

Abstract
Expressed protein ligation – which allows native proteins to be selectively linked together by a normal peptide bond in an aqueous environment – has emerged as a powerful technique. The technique requires the formation of a C-terminal α-thioester and an N-terminal Cys. An N-terminal Cys can be formed by enzymatic cleavage, commonly using the Factor Xa and TEV proteases. We show that thrombin can be used for the formation of N-terminal Cys, providing another choice of reagents for expressed protein ligation. Proteins with N-terminal Cys can be obtained by the convenient modification of vectors with the putative thrombin cleavage site LVPRG to LVPRC. Two example protein domains (Csk and Abl tyrosine kinase domain) with N-terminal Cys are demonstrated using this method. The use of thrombin protease to generate N-terminal Cys overcomes some of the limitations of existing methods, making it generally useful for expressed protein ligation and other biotechnological applications.
Keywords: Expressed protein ligation, Segmental isotopic labeling, Thrombin, Tyrosine kinase, Cysteine
1. Introduction
Expressed protein ligation (EPL) is a widely used protein-engineering approach that allows recombinant and synthetic polypeptides to be chemoselectively and regioselectively joined together [1–3]. An important application is segmental labeling especially for nuclear magnetic resonance (NMR), permitting a wide range of structural mapping studies to be conducted. In this strategy, a uniformly labeled recombinant protein fragment is ligated to an unlabeled protein fragment [4–7]. Only a portion of the newly formed protein is labeled, resulting in a NMR spectrum containing a subset of the resonances from the corresponding uniformly labeled sample. The expressed protein ligation method involves the reaction of an N-terminal protein segment having a C-terminal α-thioester with a C-terminal protein segment having a N-terminal Cys residue [8].
Current methods to generate proteins with N-terminal Cys use chemical synthesis, the self-cleaving of inteins [8] and proteolysis [4] (Fig. 1). Chemical synthesis is limited to relatively small peptides. The intein system can be used for introduction of the N-terminal Cys for the C-terminal protein fragment [4]. The intein-mediated introduction of N-terminal Cys has several limitations: (1) solution pH or temperature may need to be changed to induce the cleavage leading to protein aggregation; (2) in vivo degradation of the intein fusion protein may lead to a low yield of the target protein; (3) not all target proteins can be expressed with intein fusion and some of the fusion proteins form inclusion bodies during the expression. N-terminal Cys can also be formed by enzymatic cleavage of a proteolytic leader sequence, using enzymes like Factor Xa [4,9,10] or TEV [11,12] and such cleavage is efficient in some, but not all, cases. One of the simplest ways to generate a recombinant polypeptide containing an N-terminal Cys residue is to introduce a Cys downstream to the initiating Met residue. Once the translation step is completed, the endogeneous methionyl aminopeptidases (MAP) removes the Met residue, thereby generating in vivo a N-terminal Cys residue [13]. Some studies claimed that cleavage was only partial, and side reactions can limit the yield of target N-terminal Cys protein [13,14].
Fig. 1.
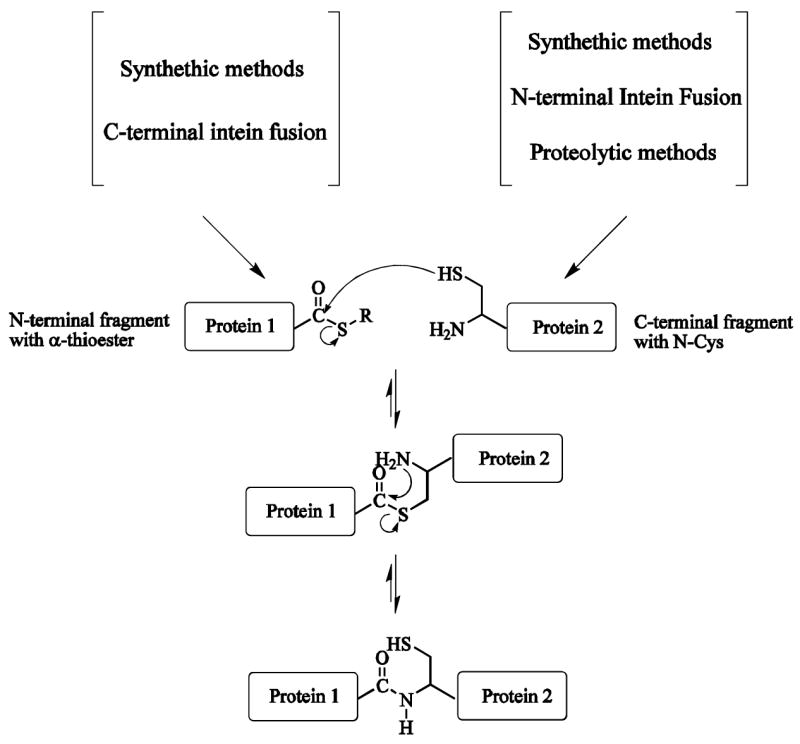
Schematic mechanism of expressed protein ligation. Expressed protein ligation is an extension of the native chemical ligation method in which two unprotected synthetic peptide are chemically ligated together. Protein 1 can be expressed as intein fusions and cleaved from the intein with a variety of thiols to give the corresponding C-terminal α-thioester. Protein 2, containing N-terminal Cys, can be made by synthetic, N-terminal intein fusion and proteolytic methods. At the ligation step, spontaneous intramolecular rearrangement generates a native peptide bond.
In this communication, we describe another choice for the formation of N-terminal Cys – demonstrating that proteins with N-terminal Cys can be produced with thrombin. Thrombin is the most widely used enzyme to cleave the target proteins from its fusion partner. Jenny et al. [15] have reviewed the use of thrombin as a tool of cleavage and have demonstrated that both thrombin and Factor Xa can hydrolyze a variety of peptide bonds in proteins. The most commonly used cutting sequences are LVPRG or LVPRGS with cleavage after the Arg residue. Proteins with N-terminal Cys can be obtained conveniently by the modification of vectors with thrombin cleavage site LVPRG to LVPRC. The use of thrombin protease to generate N-terminal Cys overcomes some of the limitations of existing methods in EPL, and should extend the applications of EPL and other biotechnological applications.
2. Materials and methods
The vector pH-MBP-T was modified from pMAL-c2X by adding His6-tag in the N-terminal of MBP and mutating Factor Xa into a thrombin cleavage site. The N-terminal His6-tag before maltose binding protein (MBP) was used to facilitate purification [16]. Human Csk kinase domain (residue 187–450) was amplified using primer 1 (5′-GAA TTC GGA TCC TGG GCC CTG AAC ATG AAG) and primer 2 (5′-TGC TAC GTC GAC CTA CAG GTG CAG CTC GTG GGT), and cloned into pH-MBP-T and further referred to as pH-MBP-T-CKinase. The plasmid with the Factor Xa cut site was obtained by primer 3 (5′-GTA GCA GGA TCC ATC GAG GGA CGT TGC GGC GGC TGG GCC CTG AAC ATG) and primer 2 and named pH-MBP-Xa-CKinase. The plasmid with an LVPRC cut site was obtained via the QuikChange [17] method using primer 4 (5′-CGG GCT GGT TCC GCG TTG TTT CTA CCG TTC CGG ATG GGC CCT G) and primer 5 (5′-CAG GGC CCA TCC GGA ACG GTA GAA ACA ACG CGG AAC CAG CCC G), and named pH-MBP-TC-CKinase. Murine ABL tyrosine kinase domain gene (protein residues 230–500) was amplified using primer 6 (5′-GGC CGG ATC CCC CAA CTA CGA CAA GTG) and primer 7 (5′-GCG CGT CGA CTT AGG ATT CCT GGA ACA TGG TTT CAA), and cloned into pH-MBP-T. This plasmid is referred to as pH-MBP-T-AKinase and was modified with an LVPRC cut site using primer 8 (5′-GCT GGT TCC GCG TTG CTC CCC CAA CTA CG) and primer 9 (5′-CGT AGT TGG GGG AGC AAC GCG GAA CCA GC) yielding H-MBP-TC-AKinase. The MBP gene was amplified and cloned into pTWIN1 (New England Biolabs Inc.) vector using the Nde I and Sap I restriction site to obtain the MBP-thioester. The plasmid was named pTWIN1-MBP and the corresponding protein product was named MBP-intein-CBD. All DNA sequences were verified using standard DNA sequencing.
BL21 cells harboring one of the plasmids (pH-MBP-T-CKinase, pH-MBP-TC-CKinase, pH-MBP-Xa-CKinase, pH-MBP-T-AKinase, pH-MBP-TC-AKinase, and pTWIN1-MBP) were inoculated in 25 ml of LB medium containing 100 μg/ml ampicillin and cultured at 37 °C until the optical density (OD600) reached 0.8. The resulting culture was then used to inoculate 11 of the LB medium. The cells were grown at 37 °C to density 0.8 and induced by adding 0.2 mM IPTG. [U-15N] H-MBP-TC-CKinase protein was produced similarly, except the cells were grown in M9 minimal media supplemented with 1 g/l 15NH4Cl as the sole nitrogen source. After overnight induction at 15 °C, the cells were harvested by centrifugation at 5000 ×g for 20 min at 4 °C. The cells were re-suspended in 35 ml of buffer A (50 mM Tris–HCl, pH 8.0, 250 mM NaCl), and lysed using French Press. The insoluble cell debris was removed by centrifugation (15000 × g) at 4 °C for 20 min. For the purification of the csk kinase fragment, the supernatant was passed through a 10 ml TALON metal affinity column (GE Healthcare) charged with Co2+ ions at allow rate of 1.0 ml/min. For the Abl kinase, Nickel-NTA beads charged with Ni2+ ions was used. The column, immobilized with the overexpressed target protein, was washed by 10× column volume of buffer A. The target fusion protein was eluted by adding 250 mM Imidazole to buffer A. All of the fractions having the target fusion protein were pooled and dialysed against the thrombin cleavage buffer (20 mM Tris–HCl, pH 8.4, 150 mM NaCl, 25 mM CaCl2) or Factor Xa cleavage buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 5 mM CaCl2), respectively. To cleave 1 mg of fusion protein with the original LVPRG site, 10 unit of human α-thrombin (Enzyme Research Laboratories) was used and for the mutated LVPRG to LVPRC site, 20 unit of human α-thrombin was used. Similarly, to cleave 1 mg of fusion protein with the Factor Xa site, 20 unit of restriction grade Factor Xa (Novagen) was added. All cleavage was conducted at 10 °C and samples were collected at different time intervals to monitor the complete cleavage of the fusion protein by running SDS–PAGE. HiTrap Q HP column was used for the purification of cleaved proteins. For the purification of MBP-intein-CBD, the clarified cell extract was loaded to 10 ml of chitin beads (New England Biolabs, S6651L) which pre-equilibrated with buffer A. The column was washed with 50 ml of buffer A to remove the unbound protein. The column bound with protein MBP-intein-CBD was stored at 4 °C for the ligation with kinase domain. The cleavage of the intein-tag was induced by equilibrating the chitin beads to buffer A with 200 mM MESNA (2-mercaptoethanesulfonic acid). After 24 h of on-column cleavage at room temperature, the target protein MBP with C-terminal thioester was eluted from the column by 40 ml of buffer A and concentrated to 0.5 mM. The protein ligation experiment was conduced at room temperature for two days in buffer A with 200 mM MESNA, 1 mM EDTA. The ligation products were checked by MALDI-TOF mass spectrum on an Applied Biosystems Voyager DE Pro machine (Table 1).
Table 1.
The molecular weight of the proteins used in the expressed protein ligation experiments
| Protein | Calculated mass (Da) | Observed mass (Da) |
|---|---|---|
| Csk CKinase | 30296 | 30308 |
| MBP (intein fusion) | 42243 | 42242 |
| MBP (kinase fusion) | 43443 | 43410 |
| Intein-CBD | 27856 | 27840 |
| MBP-intein-CBD | 70082 | 70150 |
| MBP-Ckinase ligation | 72521 | 72565 |
NMR experiments were performed at 15 °C on a Bruker 900 MHz spectrometer equipped with a triple resonance cryo-probe. Protein solutions were prepared in the following buffer conditions: 50 mM Tris–HCl (pH 7.5), 10 mM DTT, 1.0 mM EDTA, 0.01% (w/v) NaN3, 5% D2O and 0.1 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonate). 1H–15N heteronuclear single quantum coherence (HSQC) spectrum was collected with a 1H sweep width of 12626.263 Hz, with 1024 points, and at a 15N sweep width of 3284.072 Hz, 256 increments [18]. Pulsed-field gradient techniques with a WATERGATE pulse sequence were used for all H2O experiments.
3. Results
The genes of Csk and Abl kinase domain were amplified from full-length gene by PCR using primers designed to introduce BamH I and Sal I sites immediately upstream and down-stream of the gene, respectively. A similar method was used for the cloning of Csk kinase with a Factor Xa cleavage site. The cloned target gene in H-MBP-T vector is located downstream from the malE gene, which encodes maltose-binding protein (MBP). This results in the expression of a His6-tagged MBP-fusion protein. The vector uses the strong Ptac promoter and the translation initiation signals of MBP to express large amounts of the fusion protein. The fusion protein is then purified by one-step metal affinity column.
Expression of the fusion genes in Escherichia coli was analyzed by SDS–PAGE (Fig. 3) for both Csk and Abl kinase. The molecular weight of the H-MBP-T-CKinase and H-MBP-T-AKinase is ~72 kDa, which includes the molecular weight of the cleaved H-MBP (~42 kDa) and that of Csk and Abl kinase (~30 kDa). Lanes 1, 2 and 5, 6 show the SDS– PAGE before and after induction for pH-MBP-TC-Ckinase and pH-MBP-TC-Akinase with IPTG, respectively. The presence of an intense protein band around 72 kDa in lanes 2 and 6 (Fig. 3) clearly indicates that cells have expressed the fusion protein. Lanes 3, 4 and 7, 8 show the insoluble and soluble fractions in the lysate after cells were lysed using French Press for both Csk and Abl kinase fusion proteins, respectively. Presence of very intense band around 72 kDa in lanes 4 and 8 compared to lanes 3 and 7 indicates that most of the fusion proteins (H-MBP-TC-CKinase and H-MBP-TC-Akinase) are in the soluble fraction of the cell lysate. Although there are some reports that the bacterial expression of tyrosine kinase domain is difficult [19,20], our results indicate that the fusion expression system is suitable for expression of Csk kinase and Abl kinase.
Fig. 3.
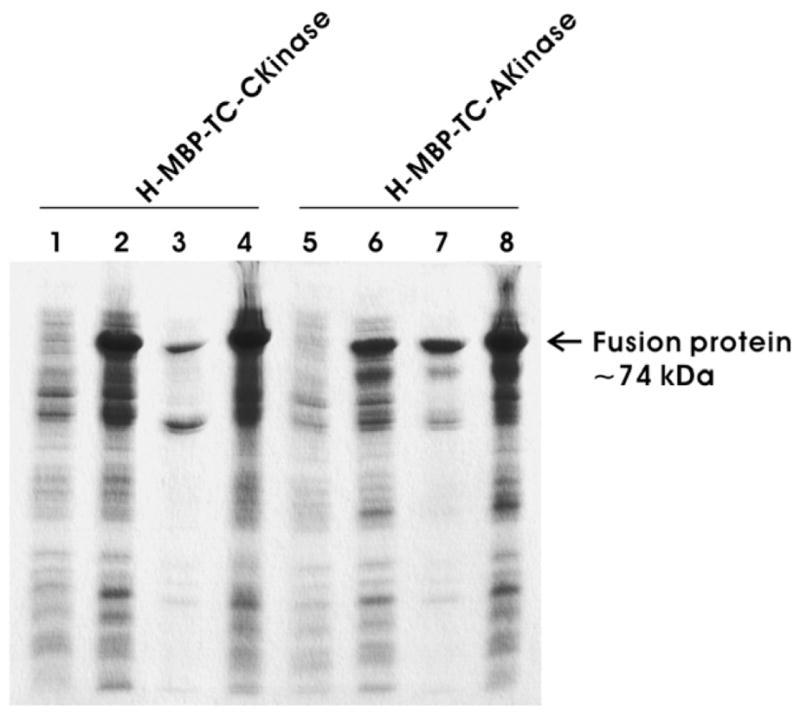
SDS–PAGE analysis of fusion expression of H-MBP-TCCKinase (lanes 1–4) and H-MBP-TC-AKinase (lanes 5–8). Lanes 1 and 5, whole cell lysate before IPTG induction; lanes 2 and 6, whole cell lysate after IPTG induction; lanes 3 and 7, pellet fraction after French Press treatment; lanes 4 and 8, soluble fraction after French Press treatment.
Since the fusion partner in the expression vector is a His6-tagged N-terminal fragment of MBP, fusion proteins can be purified by using a routine metal-chelating affinity chromatography procedure. As shown in Fig. 4, the fusion proteins were purified to apparent homogeneity by one-step purification, using chelating affinity chromatography. To test the efficiency of the mutated thrombin cleavage site, the target fusion protein with the mutated thrombin site was incubated with thrombin. A small aliquot of samples was taken at different time interval to run a SDS–PAGE. Fig. 4a shows the cleavage of H-MBP-TC-CKinase at 0, 1, 2, 4, 6, 8 and 24 h by thrombin. Lane 8 shows that the cleavage mixture after 24 h incubation with thrombin results only in specific cleavage of the kinase domain (~30 kDa), and that the complete cleavage of H-MBP-TC-CKinase can be achieved in 24 h. Thus, the target fusion protein with a mutated cut sequence (LVPRC) can be successfully cleaved without causing significant non-specific cleavage in the kinase domain. Similar results were obtained for the H-MBP-TC-AKinase. Fig. 4b summarizes the cleavage of the five fusion proteins. Note both the fusion proteins with a regular thrombin cleavage site (LVPRG) and a mutated thrombin cleavage site (LVPRC) were cleaved to completion without any detectable non-specific cleavage after 24 h. On the other hand, H-MBP-Xa-CKinase fusion protein, upon cleavage with Factor Xa, shows non-specific cleavage though the fusion protein is not fully cleaved (Fig. 4b). A 1H–15N HSQC spectrum (Fig. 5) of the Csk kinase domain with N-terminal Cys was acquired to demonstrate the folding status of this protein using the thrombin cleavage method. The HSQC spectrum is well dispersed which suggests that the isolated kinase domain is folded and well-structured.
Fig. 4.
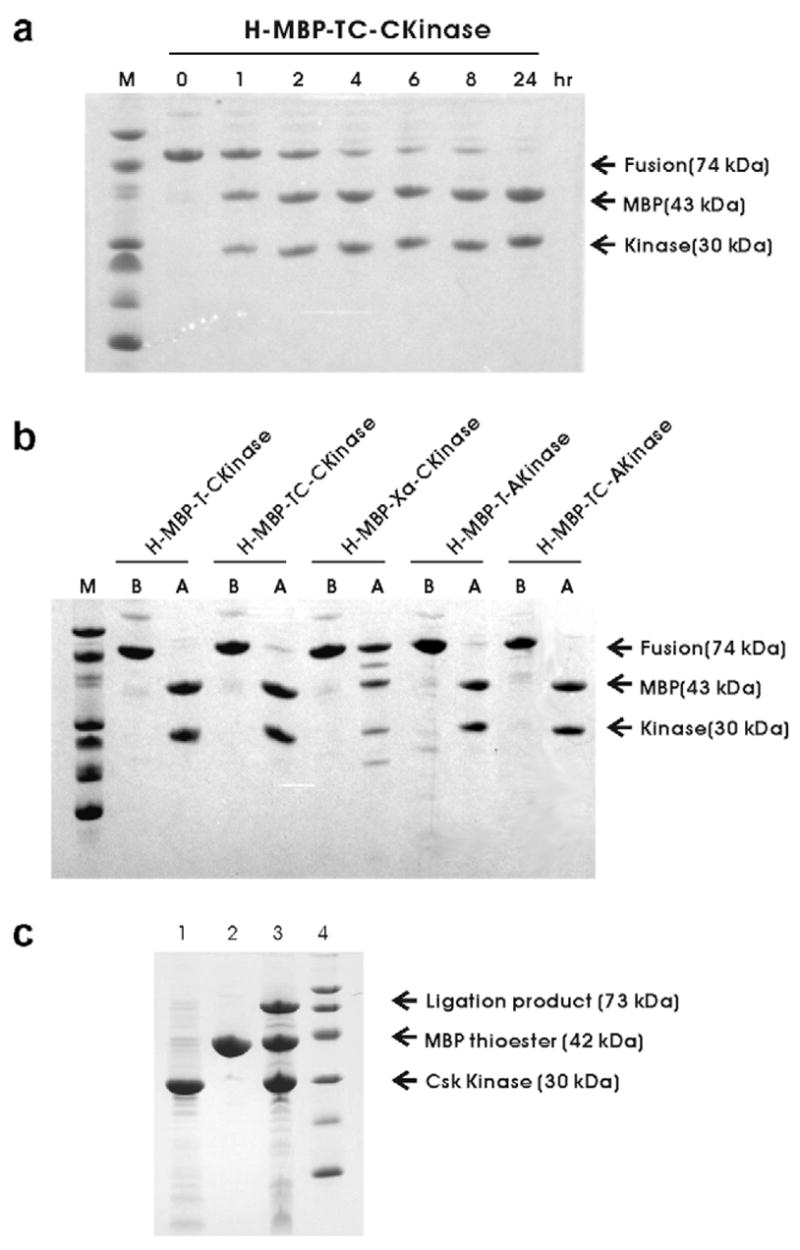
(a) SDS–PAGE analysis of H-MBP-TC-CKinase fusion protein cleaved with thrombin at different time interval. The cleavage times are indicated on the top of the gel. (b) SDS–PAGE analysis of thrombin or Factor Xa cleavage of the five fusion proteins. Protein names are indicated at the top of the gel. The lanes marked as “B” indicates the fusion protein before the enzyme cleavage whereas the lanes marked with “A” indicate after thrombin or Factor Xa cleavage. (c) SDS–PAGE analysis of protein ligation. Lane 1 shows the purified the MBP protein with C-terminal α-thioester. Lane 2 shows the purified Csk kinase domain with N-terminal Cys. Lane 3 shows the mixture of MBP with kinase domain after ligation for 24 h. A band corresponding to the ligation product appeared after 24 h of ligation. The extent of ligation was estimated as 20% by SDS–PAGE.
Fig. 5.
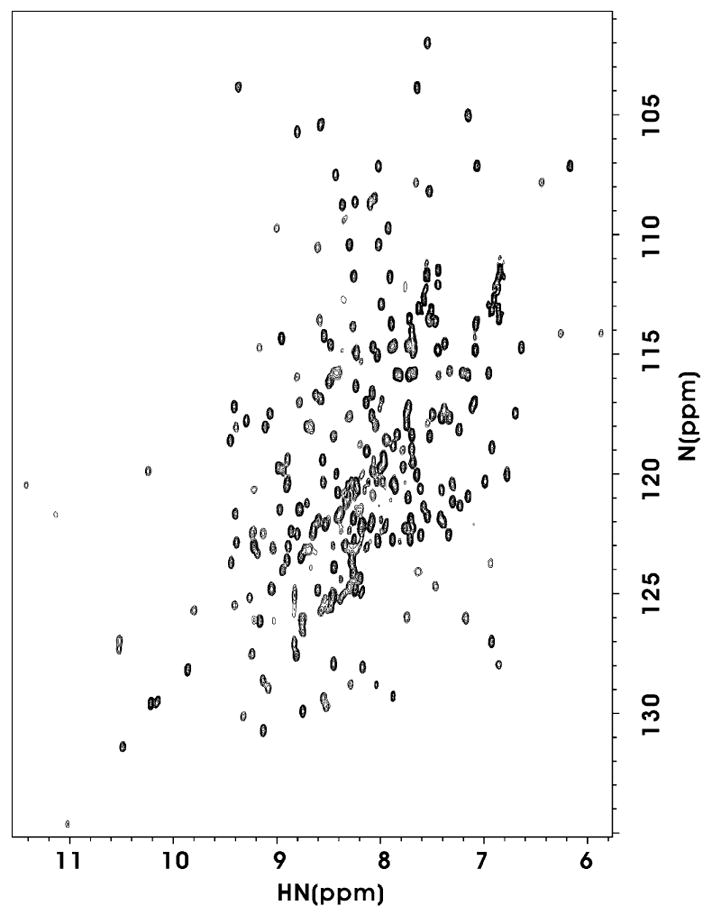
1H–15N HSQC spectrum of Csk kinase domain with Nterminal Cys. The protein sample (100 μM) was dissolved in 50 mM Tris–HCl (pH 7.5), 10 mM DTT, 1.0 mM EDTA, 0.01% (w/v) NaN3, 5% D2O, 0.1 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonate). The spectrum was recorded at 900 MHz and 15 °C.
We used MBP with C-terminal α-thioester to confirm that the protein products containing N-terminal Cys are active in ligation. MBP was expressed as an intein-chitin binding fusion protein and purified using chitin resin. Cleavage of MBP from intein was carried out at room temperature, overnight, by incubation of the fusion protein bound to chitin resin in column buffer containing buffer A and 200 mM MESNA. MBP (200 μM) and Csk kinase (400 μM) was ligated 24 h at room temperature in buffer A containing 200 mM MESNA, with 246 200 mM MESNA, 1 mM EDTA.
Fig. 4c lane 1 shows the purified the MBP protein with C-terminal α-thioester. Lane 2 shows the purified Csk kinase domain with N-terminal Cys. Lane 3 shows the mixture of MBP with Kinase domain after ligation for 24 h. A band corresponding to the ligation product appeared after 24 h of ligation. The extent of ligation was typically 20% evaluated by SDS–PAGE. The masses of the corresponding product (Table 1) are: Csk Kinase: 30 308 (30 296), MBP (from intein fusion): 42 242 (42 243), MBP (from kinase fusion): 43410 (43 443), Intein-CBD, 27 840 (27 856), MBP-intein-CBD fusion protein: 70 150 (70 082), MBP-kinase ligation product: 72565 (72 521), (calculated theoretical value indicated in ( )). These results demonstrate the N-terminal Cys prepared by thrombin cleavage can really be ligated with active α-thioester.
4. Discussion
The optimum cleavage sequence for α-thrombin could have the following nomenclature: (a) P4-P3-Pro-Arg-P1′-P2′, where P3 and P4 are hydrophobic amino acids and P1′ and P2′ are nonacidic amino acids or (b) P2-Arg-P1′, where P2 or P1′ are Gly. Though it has been shown that the hydrophobic P2 and P4 residues are essential for thrombin specificity, the apolar P1′ and P2′ residues are also crucial [21]. Thrombin prefers cleavage after P1, when the preceding residue is Arg rather than Lys [22] and cleaves efficiently when the P4 position has an aliphatic residue and the P2 position is a Pro, whereas no favorable preference is necessary at the P3 position. Jenny et al. have summarized the use of thrombin as the tool of cleavage and have demonstrated that thrombin can hydrolyze a variety of peptide bonds in proteins [15]. These findings suggest that the thrombin cleavage is not restricted to the sequence LVPRG or LVPRGS, although this sequence is optimized for the cleavage efficiency. Our results show that we can use this method to make N-terminal Cys as illustrated by Csk kinase and Abl kinase domains without significant reduction of thrombin efficiency.
Proteins with N-terminal Cys are useful in a wide range of biotechnological applications ranging from expressed protein ligation to site-specific N-terminal labeling. To the best of our knowledge, thrombin cleavage using this mutated cleavage site has not been previously reported for the generation of N-terminal Cys of the C-terminal domain for the expressed protein ligation. It is advantageous to use this mutated thrombin sequence for the generation of N-terminal Cys as thrombin cleavage sites are present in most of the commercial vectors and the thrombin protease is quite economical. The use of thrombin protease to generate N-terminal Cys overcomes some of the limitations of existing methods, and this should make it generally useful for expressed protein ligation and other biotechnological applications.
Fig. 2.
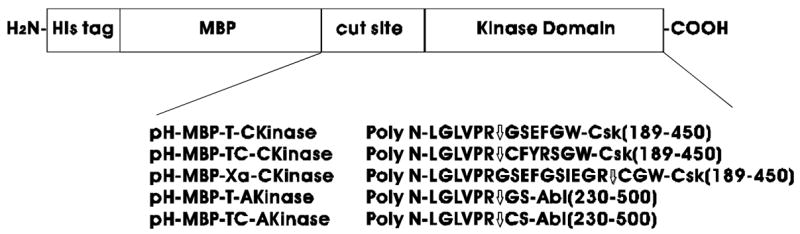
Plasmids constructed and corresponding protein sequences in this study. The enzyme cleavage site is indicated by a down arrow.
Acknowledgments
Supported by NIH Grant Nos. EB001991, GM47021, and GM66356. We thank Dr. Mary Ann Gawinowicz at Columbia University for providing the Mass Spectrometry Service.
References
- 1.Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 2.Muralidharan V, Muir TW. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 3.David R, Richter MP, Beck-Sickinger AG. Expressed protein ligation. Method and applications. Eur J Biochem. 2004;271:663–677. doi: 10.1111/j.1432-1033.2004.03978.x. [DOI] [PubMed] [Google Scholar]
- 4.Fushman D, Xu R, Cowburn D. Direct determination of changes of interdomain orientation on ligation: use of the orientational dependence of 15N NMR relaxation in Abl SH(32) Biochemistry. 1999;38:10225–10230. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]
- 5.Cowburn D, Muir TW. Segmental isotopic labeling using expressed protein ligation. Methods Enzymol. 2001;339:41–54. doi: 10.1016/s0076-6879(01)39308-4. [DOI] [PubMed] [Google Scholar]
- 6.Cowburn D, Shekhtman A, Xu R, Ottesen JJ, Muir TW. Segmental isotopic labeling for structural biological applications of NMR. Methods Mol Biol. 2004;278:47–56. doi: 10.1385/1-59259-809-9:047. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki T, Otomo T, Oda N, Kyogoku Y, Uegaki K, Ito N, Ishino Y, Nakamura H. Segmental isotope labeling for protein NMR using peptide splicing. J Am Chem Soc. 1998;120:5591–5592. [Google Scholar]
- 8.Xu MQ, Evans TC., Jr Intein-mediated ligation and cyclization of expressed proteins. Methods. 2001;24:257–277. doi: 10.1006/meth.2001.1187. [DOI] [PubMed] [Google Scholar]
- 9.Camarero JA, et al. Autoregulation of a bacterial sigma factor explored by using segmental isotopic labeling and NMR. Proc Natl Acad Sci USA. 2002;99:8536–8541. doi: 10.1073/pnas.132033899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romanelli A, Shekhtman A, Cowburn D, Muir TW. Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein–intein junction. Proc Natl Acad Sci USA. 2004;101:6397–6402. doi: 10.1073/pnas.0306616101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tolbert TJ, Franke D, Wong CH. A new strategy for glycoprotein synthesis: ligation of synthetic glycopeptides with truncated proteins expressed in E. coli as TEV protease cleavable fusion protein. Bioorg Med Chem. 2005;13:909–915. doi: 10.1016/j.bmc.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 12.Tolbert TJ, Wong CH. Conjugation of glycopeptide thioesters to expressed protein fragments: semisynthesis of glycosylated interleukin-2. Methods Mol Biol. 2004;283:255–266. doi: 10.1385/1-59259-813-7:255. [DOI] [PubMed] [Google Scholar]
- 13.Gentle IE, De Souza DP, Baca M. Direct production of proteins with N-terminal cysteine for site-specific conjugation. Bioconjug Chem. 2004;15:658–663. doi: 10.1021/bc049965o. [DOI] [PubMed] [Google Scholar]
- 14.Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci USA. 1989;86:8247–8851. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 16.Alexandrov A, Dutta K, Pascal SM. MBP fusion protein with a viral protease cleavage site: one-step cleavage/purification of insoluble proteins. Biotechniques. 2001;30:1194–1198. doi: 10.2144/01306bm01. [DOI] [PubMed] [Google Scholar]
- 17.Shen TJ, Zhu LQ, Sun X. A marker-coupled method for site-directed mutagenesis. Gene. 1991;103:73–77. doi: 10.1016/0378-1119(91)90393-p. [DOI] [PubMed] [Google Scholar]
- 18.Mori S, Abeygunawardana C, Johnson MO, Vanzijl PCM. Improved sensitivity of Hsqc spectra of exchanging protons at short interscan delays using a new fast Hsqc (Fhsqc) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 19.Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, Kuriyan J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YH, Ayrapetov MK, Lin X, Sun G. A new strategy to produce active human Src from bacteria for biochemical study of its regulation. Biochem Biophys Res Commun. 2006;346:606–611. doi: 10.1016/j.bbrc.2006.05.180. [DOI] [PubMed] [Google Scholar]
- 21.Chang JY. Thrombin specificity. requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur J Biochem. 1985;151:217–224. doi: 10.1111/j.1432-1033.1985.tb09091.x. [DOI] [PubMed] [Google Scholar]
- 22.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci USA. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]