presence of 4 μg/ml hexadimethrine bromide (Sigma) for 2 days. Fibroblasts were allowed to recover for 1 day, then infected cells were selected with 800 μg/ml Geneticin (GIBCO) for 6 days. Infected fibroblasts were either induced to differentiate (Dulbecco’s modified Eagle’s medium + 10% horse serum/2 mM l-glutamine/10 μg/ml insulin/10 μg/ml transferrin/50 units/ml penicillin/50 μg/ml streptomycin) for 2–6 days or frozen for subsequent experiments.
RNA Isolation.
For isolation of total RNA, cells were lysed in thiocyanate buffer (4 M guanidinium thiocyanate/20 mM NaOAc, pH 5.4/0.1 mM dithiothreitol/0.5% sodium lauroyl sarcosine), and RNA was pelleted through a 5.7 M CsCl cushion. Poly(A)+ RNA was purified from total RNA on a column of oligo(dT)-cellulose (Stratagene) essentially as described (11). Columns were preloaded with 100 μg of tRNA to block nonspecific hybridization.
For isolation of nuclear and cytoplasmic RNA, cells were rinsed in ice-cold PBS, scraped from culture dishes into PBS + 2 mM EDTA, and rinsed again in ice-cold PBS. Cells were incubated 5 min on ice in lysis buffer (10 mM Tris⋅HCl, pH 8.0/0.14 M NaCl/1.5 mM MgCl2/10 mM vanadyl ribonucleoside complex (GIBCO/BRL)), broken with 10 strokes of a Dounce homogenizer, and centrifuged for 15 min, 4°C, 2,500 × g, to pellet nuclei. The cytoplasmic fraction (supernatant) was removed into thiocyanate buffer. The nuclei were resuspended in lysis buffer + 0.5% Tween 40/0.5% sodium deoxycholate, incubated on ice for 3 min, and centrifuged again for 15 min, 4°C, 2,500 × g. The supernatant fractions were pooled, and the nuclei were lysed in thiocyanate buffer. Both fractions were purified through CsCl as above.
Nuclear matrices were prepared from trypsinized cells according to the protocol of He et al. (12). To purify RNA, each extraction solution was diluted 1:10 in thiocyanate buffer and spun through CsCl as above.
Northern Blotting and Probe Hybridization.
RNA samples were subjected to electrophoresis on 1% agarose/1.1% formaldehyde gels, and transferred to Hybond N (Amersham). Probes were generated by random priming of 25–50 ng of DNA. PCR for DMPK cDNA probes: DM5′A: exon 4 (DB119, 5′-GTGAGGAGAGGGACGTGTTGGTG) to exon 7 (DB120, 5′-GGACGATCTTGCCATAGGTCTCCG); DM5′B: exon 8 (DB124, 5′-CCACCGACACATGCAACTTCGAC) to exon 15 (DB100, 5′-CACGCTCGGAGCGGTTGTGAACTGG); DM3′A: 3′UTR (DB150, 5′-GCCCTGACGTGGATGGGCAAACTG) to Bluescript (Stratagene) vector (KS primer, 5′-CGAGGTCGACGGTATCG). DMPK hybridization: 7% SDS/1% BSA/0.2 M sodium phosphate, pH 7.2/1 mM EDTA/30% formamide/100 μg/ml salmon sperm DNA at 65°C. Myogenin hybridization: 5× SSC/5× Denhardt’s solution/0.5% SDS/100 μg/ml salmon sperm DNA at 65°C. Quantitative analysis was carried out on a PhosphorImager (Molecular Dynamics).
RNase H Digestion and Analysis.
Fifteen micrograms of total RNA was incubated with 100 pmol of oligonucleotide DB100 (5′-CACGCTCGGAGCGGTTGTGAACTGG; 5′ to CTG) and 100 pmol of oligonucleotide DB101 (5′-CTTCCCAGGCCTGCAGTTTGCCCATCC; 3′ to CTG) at 50°C for 3 min. An equal volume of 2× RNase H buffer (40 mM Tris⋅HCl, pH 7.5/20 mM MgCl2/200 mM KCl/0.2 mM dithiothreitol) and 40 units of rRNasin (Promega) were added, and samples were incubated at 37°C for 10 min. Then 4.4 units of RNase H (GIBCO/BRL) was added, and samples were incubated at 37°C for 1.5 hr. After digestion, RNAs were extracted, precipitated, and electrophoresed on formaldehyde gels as above. 5′ probe: DM5′A and DM5′B; 3′UTR probe: DM3′B (DB151, 5′-GCGCGATCTCTGCCTGCTTACTCGG to Bluescript KS vector primer).
RNA Half-Life.
For each cell line, equivalent plates of infected cells were allowed to differentiate for 3 days, then treated with actinomycin D (4 μg/ml; Sigma) for 0–25 hr. Cells still appeared viable after 25 hr. Nuclear and cytoplasmic RNA was isolated from each plate as above.
In Situ Hybridization and Digital Imaging Microscopy.
Myoblast and fibroblast cells from normal individuals and DM patients were hybridized with a cy3-labeled CAG-30 probe to detect mutant DMPK transcripts as described (9). DNA was visualized by inclusion of 4′,6-diamidino-2-phenylindole (DAPI) in the mounting medium. Images for each wavelength (cy3, 552 nm; DAPI, 370 nm) were acquired using a microscope equipped with epifluorescence and modified to obtain images at various planes within the cell (13). Images at each focal plane were acquired with a cooled charge-coupled device (CCD) camera (model 22; Photometrics, Tucson, AZ) and were restored to remove out-of-focus light. Camera and microscope functions were controlled by a PDP-11/73 microcomputer. Typically, images of the point fluorescent sources were obtained at 0.25-μm intervals, and 20 images were obtained for each cell. For each experiment the dark current from the camera and from a flat field were also recorded to correct for nonuniformities in the transmission characteristics of the optics and CCD detector. The optical sections taken from each cell were subjected to further processing to correct the distortion introduced by the microscope and camera system, using an algorithm described in detail in refs. 13 and 14. To quantitate nuclear fluorescence, the nuclear perimeter was traced with the mouse driver cursor; the computer then calculated the intensity of fluorescence and the average intensity per unit area within the circumscribed region. Cytoplasmic fluorescence for entire myotubes was not computed, since the boundaries of myotube cytoplasm associated with specific nuclei are indistinct. Instead, small areas of cytoplasm from different regions of each section were circled and the average intensity of fluorescence per unit area was again calculated. TFI is defined as the total light acquired from a segmented image after background is subtracted.
RESULTS
Generation of “Myoblasts” by MyoD Retroviral Infection of Fibroblasts.
To study the expression of mutant DMPK and the impact of the mutant DMPK allele on cellular processes within an affected cell type, we created “myoblasts” by infecting myotonic patient-derived and normal cultured fibroblasts with a MyoD retrovirus (10). Myoblasts expressed elevated levels of DMPK, especially after induction of differentiation (Fig. 1A). All experiments were performed on cells allowed to differentiate for at least 2 days, to allow for up-regulation and for withdrawal from the cell cycle. Infected cells also expressed myoblast-specific markers such as myogenin, myosin heavy chain, and acetylcholine receptor γ (Fig. 1B and data not shown).
Figure 1.
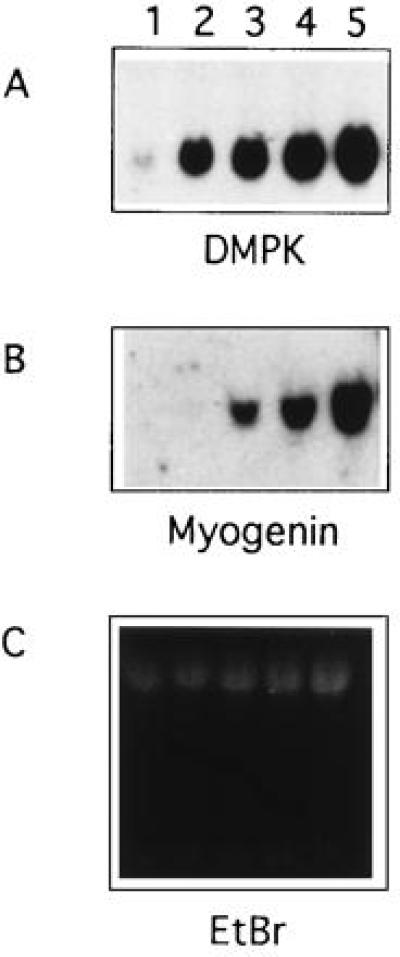
Northern analysis of DMPK and myogenin transcription in normal and MyoD-infected fibroblasts. Lane 1, fibroblast RNA; lanes 2–5, myoblast RNA after 0, 1, 2, or 3 days of differentiation; 10 μg of total RNA per lane. (A) Blot probed with DM3′A. (B) Blot probed with myogenin cDNA. (C) Ethidium bromide staining of gel prior to transfer.
Mutant DMPK Transcripts Are Less Abundant than wt Transcripts in DM Cells.
Northern analysis was used to compare the levels of wt and mutant transcripts. DMPK cDNA probes detected a single 2.8-kb transcript in RNA from wt cells, and at least one additional mRNA in all DM lines. Mutant transcripts ranged from 3 kb to greater than 7 kb, since trinucleotide expansion, which accounts for the additional message length, varies among DM patients. RNA from some DM cell lines contained more than one additional hybridizing transcript, due to repeat size heterogeneity within the cultured fibroblasts. When multiple mutant alleles were present within a sample, quantitation was based on the total of mutant transcripts.
The level of the mutant transcript was lower than that of the wt in all DM cell lines examined (n = 8). Mutant transcript abundance ranged from 32% to 71% of the level of the normal allele within the same cells (Fig. 2 and data not shown). Expression by the DM cells of the unmutated allele was roughly half that seen in control cells, suggesting that the disease does not significantly alter transcription of the normal allele. However, transcript levels of the wt allele vary among control cell lines (refs. 6 and 7 and data not shown), so a precise comparison of wt transcript levels among cell lines was not performed.
Figure 2.
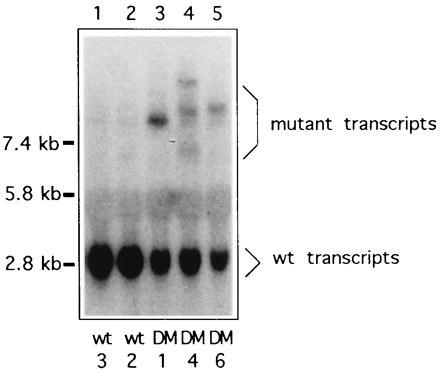
Northern analysis of DMPK transcripts in representative normal and myotonic myoblasts. Lanes 1 and 2, normal myoblast RNA; lanes 3, 4, and 5, myotonic myoblast RNA; probed with DM3′A. When multiple mutant bands were detected, all were included in calculating transcript abundance.
Mutant DMPK Transcripts Are Sequestered Within Myoblast Nuclei.
To assess the subcellular localization of DMPK transcripts, cells were fractionated and nuclear and cytoplasmic RNA was extracted. Northern analysis of fractionated myoblast RNA revealed that mutant DMPK transcripts were present only within myoblast nuclear RNAs (Fig. 3A). The 28S and 18S rRNA was almost completely absent from nuclear fractions (Fig. 3A), indicating that the nuclear samples were not contaminated by cytoplasmic RNAs. Nuclear retention of mutant DMPK transcripts was also observed in uninfected fibroblasts (Fig. 3B). However, in fibroblasts a faint cytoplasmic signal, corresponding to 15% or fewer of mutant transcripts, was also detected. In contrast, at least 43% of the wt message was exported to the cytoplasm in both control and myotonic cells. This suggests that the insertion of lengthy CUG repeats (>1 kb) into the DMPK transcript interferes with processing and/or transport of the RNA. Interestingly, transcripts with a less dramatically expanded repeat (approximately 150 CUG) showed less nuclear retention (data not shown). Localization of transcripts other than DMPK (β-actin, fibronectin, glyceraldehyde-3-phosphate dehydrogenase) did not vary between control and DM cells (data not shown).
Figure 3.
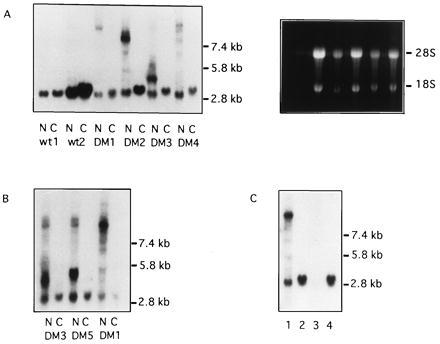
Comparison of nuclear and cytoplasmic DMPK RNAs isolated from control and myotonic cells. RNA for each nuclear (N)/cytoplasmic (C) pair was extracted from the same cells. Northern blots were probed with DM5′A/DM5′B. (A) Lanes 1–4, control myoblasts; lanes 5–12, myotonic myoblasts. Ethidium bromide staining prior to transfer is shown on the right. (B) Nuclear and cytoplasmic RNA from uninfected DM fibroblasts. (C) RNA isolated from DM myoblast nuclei by CsCl ultracentrifugation (lane 1) or acid guanidinium thiocyanate/phenol/chloroform (lane 3). Cytoplasmic RNAs (lanes 2 and 4) were purified by CsCl ultracentrifugation of cytoplasmic fractions.
To effectively isolate DMPK transcripts containing large expanded repeats, it was essential that RNAs were purified by ultracentrifugation of guanidinium thiocyanate lysates through CsCl, rather than with acid guanidinium thiocyanate/phenol/chloroform (15). Fig. 3C shows RNA extracted from differentiated DM myoblast nuclei by using the two techniques. Mutant and wt DMPK transcripts are clearly visible after CsCl ultracentrifugation of nuclei. In contrast, these transcripts are scarcely detectable in the acid guanidinium thiocyanate/phenol/chloroform preparation. This dramatic difference in purification efficiency may be responsible for some of the controversy (4–7) over mutant DMPK transcript levels.
In Situ Localization of DMPK Transcripts.
The subcellular localization of mutant DMPK transcripts in cultured myoblasts was also investigated by in situ hybridization, using a fluorescently labeled CAG-30 oligonucleotide probe. As in cultured fibroblasts and DM muscle biopsies (9), mutant transcripts were detected in discrete foci within myoblast nuclei (Fig. 4 A and B). However, hundreds of foci were detected per nucleus, rather than the 5 observed in the average fibroblast (9). Myoblast foci were brighter than those observed in fibroblasts, suggesting that myoblast foci contain more DMPK transcripts. Mutant transcripts containing relatively short (150 CUG) repeats were also found clustered in nuclear foci, but only in approximately 15% of nuclei (Fig. 4D). This probably corresponds to the incomplete nuclear retention of short repeats detected by Northern blotting. Nuclear foci were not detected when the CAG-30 probe was hybridized to control myoblast nuclei (Fig. 4C).
Figure 4.
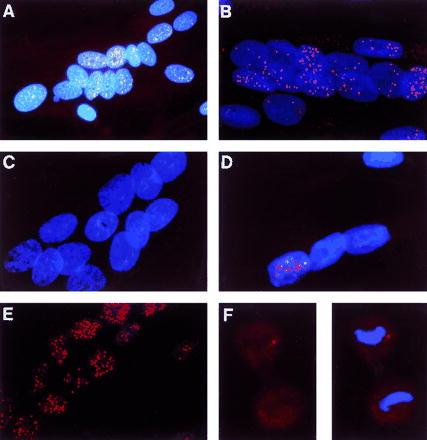
Distribution of triplet repeat transcripts in myoblasts and dividing cells. Fixed cells on coverslips were hybridized with cy3-labeled CAG-30 and counterstained with DAPI. Spots represent signal from the trinucleotide repeat. (A–E, ×225; F, ×300.) (A) Analog photomicrograph. (B–F) Digital images, generated by mathematically removing out-of-focus light from images captured by a CCD camera. Each digital image is a restored optical section through the cell(s). (A and B) DM myoblasts. (C) Normal myoblasts. (D) DM myoblasts from a minimally affected patient with approximately 150 CTG repeats. (E) DM myoblasts treated with DNase I and extracted with (NH4)2SO4. The expanded triplet repeat molecules remained in the nuclear matrix. The lack of DAPI staining indicates that the DNA has been removed. (F) Dividing fibroblasts from the same patient as in A. In late anaphase (possibly telophase), no signal was detected in the newly reformed nucleus, suggesting that transcript foci have diffused within the cytoplasm. (Left) Dividing cell without DAPI stain. (Right) DAPI shows the condensed DNA typical of anaphase.
No cytoplasmic transcripts containing an expanded repeat were detected in DM myotubes. Hybridization of the CAG-30 probe to DM myotube cytoplasm yielded only a faint signal (average TFI/area = 0.42), indistinguishable from hybridization to wt cytoplasm (average TFI/area = 0.47). This confirms the finding by Northern analysis that mutant DMPK transcripts are found only in the nuclei of differentiated myoblasts (average TFI/area = 32). Unlike in cultured fibroblasts (9), no perinuclear cytoplasmic transcripts were detected.
Transcripts were not observed to form foci in wt cells, despite the retention of approximately 50% of the DMPK transcripts in wt nuclei. wt probes hybridized to wt nuclei in a diffuse, relatively homogenous manner (data not shown). Hybridization was much weaker than to the transcript foci revealed in DM cells with the same probes (9). Thus, transcript foci are not detected in DM cells just because of the high copy number of the repeat probe’s target. It is difficult to precisely analyze the wt transcripts, as low-abundance, diffusely distributed messages cannot easily be distinguished from background hybridization.
Mutant Nuclear Transcripts Are Associated Exclusively with the Nuclear Matrix.
To investigate the nature of the DMPK foci and their relationship to the rest of the nucleus, more precise cellular fractionations were performed. Myoblasts were extracted to separate (i) soluble cytoplasmic, (ii) cytoskeletal, and (iii) chromatin-associated molecules, leaving (iv) the nuclear matrix and attached intermediate filaments (12). RNA was isolated from each fraction and subjected to Northern analysis (Fig. 5A). As expected, the cytoplasmic fractions (fractions 1 and 2) contained at least 40% of the wt message, in both control and myotonic cells, and none of the mutant message. Little wt message and no mutant message was released by chromatin extraction. Instead, all of the mutant transcripts and 40–50% of the wt transcripts were found in the nuclear matrix fraction (fraction 4). Extraction with solutions containing 130 mM KCl [an alternative method for isolation of cytoskeleton-associated mRNAs (16)] also did not release DMPK transcripts from the nucleus (data not shown). Thus, the transcripts fractionating with the nucleus are not merely associated with the surface of the nuclear envelope by means of the cytoskeleton; instead, wt and mutant transcripts are capable of strong interactions with the nuclear matrix. It seems likely that these interactions (probably RNA–protein) are disrupted by 5.7 M CsCl and ultracentrifugation, but not by acid guanidinium thiocyanate/phenol/chloroform. This would explain the loss of nuclear transcripts during extraction with acid guanidinium thiocyanate/phenol/chloroform (Fig. 3C).
Figure 5.
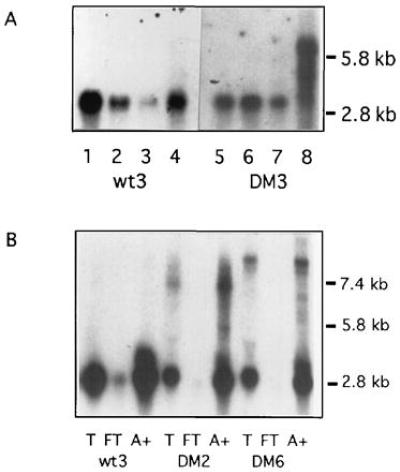
(A) Northern analysis of association of DMPK transcripts with the nuclear matrix. Some wt DMPK message is present in the cytoplasmic (lanes 1 and 5) and cytoskeletal (lanes 2 and 6) fractions, and a small amount is chromatin associated (lanes 3 and 7). All the mutant transcripts and 40–50% of the wt transcripts were retained with the nuclear matrix (lanes 4 and 8). Lanes 1–4, control cells; lanes 5–8, myotonic cells; probed with DM5′A/DM5′B. (B) Northern analysis of polyadenylylation of wt and mutant DMPK transcripts. Ten micrograms of total (T) RNA and the flowthrough (FT) and poly(A)+ (A+) fractions isolated from 20 μg of total RNA were blotted and probed with DM5′A/DM5′B. Lanes 1–3, control cells; lanes 4–9, DM cells.
Mutant DMPK transcripts were also analyzed in situ following fractionation. Transcript foci were still clearly visible in nuclei after extraction of nonmatrix components (Fig. 4E). Their number, appearance, and location were unchanged by the removal of chromatin and associated proteins. In contrast, nonspecific hybridization, both nuclear and cytoplasmic, was significantly reduced by extraction, as soluble components of the cell were removed. Thus, in situ analysis confirmed that mutant transcripts are firmly anchored to non-DNA components of the nuclear substructure. In addition, it revealed that not only the nuclear retention of trancripts but also the integrity of nuclear foci is resistant to extraction with salt, detergent, and DNase I.
Transcripts Are Released During Cell Division.
The fate of transcript foci during nuclear division was assessed in Colcemid-treated DM fibroblasts. Foci were not detected within DM fibroblasts in anaphase (Fig. 4F); instead, a low-level diffuse signal, difficult to distinguish from background hybridization, was observed. The signal was particularly faint within the nuclear region, identified by staining DNA. This suggests that DMPK transcript foci dissolve, along with the nuclear matrix, during cell division, and that mutant transcripts are left in the cytoplasm when nuclei reform. Focus dissolution and transcript exclusion probably account for the 10–15% of mutant DMPK transcripts found, by Northern blotting, in fibroblast cytoplasm.
Mutant Transcripts Are Polyadenylylated and Spliced.
Insufficient polyadenylylation or splicing of the trinucleotide-containing transcripts could theoretically induce nuclear retention of these transcripts (17). However, Northern blots of poly(A)+ RNA from infected DM cells clearly show mutant DMPK transcripts (Fig. 5B). The ratio of wt to mutant transcripts in poly(A)+ RNA is comparable to the ratio within total RNA, suggesting that the DM mutation does not interfere with polyadenylylation. In addition, wt poly(A)+ transcripts were recovered with similar efficiency from control and DM cells; unlike in ref. 7, no “trans” effect of the DM mutation on the wt allele was observed.
To examine DMPK transcript splicing, total RNA blots were rehybridized with intron-derived probes. Probes from between exons 9 and 10 and between exons 12 and 13 were tested independently. Neither probe specifically hybridized to either DMPK transcript, nor did they detect larger, unspliced precursor transcripts. Instead, they showed equivalent nonspecific nuclear hybridization in wt and mutant cells (data not shown). These results suggest that unspliced transcripts do not accumulate in DM cells, and that mutant transcripts are not retained in the nucleus as a result of insufficient splicing.
Oligonucleotide-directed cleavage of DMPK transcripts by RNase H confirmed that mutant transcripts do not contain intronic sequences. Total DM and control RNA was annealed to oligonucleotides complementary to sequences flanking the trinucleotide repeat, in the 3′UTR. DNA⋅RNA hybrids were then digested with RNase H, yielding three DMPK transcript fragments: a 5′ fragment, which could theoretically contain introns; a repeat-containing fragment, with about 170 bp of flanking sequences; and a 3′ fragment, containing the poly(A) tail. Northern analysis of these fragments revealed that control and DM cells contain identical 5′ and 3′ fragments (Fig. 6). Therefore, the 5′ fragments of mutant transcripts cannot contain unspliced introns. The 3′ fragments have a broad size range in both control and DM cells, probably due to variation in the length of the poly(A) tail.
Figure 6.
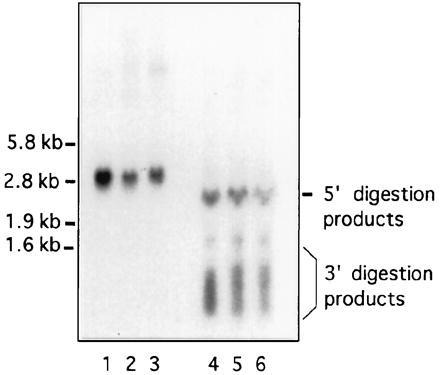
Northern analysis of oligonucleotide-directed cleavage of DMPK transcripts with RNase H. Undigested (lanes 1–3) and digested (lanes 4–6) total RNA, 15 μg per lane; probed with DM5′A, DM5′B, and DM3′B. Lanes 1 and 4, wt2; lanes 2 and 5, DM4; and lanes 3 and 6, DM1.
Mutant Transcripts and Nuclear Foci Are Not Unstable.
The stability of mutant transcripts and the possibility that foci might be sites of transcript degradation were also analyzed. Infected control and myotonic cells were treated with actinomycin D (4 μg/ml) to block RNA synthesis, and DMPK mRNA remaining was quantitated over the following 25-hr period. Both messages were still readily detected by Northern analysis after 25 hr of actinomycin D exposure (Fig. 7). The half-lives of the wt and mutant messages were roughly equivalent, both ranging from 13 to 17 hr (Fig. 7 and data not shown). As expected, all mutant transcripts remained in the nucleus. Some wt transcripts were also still within the nucleus after 25 hr, suggesting that either (i) actinomycin D disrupted export of some nuclear transcripts (see ref. 18) or (ii) a subpopulation of transcripts were not destined for export. In situ hybridization with the CAG-30 probe showed that the number and distribution of nuclear foci was not altered after 15 hr of actinomycin D treatment (data not shown), though foci did appear slightly less bright. Thus, the lower levels of mutant transcripts are not due to decreased message stability, and foci are not sites of rapid message degradation.
Figure 7.
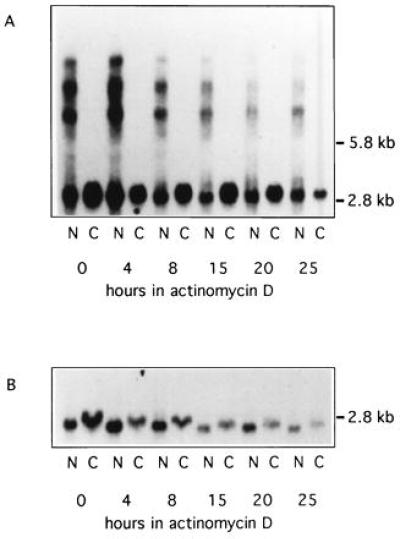
Northern analysis of DMPK mRNA stability. Nuclear (N) and cytoplasmic (C) RNA was probed with DM3′A. (A) DM cells (DM4). (B) Control cells (wt3).
DISCUSSION
We used Northern blotting and in situ hybridization to assess the abundance, localization, and processing of DMPK transcripts in normal and DM patient-derived myoblasts. The two techniques yielded entirely consistent results with intriguing implications for the etiology of DM and for analysis of nuclear RNAs. We found that mutant DMPK transcripts form extremely stable, extraction-resistant nuclear foci that are firmly anchored to the nuclear matrix. wt nuclear transcripts are also matrix associated, but they do not form clusters in wt cells. Aggregated mutant transcripts are quantitatively retained within the nuclei of nondividing myoblasts. Thus, although mutant DMPK transcripts are 32–71% as abundant as wt transcripts in myoblasts (consistent with refs. 8 and 9), there is a 50% reduction in DMPK mRNA in myoblast cytoplasm; a parallel reduction of DMPK protein is expected. Recent analyses suggest that DMPK levels are in fact reduced in DM patients (8) and MyoD-infected DM patient fibroblasts (B.M.D., unpublished data). Incomplete nuclear retention of a short (150 CTG) repeat from a minimally affected patient, presumably causing an intermediate reduction in DMPK protein levels, is consistent with observed correlations (19) between repeat size and disease severity. Formation of nuclear foci is a novel mechanism for inducing a loss of gene function in these and probably other types of nondividing cells. Since the tissues most severely affected by DMPK triplet expansion—skeletal muscle, cardiac muscle, lens, and potentially brain—are predominantly or completely postmitotic, haploinsufficiency of DMPK is probably a significant component of DM induction.
A connection between reduced DMPK levels and disease development is also suggested by a recent study of “knock-out” mice by Reddy et al. (20). DMPK (−/−) mice develop a late onset progressive skeletal myopathy with many features reminiscent of DM. Decreased muscle force generation, variations in fiber size, increased fiber degeneration and regeneration, and fibrosis are observed both in (−/−) mice and in DM patients. Comparable ultrastructural changes, including Z line distortion and mitochondrial abnormalities, are also observed. DMPK (+/−) mice have a subtle, less severe phenotype, marked by smaller increases in regenerative activity and varying decreases in force production. Alterations in force production and gene expression suggest a phenotype intermediate to that of wt and (−/−) mice. The similarities between microscopic defects in DM patients and DMPK-deficient mice support the hypothesis that reduced kinase levels contribute to DM development.
The cause of nuclear retention of the mutant DMPK mRNA has not yet been identified. We analyzed several of the early steps in transcript processing but found no differences between processing of the wt and mutant DMPK transcripts. We could easily detect spliced and polyadenylylated mutant transcripts, suggesting that these processing steps are not blocked by the expanded trinucleotide. Our data also clearly show (contradicting ref. 7) that dominance in DM is not due to an effect in “trans” upon polyadenylylation of the wt allele. In addition, it is unlikely that expansion of the trinucleotide repeat disrupts a regulatory unit or creates a novel signal for transcript degradation, as the wt and mutant DMPK messages have comparable half-lives. Slight impairment of splicing, polyadenylylation, and/or stability by the expanded repeat cannot be ruled out, and might account for the relative reduction in mutant transcripts. However, DM cells are clearly capable of fully processing a significant fraction of mutant transcripts, suggesting that nuclear retention is probably due to disruption of a later stage of the export pathway. Perhaps novel RNA-binding proteins that interact preferentially with expanded triplet repeats (21, 22) will prove to be significant for the nuclear retention of mutant DMPK transcripts.
It is possible that nuclear retention of transcripts and DMPK insufficiency are not the only consequences of trinucleotide expansion. The trinucleotide might also interfere with processes unrelated to DMPK. For instance, transcript foci might disrupt transport and/or processing of other mRNAs. However, since foci have never been observed in cells with normal DMPK dosage, an independent assessment of their role has not been performed. Expanded trinucleotides could also have significance at the DNA level. Trinucleotides are very strong nucleosome positioning elements (23); related alterations in chromatin structure could influence regulation of surrounding genes. There is speculation that DMHAP, a homeodomain-encoding gene 3′ to the expansion that is expressed in skeletal muscle, heart, and brain, might be influenced by the trinucleotide (24). Discovery of such additional roles for the expanded trinucleotide would explain why DM patients with simple loss-of-function (i.e., deletion, frame-shift) mutations have not been identified. Perhaps the full range of DM symptoms can develop only in patients with both a DMPK deficiency and a disruption of another, as yet unidentified, triplet-dependent cellular process.
Acknowledgments
We thank Dr. J. David Brook and Dr. Marcy MacDonald for providing fibroblast cell lines, and Dr. A. Dusty Miller for providing the LMD25N retrovirus producer cell line. We also thank Dr. Benjamin Blencowe, Dr. Scott Lowe, and Dr. Anthony Albino for helpful discussions and technical suggestions. This work was supported by Grant 5R37-CA 17575–16 from the National Cancer Institute to D.E.H., Grant GM54577–13 from the National Institute of General Medical Sciences to R.H.S., Grant 522010 from the Muscular Dystrophy Association to K.L.T., and Grant PO1-HL41484 from the National Heart, Lung and Blood Institute to Massachusetts Institute of Technology.
ABBREVIATIONS
- DM
myotonic dystrophy
- DMPK
myotonic dystrophy protein kinase
- UTR
untranslated region
- wt
wild type
- DAPI
4′,6-diamidino-2-phenylindole
- CCD
charge-coupled device
- TFI
total fluorescence intensity (corrected for background)
References
- 1.Brook J D, McCurrach M E, Harley H G, Buckler A J, Church D, Aburatani H, Hunter K, Stanton V P, Thirion J P, Hudson T, Sohn R, Zemelman B, Snell R G, Rundle S A, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juxenon V, Johnson K, Harper P S, Shaw D J, Housman D E. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 2.Fu Y-H, Pizzuti A, Fenwick R G, King J, Jr, Rajnarayan S, Dunne P W, Dubel J, Nasser G A, Ashizawa T, de Jong P J, Wieringa B, Korneluk R, Perryman M B, Epstein H F, Caskey C T. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 3.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O’Hoy K, Leblond S, Earle-MacDonald J, de Jong P G, Wieringa B, Korneluk R G. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 4.Sabouri L A, Mahadevan M S, Narang M, Lee D S C, Suhr L C, Korneluk R G. Nat Genet. 1993;4:233–238. doi: 10.1038/ng0793-233. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y-H, Freidman D L, Richards S, Pearlman J A, Gibbs R A, Pizzuti A, Ashizawa T, Perryman M B, Scarlato G, Fenwick R G, Jr, Caskey C T. Science. 1993;260:235–238. doi: 10.1126/science.8469976. [DOI] [PubMed] [Google Scholar]
- 6.Krahe R, Ashizawa T, Abbruzzese C, Roeder E, Carango P, Giacanelli M, Funanage V L, Siciliano M J. Genomics. 1995;28:1–14. doi: 10.1006/geno.1995.1099. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Pegoraro E, Menegazzo E, Gennarelli M, Hoop R C, Angelini C, Hoffman E P. Hum Mol Genet. 1995;4:599–606. doi: 10.1093/hmg/4.4.599. [DOI] [PubMed] [Google Scholar]
- 8.Maeda M, Taft C S, Bush E W, Holder E, Bailey W M, Neville H, Perryman M B, Bies R D. J Biol Chem. 1995;270:20246–20249. doi: 10.1074/jbc.270.35.20246. [DOI] [PubMed] [Google Scholar]
- 9.Taneja K L, McCurrach M, Schalling M, Housman D E, Singer R H. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weintraub H, Tapscott S J, Davis R L, Thayer M J, Adam M A, Lassar A B, Miller A D. Proc Natl Acad Sci USA. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingston R E. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Mith J A, Struhl K, editors. New York: Wiley; 1991. pp. 4.5.1–4.5.3. [Google Scholar]
- 12.He D, Nickerson J A, Penman S. J Cell Biol. 1990;110:569–580. doi: 10.1083/jcb.110.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fay F S, Carrington W, Fogarty K E. J Microsc (Oxford) 1989;153:133–149. [PubMed] [Google Scholar]
- 14.Carrington W A, Fogarty K E, Fay F S. In: Non-invasive Techniques in Cell Biology. Fosket K, Grinstein S, editors. New York: Wiley–Liss; 1990. pp. 53–72. [Google Scholar]
- 15.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 16.Hesketh J, Campbell G, Piechaczky M, Blanchard J M. Biochem J. 1994;298:143–148. doi: 10.1042/bj2980143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krug R M. Curr Opin Cell Biol. 1993;5:944–949. doi: 10.1016/0955-0674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence J, Singer R H, Marselle L M. Cell. 1989;57:493–502. doi: 10.1016/0092-8674(89)90924-0. [DOI] [PubMed] [Google Scholar]
- 19.Tsilfidis C, MacKenzie A E, Mettler G, Barceló J, Korneluk R G. Nat Genet. 1992;49:961–965. doi: 10.1038/ng0692-192. [DOI] [PubMed] [Google Scholar]
- 20.Reddy S, Smith D B J, Rich M, Leferovich J M, Reilly P, Davis B M, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon R, Housman D. Nat Genet. 1996;13:325–334. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 21.Timchenko L T, Timchenko N A, Caskey C T, Roberts R. Hum Mol Gen. 1996;5:115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]
- 22.Timchenko L T, Miller J W, Timchenko N A, DeVore D R, Datar K V, Lin L, Roberts R, Caskey C T, Swanson M S. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y-H, Amirhaeri S, Kang S, Wells R D, Griffith J D. Science. 1994;265:669–671. doi: 10.1126/science.8036515. [DOI] [PubMed] [Google Scholar]
- 24.Boucher C A, King S K, Carey N, Krahe R, Winchester C L, Rahman S, Creavin T, Meghji P, Bailey M E, Chartier F L, Brown S D, Siciliano M J, Johnson K J. Hum Mol Gen. 1995;4:1919–1925. doi: 10.1093/hmg/4.10.1919. [DOI] [PubMed] [Google Scholar]