

Abstract
Myotonic dystrophy (DM) is caused by the expansion of a trinucleotide repeat, CTG, in the 3′ untranslated region of a protein kinase gene, DMPK. We set out to determine what effect this expanded repeat has on RNA processing. The subcellular fractionation of RNA and the separate analysis of DMPK transcripts from each allele reveals that transcripts from expanded DMPK alleles are retained within the nucleus and are absent from the cytoplasm of DM cell lines. The nuclear retention of DMPK transcripts occurs above a critical threshold between 80 and 400 CTGs. Further analysis of the nuclear RNA reveals an apparent reduction in the proportion of expansion-derived DMPK transcripts after poly(A)+ selection. Quantitative analysis of RNA also indicates that although the level of cytoplasmic DMPK transcript is altered in DM patients, the levels of transcripts from 59 and DMAHP, two genes that immediately flank DMPK, are unaffected in DM cell lines.
Myotonic dystrophy (DM) is the most common form of muscular dystrophy affecting adults. It is dominantly inherited and involves many systems, including endocrine, heart, and brain, though principally it is a disease of muscle characterized by myotonia with muscle weakness and wasting (1). DM is associated with the expansion of a CTG repeat located in the 3′ untranslated region (UTR) of a protein kinase gene, DMPK (2–4) (See Fig. 1). It is not known how this mutation, in a noncoding part of the gene, exerts an effect at the cellular level, and conflicting data have been published about the effect of repeat expansion on DMPK RNA levels in DM tissues and cell lines (5–8). Three models have been proposed to explain the molecular mechanism of DM (9). First, the mutation may directly effect the level of DMPK protein. Second, the mutation may affect the higher-order structure of DNA around the repeat, altering the level of expression of neighboring genes in a field effect (10–13). Third, the repeat expansion may produce a gain-of-function mutation at either the DNA or RNA level. Recently, foci of DMPK transcripts containing expanded repeats have been reported in the nuclei of DM muscle specimens and fibroblast cell lines (14), and other studies have suggested that a disruption of RNA processing may be critical for the development of DM (15, 16). In view of these findings, we have performed a series of experiments to compare the distribution of expanded and wild-type DMPK alleles in RNA from nuclear and cytoplasmic fractions of DM cells. We have also examined the expression of genes 59 (10, 17) and DMAHP (12), which flank DMPK, to establish whether this is affected by repeat expansion.
Figure 1.
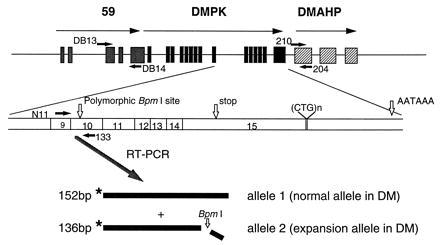
The exon/intron organization of genes surrounding the DM-associated triplet. Gray boxes depict gene 59, black boxes DMPK, and the hatched boxes the putative exons of DMAHP. The transcriptional orientation is indicated with arrows. The positions of the polymorphic BpmI recognition site in exon 10 of DMPK, the stop codon, the (CTG)n repeat, and the polyadenylylation signal are shown in relation to the DMPK transcript. The positions of the oligonucleotide primers are indicated by short black arrows. The two isoforms of DMPK are indicated as allele 1 and allele 2.
METHODS
Patient Cell Lines.
Fibroblast cell lines were established from DM patients and normal controls. To allow the separate quantification of RNA from each copy of the DMPK gene (allele 1 and allele 2), six DM cell lines and four normal control lines that were informative for a Bpm1 polymorphism in exon 10 have been used. The segregation of allele 1 with the normal chromosome for five of the six DM patients (DM A, DM C, DM D, DM E, and DM F) has been demonstrated by analysis of DNA from their relatives (data not shown); no DNA was available from the family of DM B. However, as allele 1 is rare and has always been shown to associate with the normal allele in DM, patients heterozygous for this polymorphism (this report and refs. 8, 15, and 16), it is very likely that allele 1 in DM B is also associated with the normal allele.
Clinical Details of DM Patients.
Patient DM A was a minimally affected male aged 56, detected on the basis of family history, with a repeat expansion size of 80 triplets. Clinically he was asymptomatic except for bilateral cataracts, which were diagnosed recently. DM B was an affected male with a repeat expansion of 3.5 kb. He had classical symptoms of proximal muscle wasting, myotonia, and myotonic facies. DM C, an adult male, had a repeat expansion size of 1.2 kb. Clinically he had classical symptoms of proximal muscle wasting, myotonia, and myotonic facies and bilateral cataracts. DM D was an affected male with a repeat expansion of 3.0 kb. Clinically he showed classical symptoms of myotonia, proximal muscle wasting, myotonic facies, and bilateral cataracts. DM E was an affected female with a repeat expansion of 5.5 kb. Clinically, she exhibited myotonic facies, proximal muscle weakness, and wasting with myotonia (clinically and by electromyogram). DM F was an affected female with an expansion of 5.5 kb. Clinical symptoms included mild proximal muscle wasting, myotonia, and myotonic facies. Approval for this study was granted by the Ethics Committee of the City Hospital National Health Service Trust, Nottingham, and informed consent was obtained from all participating individuals.
RNA Extraction.
Cells were washed in cold PBS and 1 ml of 0.65% Nonidet P-40 in 0.01 M Tris⋅HCl, pH 7.9/0.15 M NaCl/1.5 mM MgCl2 was added. Nuclei from the lysed cells were pelleted and the supernatant containing the cytoplasmic fraction of the cells was removed. The nuclear pellet was resuspended in 400 μl of water. Then 100 μl of 0.5 M Tris⋅HCl, pH 9.0/0.05M EDTA/2.5% SDS was added to the nuclear pellet and 200 μl was added to 800 μl of the cytoplasmic fraction. RNA was purified by two extractions with phenol/chloroform, and the RNA was precipitated by the addition of ethanol/sodium acetate (pH 5.2). RNA samples were examined on ethidium bromide-stained agarose gels. Within the nuclear RNA preparations, tRNA bands were detectable, indicating that some contamination from the cytoplasmic fraction had occurred. This was estimated to be less than 5%. In the cytoplasmic fractions, genomic DNA was not seen; however, preliminary PCR analysis with genomic primers generated products from reactions without reverse transcriptase (RT), indicating that a small proportion of nuclei were disrupted by the RNA preparation method. Therefore, all RNA samples were treated with DNase (Promega), following supplier’s specifications.
RT-PCR.
One microgram of RNA was incubated in a total reaction volume of 20 μl containing 0.3 μg of random hexamers (Pharmacia), and 200 units of Moloney murine leukemia virus RT (GIBCO/BRL), following the supplier’s instructions. First-strand synthesis was performed in duplicate on all samples, and parallel samples were analyzed without the addition of RT to establish that products were derived from RNA and not DNA. RT reactions could be primed with random hexamers, as identical ratios for the two alleles in DM patient RNA were obtained whether random or sequence-specific (using oligonucleotide 133) priming was used (data not shown). Subsequently, 1/20th of each duplicated RT reaction (and the minus RT control) was used as a template for two independent PCR analyses (four replicates in total). One oligonucleotide primer of each pair was end labeled with [γ-32P]ATP by using polynucleotide kinase (NBL Gene Sciences). Standard PCR conditions of 20-μl reaction mixtures containing 200 μM dNTPs, 10 pmol of each oligonucleotide, and 0.5 unit of Taq DNA polymerase (Boehringer Mannheim) with buffer supplied, were used. Twenty-seven cycles of 94°C, 1 min; 58°C, 1 min; 72°C, 1 min were employed. Control for RNA input was achieved by the addition of oligonucleotides for glyceraldehyde-3-phosphate dehydrogenase (GAPDH): after 4 cycles of PCR (23 cycles being within the linear range for GAPDH). For DMPK analysis, to distinguish the two alleles of DMPK, 3 μl of the PCR mixture was taken into a 25-μl digestion mixture containing 2 units of BpmI (New England Biolabs) with the supplier’s buffer and 0.1 mg/ml BSA and incubated overnight at 37°C. Three microliters of this digest was loaded onto 8% polyacrylamide/urea gels. Quantification of PCR products was performed on PhosphorImager scans (Molecular Dynamics) using the imagequant version 1.1 image analysis program (Molecular Dynamics). Prior to the quantification analysis, a comprehensive series of control experiments was performed with six replicates using time points between 22 and 32 cycles. The optimal number of cycles was established as 27, for which the PCR was within the linear range, with no heteroduplex formation, hence BpmI digestion was complete (data not shown).
Oligonucleotide Primer Sequences.
Sequences were as follows: N11, 5′-CACTGTCGGACATTCGGGAAGGTGC-3′; 133, 5′-GCTTGCACGTGTGGCTCAAGCAGCTG-3′; GAPDH-2, 5′-GATGACAAGCTTCCCGTTCTCAGCC-3′; GAPDH-5′, 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′; DB13, 5′-CCCCTTGTGTGCAAGAAGATCGCCC-3′; DB14, 5′-CCATGGCTTCACACCACTGTGCCAC-3′; 204, 5′-GTCGCCGGTTCTTGAACCTGTTGC-3′; 210, 5′-CAGTGGACAAGTATCGACTGC-3′).
RESULTS
Subcellular Localization of DMPK Transcripts.
Fibroblast cell lines have been established from six DM patients (see Methods for clinical details) and four unaffected individuals who are informative for a BpmI restriction site polymorphism (8) located within exon 10 of DMPK (see Fig. 1). Nuclear and cytoplasmic RNA fractions from these fibroblast cell lines were analyzed by quantitative RT-PCR using oligonucleotides for DMPK. The presence of a BpmI recognition site in only one of the alleles, the expansion allele in the DM patients, enabled the assessment of spliced transcript levels from each copy of DMPK to be determined (see Fig. 1). In the nuclear fractions, it is clear that both alleles were transcribed. However, in five of the six DM lines only the normal allele (allele 1) was present in the cytoplasm (Fig. 2). The RNA levels for each allele have been measured and compared with an internal standard, GAPDH, using replicate RT-PCR analysis (see Fig. 3). The levels of allele 1 in the nucleus were the same in the DM and control cell lines (Fig. 3A), but those of allele 2 were significantly different (P = 0.04 by Mann–Whitney test). As shown in Fig. 3B, the levels of DMPK allele 2 were higher in the DM group. For the cytoplasmic fraction, the levels of allele 1 were again the same in DM and control cell lines (Fig. 3C), but the levels of allele 2 were significantly lower in the DM cell lines and were close to the limit of detection in five of the six cases (P = 0.01 by Mann–Whitney) (Fig. 3D). These nuclear and cytoplasmic results were reproduced in replicate with an alternative standard RNA (TFIIS) (data not shown). In the cytoplasmic fraction from DM patient A the proportion of the two alleles was very similar to that in the normal control lines. Comparison of repeat lengths in each of the cell lines and clinical details of the patients revealed that DM A has a repeat size of 80 CTGs and is minimally affected. The other five DM patients have classic DM features with repeat lengths in their fibroblasts ranging from 1.2 to 5.5 kb. Thus, the DM expansion allele does not appear in the cytoplasm and is retained at an increased level in the nuclei of classically affected DM patients.
Figure 2.
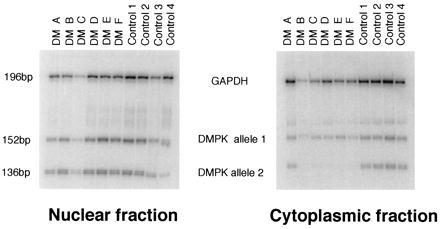
Multiplex RT-PCR analysis of DMPK transcripts compared with those for GAPDH. The phosphor image shows RT-PCR products of DMPK allele 1 and allele 2 in the nuclear and cytoplasmic fractions. Bands were visualized by overnight exposure to a PhosphorImager screen. After scanning, the image range was set at 1.0–255.
Figure 3.
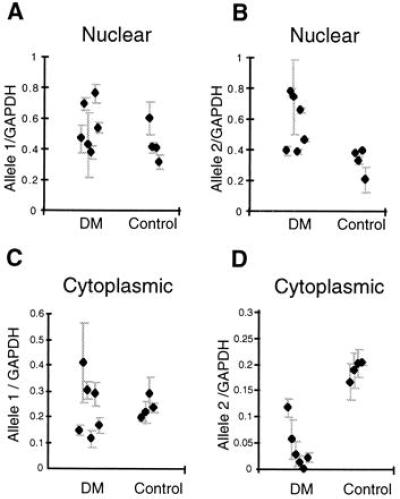
Analysis of multiplex RT-PCR analysis of DMPK transcripts compared with those for GAPDH. (A) Results of quantitative analysis for the levels of allele 1 of DMPK in nuclear fractions from six DM cell lines and four controls. The means and errors for replicates on each sample are shown (four replicates were produced by two independent RT reactions, which were then used as substrates for two independent PCRs). (B) Results for mean levels of DMPK allele 2 in nuclear fractions. (C) Mean levels of DMPK allele 1 in cytoplasmic fractions. (D) Levels of DMPK allele 2 in cytoplasmic fractions. All RT-PCRs were repeated using an alternative RNA control (TF11S) and were shown to give similar results (data not shown).
Expression of Neighboring Genes 59 and DMAHP.
The DMPK locus is flanked by genes 59 and DMAHP (see Fig. 1), and it has been suggested that these genes may be involved in the etiology of DM (12). Under a field-effect model for DM, expansion of the triplet would cause an alteration in level of expression from several surrounding genes (11, 13, 18), possibly because of gross distortions in chromatin structure. To test this hypothesis we have analyzed the levels of expression from DMPK compared with those from DMAHP and 59, the two genes that immediately flank DMPK. Fig. 4A shows that, compared with the control group, DM cell lines show a significant drop in the total level of cytoplasmic DMPK RNA (due to absence of allele 2) (P = 0.01 by Mann–Whitney). Thus, an alteration in the cytoplasmic levels of 59 or DMAHP would be expected if their expression from the expansion allele was similarly affected. Fig. 4B shows that the cytoplasmic levels of RNA from DMAHP corrected to the level of GAPDH were no different from those of the control group (analysis by Mann–Whitney test). The primers for the analysis of 59 flank an alternatively spliced exon (exon 4), consequently two different PCR products, 59(A) and 59(B), are made, and these have been analyzed separately. Fig. 4 C and D shows that the levels of RNA from each of these forms compared with GAPDH are no different in the DM and control group (by Mann–Whitney). These data indicate that, whereas the level of DMPK transcripts in the cytoplasmic RNA fraction of DM cell lines was reduced, expression from 59 and DMAHP was unaltered, thus a mechanism for DM based on a field effect is unlikely.
Figure 4.
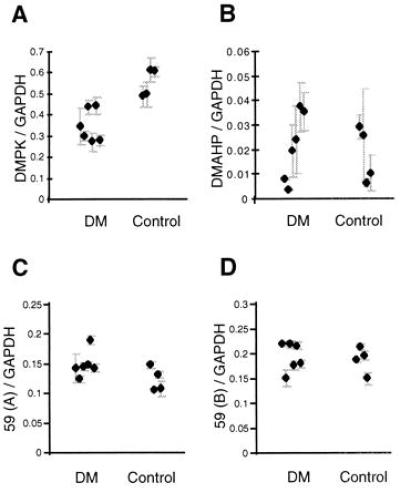
Quantitative analysis of cytoplasmic RNA from genes around the DM triplet expansion, corrected for input amount of RNA by comparison to levels of GAPDH. (A) The means and errors for quantitative analysis of DMPK RNA in DM and Control cell lines. (B) Quantitatve analysis for the levels of DMAHP in DM and Control cell lines. (C and D) Analysis of levels of expression from 59(A) and 59(B) respectively in DM and Control cell lines. The results have been confirmed by an alternative RT-PCR in which the control RNA was TF11S (data not shown).
Further Analysis of Expanded DMPK Transcripts.
To establish why expanded DMPK transcripts fail to appear in the cytoplasm, we have performed poly(A)+ selection of nuclear RNA from cell lines of two patients (DM D and DM E) and one unaffected individual (control 4) using Oligotex columns (Qiagen) with RT-PCR analysis to compare the abundance of each DMPK allele (Fig. 5). For the unaffected individual the allele ratios were equivalent in the nuclear, nuclear poly(A)+, and cytoplasmic fractions. In the DM patients, however, the expanded alleles were significantly under-represented in the nuclear poly(A)+ fractions.
Figure 5.
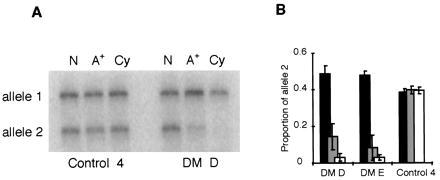
Analysis of nuclear DMPK transcripts. (A) Phosphor image showing the amounts of each DMPK allele in total nuclear RNA (N), poly(A)+-selected nuclear RNA (A+), and cytoplasmic RNA (Cy) from DM patient D and control cell line 4. (B) Histogram showing the proportion of DMPK allele 2 in nuclear RNA (black bars), poly(A)+-selected nuclear RNA (gray bars), and cytoplasmic RNA (open bars) from patients DM D and DM E and a normal control.
DISCUSSION
DM is one of a number of disorders associated with the expansion of a repeated trinucleotide DNA sequence. For many of these conditions the underlying molecular mechanism is becoming clearer (19–24). For DM the molecular basis has remained unresolved, and previous studies on DMPK RNA levels in DM patients have been equivocal. The nuclear retention of expanded DM transcripts reported here may explain discrepancies in previous RNA quantitation experiments in which raised, lowered, and unaltered levels of total DMPK RNA have been reported (5–8, 15, 16). As those analyses used intact cells or biopsied tissue from DM patients for RNA extraction, absence of the expanded allele from the cytoplasm may have been masked by increased levels in the nuclei. Different methods for RNA extraction produce variable ratios of nuclear and cytoplasmic RNA in the sample (data not shown), and if the extent of nuclear retention varies between tissues in line with DMPK expression, the disparate results noted elsewhere can be explained. Analysis of our data indicates that if cytoplasmic and nuclear levels are combined the total amounts of DMPK RNA, when measured 5′ of the repeat, in the DM patient cell lines are indistinguishable from those of the controls (by Mann–Whitney).
Contrary to reports elsewhere (15), there is no evidence of a change in the level of expression of the normal allele in any of the DM patient cell lines in our study. However, our observations have been made on fibroblasts, and it will be necessary to study further the expression in muscle and other cell lines before it is certain that there is no trans effect due to sequestration of a nuclear factor in these tissues. If the findings reported here are found in other tissues, the level of DMPK protein in classically affected DM patients is likely to be reduced to about 50% of wild-type level, lending support to a loss-of-function model for DM. Certainly some of the features of DM such as calcifying epitheliomas (1) may be attributable to a loss of DMPK function (25). However, three observations are difficult to reconcile with this being the primary mechanism underlying most features of DM. First, the other triplet-repeat disorders (reviewed in refs. 26 and 27) caused by loss-of-function mutations are recessive. Second, no DM patient has been identified with an inactivating point mutation in DMPK. Third, DMPK knock-out studies reveal that hemizygous DMPK +/− mice are virtually indistinguishable from wild type (28, 29). Thus it would seem unlikely that the reduction in DMPK RNA levels per se, with consequent reduction in DMPK protein, could account for all of the pleiotropic effects seen in DM.
It has been suggested that expansion of the CTG repeat may affect the higher-order structure of DNA, thus altering the level of expression of neighboring genes in a field effect (11–13, 18). To investigate this possibility we have examined the level of expression of 59 and DMAHP, the two genes that immediately flank DMPK, in cytoplasmic fractions of DM and control cell lines. We find that, unlike the situation for DMPK, the expression of 59 and DMAHP is not altered in DM cell lines, and a mechanism for DM based on a field effect is, therefore, unlikely.
A number of possible mechanisms might account for the nuclear retention of expanded DMPK transcripts. For example, premature transcriptional termination from the expanded DMPK allele would produce truncated transcripts that lack polyadenylylation signals in their 3′ untranslated regions. Such a mechanism is consistent with the data of Wang et al. (30), who have reported preferential nucleosome assembly at CTG repeats and suggested that this may result in transcriptional repression. Other studies have also revealed that expanded trinucleotide repeats form structures that may result in a transcriptional blockage (31, 32). Presumably truncated DMPK transcripts would be degraded in the nucleus in a similar manner to other transcripts showing premature termination, and their possible contribution to the underlying pathology of DM is unclear. Alternative explanations for the observed lack of poly(A)+ transcripts from the expansion allele in the nucleus are either that poly(A) tails on the DM allele transcripts are very short and do not bind to the Oligotex beads, or that preferential degradation of full-length and polyadenylylated DMPK transcripts, from the expanded allele, is occurring. This degradation could be either in vitro during RNA preparation (although analysis of RNA on ethidium-stained gels showed no evidence of degradation), or in vivo as part of a discard pathway for retained nuclear transcripts. However, under this model, why full-length poly(A)+ expansion transcripts should fail to be exported from the nucleus requires explanation. It is possible that the repeat expansion causes incorrect trafficking of these transcripts within the nucleus, or perhaps a conformational change at a critical length of CUG prevents nuclear export or promotes interactions with nuclear proteins which in turn prevent export.
The “foci” of expansion repeat transcripts which have been observed in the nuclei of DM cells (14) are consistent with the nuclear retention reported here. Unlike this previous study, however, we find no evidence of the expanded allele in the cytoplasm of cell lines from classical DM patients. Whatever the underlying mechanism of nuclear retention for expanded DMPK transcripts, it would seem to occur within a relatively narrow threshold of CTG length. Our data indicate that in the cell line from DM A, with an expanded repeat of 80 CTGs, the relative amounts of cytoplasmic RNA from the two DMPK alleles are virtually the same, whereas in the cytoplasm of the cell line with 400 CTGs, RNA from the expanded allele is absent. This implies that there is a threshold between 80 and 400 CTGs at which nuclear retention occurs.
In the other dominantly inherited expansion disorders, such as Huntington disease, a CAG repeat encodes an expanded polyglutamine tract that confers a gain-of-function mutation, probably through interactions with other cellular proteins (21, 22). The complete retention of expanded DMPK transcripts within the nucleus provides a vital clue to the molecular pathology of DM. Accumulation of expanded DMPK transcripts may produce a similar gain-of-function mutation, leading to inappropriate interactions within the nucleus such as the binding and sequestration of nuclear factors to the RNA. The recent identification of proteins that bind to CUG and CTG repeats could provide candidates for such interactions (33).
Acknowledgments
We thank J. A. L. Armour and R. G. Lloyd for helpful discussions and critical reading of the manuscript, M. Sibbering for help in the collection of skin biopsies, The Myotonic Dystrophy Patient Group (U.K.) for their continued support, and particularly the patients who donated skin biopsies. This work was funded by the Muscular Dystrophy Association (U.S.A.) and the University of Nottingham.
ABBREVIATIONS
- DM
myotonic dystrophy
- RT
reverse transcriptase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
References
- 1.Harper P. Myotonic Dystrophy. London: Saunders; 1989. [Google Scholar]
- 2.Brook J D, McCurrach M E, Harley H G, Buckler A J, Church D, Aburatani H, Hunter K, Stanton V P, Thirion J P, Hudson T, Sohn R, Zemelman B, Snell R G, Rundle S A, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper P S, Shaw D J, Housman D E. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 3.Fu Y-H, Pizzuti A, Fenwick R G, King J, Rajnarayan S, Dunne P W, Dubel J, Nasser G A, Ashizawa T, Dejong P, Wieringa B, Korneluk B, Perryman M B, Epstein H F, Caskey C T. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, Ohoy K, Leblond S, Earle-Macdonald J, Dejong P J, Wieringa B, Korneluk R G. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 5.Carango P, Noble J E, Marks H G, Funanage V L. Genomics. 1993;18:340–348. doi: 10.1006/geno.1993.1474. [DOI] [PubMed] [Google Scholar]
- 6.Fu Y H, Friedman D L, Richards S, Pearlman J A, Gibbs R A, Pizzuti A, Ashizawa T, Perryman M B, Scarlato G, Fenwick R G, Caskey C T. Science. 1993;260:235–238. doi: 10.1126/science.8469976. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann-Radvayi H, Lavedan C, Rabes J-P, Savoy D, Duros C, Johnson K, Junien C. Hum Mol Genet. 1993;2:1263. doi: 10.1093/hmg/2.8.1263. [DOI] [PubMed] [Google Scholar]
- 8.Sabouri L A, Mahadevan M S, Narang M, Lee D S C, Surh L C, Korneluk R G. Nat Genet. 1993;4:233–238. doi: 10.1038/ng0793-233. [DOI] [PubMed] [Google Scholar]
- 9.Hamshere M G, Brook J D. Trends Genet. 1996;12:332–334. doi: 10.1016/s0168-9525(96)80002-3. [DOI] [PubMed] [Google Scholar]
- 10.Jansen G, Mahadevan M, Amemiya C, Wormskamp N, Segers B, Hendriks W, O’Hoy K, Baird S, Sabourin L, Lennon G, Jap P L, Iles D, Coerwinkel M, Hofker M, Carrano A V, de Jong P J, Korneluk R G, Wieringa B. Nat Genet. 1992;1:261–266. doi: 10.1038/ng0792-261. [DOI] [PubMed] [Google Scholar]
- 11.Wieringa B. Hum Mol Genet. 1994;3:1–7. doi: 10.1093/hmg/3.1.1-a. [DOI] [PubMed] [Google Scholar]
- 12.Boucher C A, King S K, Carey N, Krahe R, Winchester C L, Rahman S, Creavin T, Meghji P, Bailey M E S, Chartier F L, Brown S D, Siciliano M J, Johnson K J. Hum Mol Genet. 1995;4:1919–1925. doi: 10.1093/hmg/4.10.1919. [DOI] [PubMed] [Google Scholar]
- 13.Harris S, Moncrieff C, Johnson K. Hum Mol Genet. 1996;5:1417–1423. doi: 10.1093/hmg/5.supplement_1.1417. [DOI] [PubMed] [Google Scholar]
- 14.Taneja K L, McCurrach M, Schalling M, Housman D, Singer R H. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J Z, Pegoraro E, Menegazzo E, Gennarelli M, Hoop R C, Angelini C, Hoffman E P. Hum Mol Genet. 1995;4:599–606. doi: 10.1093/hmg/4.4.599. [DOI] [PubMed] [Google Scholar]
- 16.Krahe R, Ashizawa T, Abbruzzese C, Roeder E, Carango P, Giacanelli M, Funanage V L, Siciliano M J. Genomics. 1995;28:1–14. doi: 10.1006/geno.1995.1099. [DOI] [PubMed] [Google Scholar]
- 17.Shaw D J, McCurrach M, Rundle S A, Harley H G, Crow S R, Sohn R, Thirion J P, Hamshere M G, Buckler A J, Harper P S, Housman D E, Brook J D. Genomics. 1993;18:673–679. doi: 10.1016/s0888-7543(05)80372-6. [DOI] [PubMed] [Google Scholar]
- 18.Otten A D, Tapscott S J. Proc Natl Acad Sci USA. 1995;92:5465–5469. doi: 10.1073/pnas.92.12.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pieretti M, Zhang F, Fu Y-H, Warren S T, Oostra B A, Caskey C T, Nelson D L. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 20.Campuzano V, Montermini L, Molto M D, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani S I, Gellera C, Brice A, Trouillas P, Demichele G, Filla A, Defrutos R, Palau F, Patel P I, Didonato S, Mandel J L, Cocozza S, Koenig M, Pandolfo M. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 21.Li X J, Li S H, Sharp A H, Nucifora F C, Schilling G, Lanahan A, Worley P, Snyder S H, Ross C A. Nature (London) 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 22.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Agid Y, Brice A, Mandel J-L. Nature (London) 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 23.Burke J R, Enghild J J, Martin M E, Jou Y S, Myers R M, Roses A D, Vance J M, Strittmatter W J. Nat Med. 1996;2:347–350. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg Y P, Nicholson D W, Rasper D M, Kalchman M A, Koide H B, Graham J P, Bromm M, Kazemi-Esfarjani P, Thornberry N A, Vaillancourt J P, Hayden M R. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 25.Justice R W, Zilian O, Woods D F, Noll M, Bryant P J. Genes Dev. 1995;9:534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- 26.Warren S T. Science. 1996;271:1374–1375. doi: 10.1126/science.271.5254.1374. [DOI] [PubMed] [Google Scholar]
- 27.Willems P J. Nat Genet. 1994;8:213–215. doi: 10.1038/ng1194-213. [DOI] [PubMed] [Google Scholar]
- 28.Jansen G, Groenen P J T A, Bachner D, Jap P H H, Coerwinkel M, Oerlemans F, van der Broek W, Gohlsch B, Pette D, Plomp J J, Molenaar P C, Nederhoff M G J, van Echteld C J A, Dekker M, Berns A, Hameister H, Wieringa B. Nat Genet. 1996;13:316–323. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 29.Reddy S, Smith D B J, Rich M M, Leferovich J M, Reilly P, Davis B M, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon R J, Housman D. Nat Genet. 1996;13:324–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y H, Amirhaeri S, Kang S, Wells R D, Griffith J D. Science. 1994;265:669–671. doi: 10.1126/science.8036515. [DOI] [PubMed] [Google Scholar]
- 31.Gacy A M, Goellner G, Juranic N, Macura S. Cell. 1995;81:533–535. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 32.Pearson C E, Sinden R R. Biochemistry. 1996;35:5041–5053. doi: 10.1021/bi9601013. [DOI] [PubMed] [Google Scholar]
- 33.Timchenko L T, Timchenko N A, Caskey C T, Roberts R. Hum Mol Genet. 1996;5:115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]