

Abstract
Recent studies have demonstrated that reactive oxygen species (ROS) mediate myocardial ischemia-reperfusion (I/R) and angiogenesis via the mitogen-activated protein kinases and the serine-threonine kinase Akt/protein kinase B pathways. NADPH oxidases are major sources of ROS in endothelial cells and cardiomyocytes. In the present study, we investigated the role of NADPH oxidase-derived ROS in hypoxia-reoxygenation (H/R)-induced Akt and ERK1/2 activation and angiogenesis using porcine coronary artery endothelial cells (PCAECs) and a mouse myocardial I/R model. Our data demonstrate that exposure of PCAECs to hypoxia for 2 h followed by 1 h of reoxygenation significantly increased ROS formation. Pretreatment with the NADPH oxidase inhibitors, diphenyleneiodonium (DPI, 10 μM) and apocynin (Apo, 200 and 600 μM), significantly attenuated H/R-induced ROS formation. Furthermore, exposure of PCAECs to H/R caused a significant increase in Akt and ERK1/2 activation. Exposure of PCAEC spheroids and mouse aortic rings to H/R significantly increased endothelial spheroid sprouting and vessel outgrowth, whereas pharmacological inhibition of NADPH oxidase or genetic deletion of the NADPH oxidase subunit, p47phox (p47phox−/−), significantly suppressed these changes. With the use of a mouse I/R model, our data further show that the increases in myocardial Akt and ERK1/2 activation and vascular endothelial growth factor (VEGF) expression were markedly blunted in the p47phox−/− mouse subjected to myocardial I/R compared with the wild-type mouse. Our findings underscore the important role of NADPH oxidase and its subunit p47phox in modulating Akt and ERK1/2 activation, angiogenic growth factor expression, and angiogenesis in myocardium undergoing I/R.
Keywords: mouse model of ischemia-reperfusion, reduced nicotinamide adenine dinucleotide phosphate oxidase-derived reactive oxygen species, serine-threonine kinase Akt/protein kinase B, extracellular signal-regulated kinase, p47phox mouse
EARLY REPERFUSION of the ischemic myocardium is a key clinical event after myocardial infarction, and angiogenesis is a critical process in the restoration of coronary blood flow and the repair of the ischemic myocardium (18, 27, 31). Myocardial angio-genesis and collateral blood vessel growth are adaptive responses to hypoxia-reoxygenation (H/R) and ischemia-reperfusion (I/R). H/R and I/R are known stimuli for myocardial angiogenesis (8, 35–38, 49). In vitro, exposure of human coronary microvascular endothelial cells to H/R has been shown to increase endothelial cell tubule formation and angiogenesis (22, 28). In rats, exposure of the myocardium to I/R has been reported to cause significant increases in angiogenic growth factor expression, such as vascular endothelial growth factor (VEGF), and myocardial capillary density (26, 34, 37, 45).
Reactive oxygen species (ROS) are essential signaling intermediates in hypoxia-induced endothelial cell proliferation, angiogenic factor expression, and angiogenesis (15, 16, 22, 26, 28, 39). Exposure of human microvascular endothelial cells to H2O2 results in induction of endothelial tubule formation and angiogenesis (44). Generation of ROS is increased during H/R and I/R in the heart and in cardiomyocytes (10, 11, 30, 50). NADPH oxidases are major sources of ROS in vascular cells and cardiomyocytes. Many components of the phagocytic NADPH oxidase complex, including p22phox, p47phox, p67phox, gp91phox, and Rac1, have been identified in endothelial cells, cardiomyocytes, and vascular smooth muscle cells (14–16, 33, 39). Activation of NADPH oxidase and production of ROS by this enzyme system has been implicated in the pathogenesis of numerous cardiovascular diseases and has been linked to cardiomyocyte hypertrophy, myocardial remodeling, and heart failure (14, 16). Our previous study and data from other investigators clearly demonstrate that endothelial NADPH oxidase plays a critical role in the regulation of angiogenic growth factor (VEGF and angiopoietin-1) signaling and hind-limb ischemia-induced angiogenesis (1, 7, 20, 46, 48). However, the role of myocardial endothelial NADPH oxidase in H/R and I/R-induced myocardial angiogenesis has not previously been explored.
NADPH oxidase-derived ROS have been implicated as key players in myocardial I/R. Recently, ROS generated through NADPH oxidase were found to be responsible for I/R preconditioning (3, 8, 21, 27, 32). Pharmacological inhibition of NADPH oxidase activity by apocynin (Apo), an agent blocking p47phox assembly, or genetic deletion of the NADPH oxidase subunit, NAPDH oxidase (NOX)2 (gp91phox), significantly diminished I/R preconditioning (3, 8, 21). In addition, it has been reported that I/R preconditioning is mediated by the activation of the mitogen-activated protein kinases (MAPKs) and the serine-threonine kinase Akt/protein kinase B signaling pathways (9, 17, 21). The extracellular signal-regulated kinase (ERK1/2) and Akt have been reported to be activated by I/R in vivo. However, the signaling pathways involved in activation of these kinases are not fully understood in the I/R myocardium. Our previous study showed that ERK1/2 and Akt activities were modulated by NADPH oxidase in myocardial endothelial cells (7).
The present study utilizes porcine coronary artery endothelial cells (PCAECs), mouse heart microvascular endothelial cells (MHMECs) isolated from the p47phox−/− mouse, and a mouse I/R model to dissect the possible role of endothelial NADPH oxidase and its p47phox subunit in H/R (an in vitro model of I/R), I/R-stimulated Akt and ERK1/2 signaling, and angiogenesis. Our data demonstrate that exposure to either H/R or I/R leads to striking activation of ERK1/2 and Akt and to increased VEGF expression and angiogenesis. The H/R and I/R-induced ERK1/2 and Akt activation, VEGF expression, and angiogenesis were significantly suppressed by pharmacological inhibition of NADPH oxidase or genetic blockade of the NADPH oxidase subunit p47phox. Our data indicate that NADPH oxidase and its p47phox subunit are both necessary and required for myocardial Akt and ERK1/2 activation, angiogenic growth factor expression, and angiogenesis in response to H/R and I/R.
MATERIALS AND METHODS
PCAECs
Endothelial cells were carefully removed from the luminal surface of normal porcine coronary arteries and cultured as previously described (5, 6). Primary cultures of PCAECs, between passages 5 and 10, were used in all experiments.
p47phox-deficient mouse heart microvascular endothelial cells
The p47phox knockout (p47phox−/−) mice were provided by Dr. Steve Holland at National Institute of Allergy and Infectious Diseases. p47phox-deficient MHMECs were isolated and cultured as previously described (7). Primary cultures of p47phox-deficient MHMECs, between passages 4 and 10, were used in all experiments. p47phox protein expression was absent in these cells as assessed by Western blot analysis.
H/R
Before initiation of hypoxia, the culture medium was bubbled with 5% CO2-95% N2 gas mixture for 15 min to allow the medium to equilibrate. The cultures were then placed in a modular incubator chamber (Billups-Rothenburg) and flushed with a gas mixture of 5% CO2-95% N2 for 15 min at 3 l/min (6). Using a standard blood-gas analyzer, we have verified that the PO2 of the media was 30 mmHg. After exposure to hypoxia for 2 h, cell cultures were reoxygenated under normoxic conditions for the indicated times.
Pharmacological interventions
Before exposure to H/R, PCAECs were pretreated for 30 min with the NADPH oxidase inhibitors diphenyleneiodonium (DPI, 10 μM Calbiochem, San Diego, CA) and apocynin (Apo, 200 and 600 μM, CalBiochem). These concentrations were chosen because we have previously shown that they have no effect on basal levels of ROS and superoxide formation (7).
Measurement of intracellular ROS
Intracellular ROS were determined by oxidative conversion of cell-permeable chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Molecular Probes; Eugene, OR) to fluorescent dichlorofluorescein (DCF) as previously described (7). Briefly, endothelial cells, cultured in two-well chamber slides, were pretreated with the NADPH oxidase inhibitors DPI (10 μM) and Apo (200 and 600 μM) for 30 min and then exposed to hypoxia for 2 h followed by 60 min of reoxygenation. The cells were then incubated with 10 μM CM-H2DCFDA in PBS for 30 min. DCF fluorescence was measured over the whole field of vision using a Zeiss fluorescence microscope connected to an imaging system.
Measurement of superoxide
The cells were washed twice with ice-cold PBS, scraped, and suspended in 500 μl of homogenization buffer containing 20 mmol/l K2HPO4, 1 mmol/l EGTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mmol/l phenylmethylsulfonyl fluoride. The cell suspension was then homogenized on ice in a Dounce homogenizer. Superoxide was determined by lucigenin-dependent chemiluminescence. Briefly, 900 μl of assay buffer containing 50 mmol/l NaHPO4, 1 mmol/l EGTA, 150 mmol/l sucrose, 5 μmol/l lucigenin (Sigma), and 100 μmol/l NADPH were added to 100 μl of the cell homogenate. Chemiluminescent photoemission was determined in relative light units using a luminometer (7).
Western blot analysis
PCAECs and MHMECs were lysed in 300 μl of lysis buffer, and total protein concentrations were determined using a bicinchoninic acid protein assay kit (Pierce). Fifty micrograms of protein were subjected to SDS-PAGE on 10% polyacrylamide gels and transferred to a nitrocellulose membrane. The blot was probed with rabbit anti-phospho-Akt, phospho-ERK1/2, or VEGF (1:1,000 dilution, Santa Cruz Biotech) for 1 h followed by incubation with horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000 dilution, Promega) for 1 h at room temperature. Total levels of Akt and ERK1/2 were detected using anti-Akt and anti-ERK1/2 (1:1,000, Cell Signaling Technology) on the same nitrocellulose blots after the stripping.
Endothelial spheroid angiogenesis assay
Endothelial spheroids were generated as previously described (5, 7). The endothelial spheroids were harvested and evenly dispersed in 0.2% methocell-10% FBS in endothelial growth medium (EGM). For the pharmacological intervention experiments, each pharmacological intervention was mixed with 0.5 ml of the spheroid suspension and collagen gel solution and then added to a 24-well culture plate. The gel was solidified at 37°C for 30 min, and 0.5 ml 20% FBS EGM with or without each pharmacological intervention was added to the top of the gel before exposure to H/R. After exposure to H/R for 24 h, the mean length of capillary sprouts from each spheroid was measured with image acquisition and analysis software.
Mouse aortic ring assay
Wild-type (WT; C57BL/6J) and p47phox−/− mice at 8 wk of age were used for these experiments. Mouse aortas were isolated under aseptic conditions. Cleaned arteries were cut into small rings ∼1 mm in thickness and placed in the middle of a 24-well plate. The rings were overlaid with 300 μl of Matrigel and left to polymerize for 1 to 2 h at 37°C before adding 10% FBS EGM. The rings were exposed to hypoxia for 2 h followed by reoxygenation for 5−7 days. Vessel sprouts were observed by phase contrast microscopy, and pictures were taken after 5−7 days. The areas of sprouts were measured using image acquisition and analysis software (7).
In vivo mouse myocardial I/R
WT and p47phox−/− mice (8 wk of age) were anesthetized with ketamine (100−120 mg/kg) plus xylazine (15 mg/kg), intubated, and artificially ventilated with room air. A left thoracotomy was performed, and the left anterior descending coronary artery (LAD) was exposed. An 8−0 nylon suture was placed around the LAD. Coronary I/R was achieved by tightening the suture for 2 h followed by either 45 min, 2 h, or 24 h of reperfusion. The sham-operated control underwent the surgery without coronary occlusion and reperfusion. After coronary I/R, the hearts were harvested. Mouse left ventricle was dissected and homogenized in lysis buffer (100 mmol/l K2HPO4, 1 mmol/l phenylmethylsulfonyl fluoride, and 0.2% Triton X-100), subjected to SDS-PAGE on 10% polyacrylamide gels, and transferred to a nitrocellulose membrane. The membranes were immunoblotted with rabbit anti-phospho-Akt, anti-phospho-ERK1/2, and anti-VEGF (1:1,000 dilution) for 1 h, followed by incubation with horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000 dilutions) for 1 h at room temperature. Total levels of Akt and ERK1/2 were detected by using anti-Akt and anti-ERK1/2 (1:1,000, Cell Signaling Technology) on the same nitrocellulose blots after the stripping.
All procedures were in conformance with the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Statistical analysis
The results are expressed as means ± SD. Statistical analysis was performed using a one-way ANOVA followed by the Duncan's multiple-comparison test. A value of P < 0.05 was considered significant.
RESULTS
H/R-mediated ROS and superoxide formation are dependent on NADPH oxidase activity
Exposure of PCAECs to hypoxia for 2 h followed by 15, 30, and 60 min of reoxygenation caused a significant increase in intracellular ROS formation (Fig. 1A). Exposure to hypoxia for 2 h followed by 60 min of reoxygenation also resulted in a twofold increase in intracellular super-oxide formation measured by lucigenin-dependent chemiluminescence (Fig. 1C). After 60 min of reoxygenation, pretreatment with the NADPH oxidase inhibitors DPI (10 μM) and Apo (200 and 600 μM) for 30 min completely abolished H/R-induced ROS and superoxide formation (Fig. 1, B and C). Treatment with DPI or Apo alone had no effect on basal superoxide production (Fig. 1C).
Fig. 1.
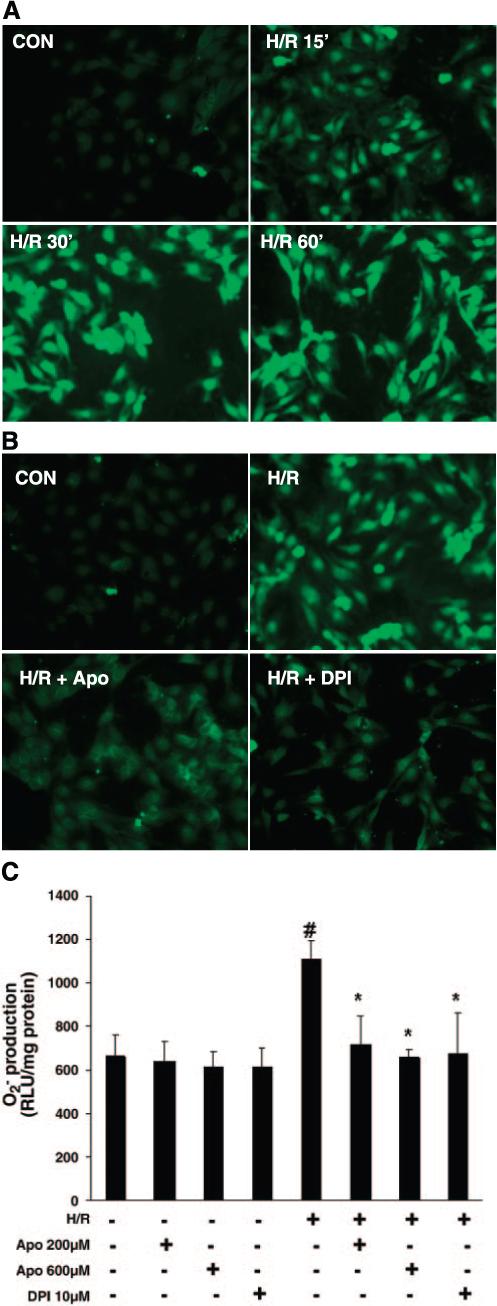
A: time course of hypoxia-reoxygenation (H/R)-stimulated generation of reactive oxygen species (ROS) in porcine coronary artery endothelial cells (PCAECs). Representative images from 3 separate studies of dichlorofluorescein fluorescence in chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate-loaded PCAECs taken at baseline and after exposure to 2 h of hypoxia followed by 15, 30, or 60 min of reoxygenation. B: pretreatment of PCAECs with either diphenyleneiodonium (DPI; 10 μM) or apocynin (Apo; 600 μM) caused an inhibition of H/R-induced ROS generation. C: pretreatment of PCAECs with either the NADPH oxidase inhibitors DPI (10 μM) or Apo (200 and 600 μM), followed by exposure to hypoxia for 2 h and reperfusion for 60 min, resulted in significant inhibition of H/R-induced superoxide formation as measured by lucigenin-dependent chemiluminescence (n = 3−4 cell lines, data are means ± SD. *P < 0.05 compared with H/R; #P < 0.05 compared with baseline). RLU, relative light units; Con, control.
H/R stimulates Akt and ERK1/2 kinase phosphorylation in a NADPH oxidase-dependent manner
Exposure to hypoxia for 2 h followed by various periods of reoxygenation (5, 15, 30, 60, and 120 min) resulted in a gradual increase in Akt phosphorylation at Ser473 (p-Akt). Akt phosphorylation peaked at 15 and 30 min of reoxygenation and, by 60 min, had declined and but remained above control levels (Fig. 2A). Pretreatment with the NADPH oxidase inhibitors DPI (10 μM) and Apo (200 and 600 μM) for 30 min completely abolished H/R (2 h of hypoxia and 30 min of reoxygenation)-induced Akt phosphorylation (Fig. 2, B and C). Treatment with DPI and Apo alone had no effect on basal Akt phosphorylation (Fig. 2, B and C).
Fig. 2.
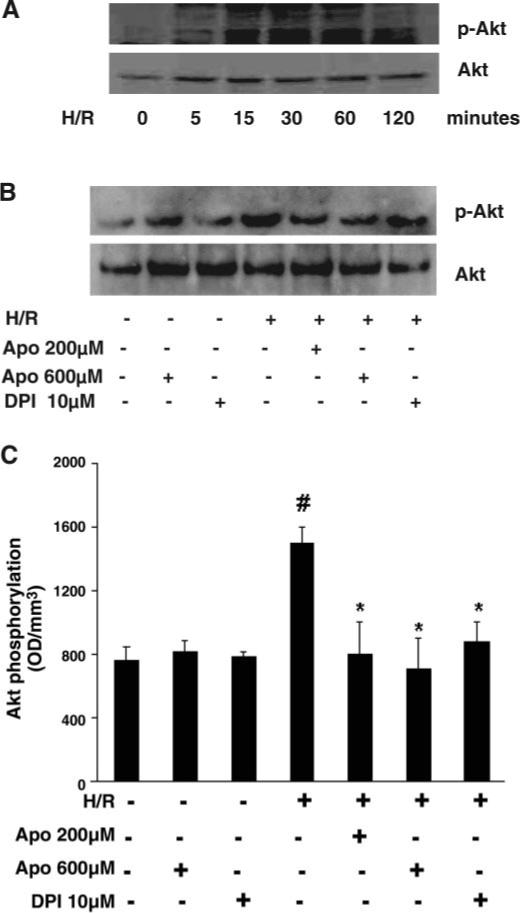
A: Western blot analysis showing time course of H/R-stimulated Akt phosphorylation in PCAECs. Phosphorylation of Akt was increased and peaked at 15 and 30 min; by 120 min phospho (p)-Akt had declined toward normal. Total Akt levels were unaltered over 120 min of reoxygenation. B: effect of NADPH oxidase inhibitors DPI (10 μM) or Apo (200 and 600 μM) on H/R-stimulated Akt phosphorylation. C: densitometric data from Western blot analyses of PCAECs showing that pretreatment with DPI and Apo significantly suppressed H/R-induced Akt phosphorylation (n = 3−4 cell lines, data are means ± SD, *P < 0.05 compared with H/R; #P < 0.05 compared with baseline). OD, optical density.
Exposure to hypoxia for 2 h followed by reoxygenation for 5, 15, 30, 60, and 120 min resulted in a significant increase in phosphorylated ERK1/2. Increased phosphorylation was apparent at 5 min of reoxygenation and remained high over the 120 min of reoxygenation (Fig. 3A). Pretreatment with DPI (10 μM) and Apo (600 μM) significantly suppressed H/R (2 h of hypoxia and 30 min of reoxygenation)-induced ERK1/2 phosphorylation (Fig. 3, B and C). Pretreatment with Apo (200 μM) had little effect on H/R-stimulated ERK1/2 phosphorylation. Treatment with DPI and Apo in the absence of H/R had no effect on basal ERK1/2 phosphorylation (Fig. 3, B and C).
Fig. 3.

A: Western blot analysis showing the time course of H/R-stimulated ERK1/2 phosphorylation in PCAECs. Increased phosphorylation was apparent at 5 min, peaked at 15 min, and remained high over the 120 min of reoxygenation. B: effect of NADPH oxidase inhibitors DPI (10 μM) or Apo (200 and 600 μM) on H/R-stimulated ERK1/2 phosphorylation. Both DPI (10 μM) and Apo (600 μM) but not 200 μM Apo suppressed H/R-stimulated ERK1/2 phosphorylation. C: densitometric data from Western blot analyses of PCAECs showing that pretreatment with DPI (10 μM) and Apo (600 μM), but not Apo (200 μM), significantly suppressed H/R-induced ERK1/2 phosphorylation (n = 4 cell lines, data are means ± SD, *P < 0.05 compared with H/R; #P < 0.05 compared with baseline).
H/R fails to stimulate Akt and ERK1/2 phosphorylation in p47phox−/− MHMECs
Using MHMECs from p47phox−/− mice, we examined the potential role of the NADPH oxidase subunit p47phox in the regulation of Akt and ERK1/2 phosphorylation in response to H/R. In contrast to WT MHMECs, exposure of p47phox−/− MHMECs to hypoxia for 2 h followed by 5, 15, 30, 60, and 120 min of reoxygenation caused little change in Akt and ERK1/2 phosphorylation. Total Akt and ERK1/2 levels were also unaltered over the 2 h of hypoxia and 120 min of reoxygenation (Fig. 4, A and B). In contrast to results in MHMECs from WT mice, exposure of p47phox−/− MHMECs to hypoxia for 2 h followed by 60 min of reoxygenation failed to induce a significant increase in intracellular ROS and super-oxide formation (Fig. 4, C and D).
Fig. 4.

Effect of H/R on Akt, ERK1/2 phosphorylation, and ROS formation in wild-type (WT) and p47phox−/− mouse heart microvascular endothelial cells (MHMECs). A: exposure of WT MHMECs to hypoxia for 2 h followed by reoxygenation for 5, 15, 30, 60, and 120 min resulted in a gradual increase in Akt phosphorylation. p47phox−/− MHMECs failed to show an increase in Akt phosphorylation in response to H/R (n = 3 cell lines). B: time course of H/R-stimulated ERK1/2 phosphorylation in WT and p47phox−/− MHMECs. Exposure of WT MHMECs to H/R resulted in a significant increase in phosphorylated ERK1/2. Cells isolated from p47phox−/− mice failed to show an increase in ERK1/2 phosphorylation (n = 3 cell lines). C: H/R- induced intracellular ROS formation was markedly attenuated in p47phox−/− MHMECs compared with WT MHMECs (n = 4 −5 cell lines). D: H/R-induced superoxide production was significantly suppressed in p47phox−/− MHMECs (black bars) as compared with WT MHMECs (white bars) (n = 5 cell lines, data are means ± SD, #P < 0.05 compared with baseline). E and F: time course and patterns of I/R-induced ERK1/2 (E) and Akt (F) phosphorylation in WT (△) and p47phox−/− (▲) mice [n = 4−5 mice, data are means ± SD, *P < 0.05 compared with ischemia-reperfusion (I/R); #P < 0.05 compared with control]. KO, knockout.
p47phox regulates I/R-induced myocardial Akt and ERK1/2 activation
To confirm our in vitro findings, we examined myocardial Akt and ERK1/2 activation in response to I/R in WT and p47phox−/− mice. Western blot analysis showed that WT mouse heart exposed to ischemia for 2 h followed by 45 min, 2 h, and 24 h of reperfusion resulted in a biphasic increase in ERK1/2 phosphorylation at 45 min and 24 h of reperfusion. Similarly, exposure of WT mouse heart to ischemia for 2 h followed by 45 min and2hof reperfusion increased Akt phosphorylation. Our densitometric data further revealed that in contrast to WT mice, the p47phox−/− mouse heart subjected to ischemia for 2 h followed by 45 min, 2 h, and 24 h of reperfusion had a gradual decline in ERK1/2 phosphorylation compared with baseline, but this did not reach statistical significance (Fig. 4E). Our densitometric data also showed that no statistically significant changes in Akt phosphorylation were detected in the p47phox−/− mouse hearts subjected to I/R at any time point (Fig. 4F).
H/R stimulates sprouting from collagen gel-embedded endothelial spheroids
Using endothelial spheroid sprouting, a model of angiogenesis in vitro, we examined the angiogenic activity of PCAECs in response to H/R. As shown in Fig. 5A, collagen gel-embedded spheroids under control conditions had a low level of spontaneous sprouting. Exposure of PCAECs spheroids to2hof hypoxia followed by 24 h of reoxygenation led to a significant increase in the sprout length from the spheroids (Fig. 5B). Exposure to 2 h of hypoxia followed by 48−72 h of reoxygenation resulted in a significant network of sprouts and branch formations from the spheroids (Fig. 5, C and D).
Fig. 5.
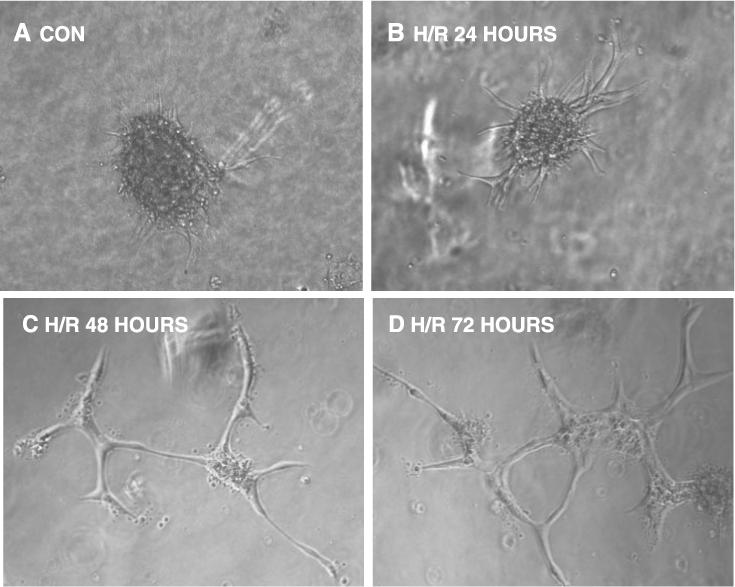
Representative images showing the sprouting of capillary-like structures from collagen-embedded PCAEC spheroids over time following exposure to H/R. A: 24-h control. B: H/R, 24 h. C: H/R, 48 h. D: H/R, 72 h. Untreated PCAEC spheroids had a low level of spontaneous sprouting. Twenty-four hours of exposure to H/R led to growth of sprouts from the spheroids. Exposure to H/R for 48−72 h resulted in a complex network of tubules sprouting from the spheroids.
H/R-stimulated endothelial cell spheroid sprouting is dependent on NADPH oxidase
Exposure of endothelial spheroids to either H/R + DPI (10 μM) or H/R + Apo (200 and 600 μM) for 24 h of reoxygenation resulted in a significant suppression of H/R-induced endothelial cell spheroid sprouting (Fig. 6, A and B). In addition, treatment with phosphatidylinositol 3-kinase (PI3K) inhibitor wortmannin (500 nM) or the ERK1/2 inhibitor PD-98059 (25 μM) led to a dramatic inhibition of H/R-stimulated PCAEC spheroid sprouting (Fig. 6, A and B). Treatment with DPI, Apo, wortmannin, or PD-98059 alone had little effect on basal endothelial spheroid sprouting (Fig. 6, A and B). Exposure of p47phox−/− endothelial spheroids to H/R for 24 h led to a smaller increase in endothelial spheroid sprouts compared with that in WT MHMECs (Fig. 6, C and D).
Fig. 6.

A: representative images of the effects of inhibitors of NADPH oxidase, phosphatidylinositol 3-kinase (PI3K), and ERK1/2 on H/R-stimulated sprouting from PCAEC spheroids at 24 h. B: quantitative analysis of sprout length from PCAEC spheroids exposed to H/R. Treatment with DPI (10 μM), Apo (200 μm and 600 μM), Wortmannin (Wort; 500nM), or PD-98059 (25 μM) suppressed the H/R-stimulated increase in sprout length (n = 4 cell lines, data are means ± SD; *P < 0.05 compared with H/R; #P < 0.05 compared with baseline). C: images of WT and p47phox−/− MHMEC spheroids exposed to H/R for 24 h. D: quantitative analysis of sprout length from WT (white bars) and p47phox−/− (black bars) MHMEC spheroids exposed to H/R. H/R was less effective in stimulating spheroid spourting in p47phox−/− MHMECs compared with WT MHMECs (n = 3 cell lines, data are means ± SD, *P < 0.05 compared with H/R; #P < 0.05 compared with non-H/R).
H/R-induced vascular sprouting and VEGF expression were attenuated in p47phox−/− mice
The effect of the NADPH oxidase subunit p47phox on H/R-mediated angiogenesis was further examined by using the aortic ring assay in WT and p47phox−/− mice. WT mouse aortic rings exposed to H/R showed dramatic increases in vessel outgrowths by day 5−7 (Fig. 7, A and B). Outgrowth area was also increased in p47phox−/− mouse aortic rings exposed to H/R compared with baseline, but the H/R-induced increase was greatly diminished compared with that induced by H/R in WT aortic rings (Fig. 7, A and B). Exposure of WT and p47phox−/− MHMECs to hypoxia for 2 h followed by 4, 8, and 24 h of reoxygenation led to a dramatic induction of VEGF expression in WT MHMECs but not in p47phox−/− MHMECs exposed to H/R (Fig. 8A). Similarly, exposure to ischemia for 2 h followed by 24 h of reperfusion resulted in an increase in VEGF expression in WT mouse heart, whereas I/R had little effect on VEGF expression in the p47phox−/− mouse heart (Fig. 8B).
Fig. 7.
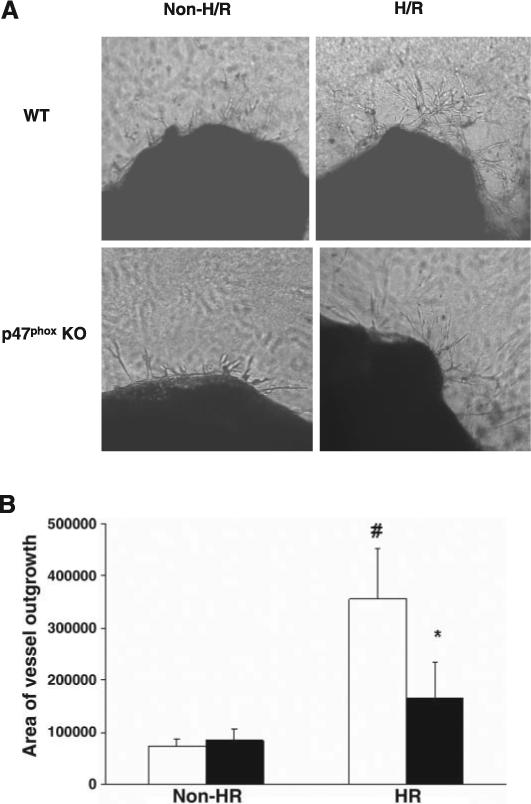
A: images of the effects of NADPH oxidase inhibitors on H/R-stimulated vessel outgrowth from WT and p47phox−/− mouse aortic rings at days 5−7. WT aortic rings exposed to H/R demonstrated increases in vessel outgrowth at 5−7 days, whereas vessel outgrowth from p47phox−/− mouse aortic rings was minimal at days 5−7. B: quantitative analysis of the area of vessel outgrowth (in arbitrary units) from WT (white bars) and p47phox−/− (black bars) mouse aortic rings. Exposure to H/R significantly increased the outgrowth area in WT mouse aortic rings. The outgrowth area was also increased in the p47phox−/− mouse aortic rings exposed to H/R compared with baseline, but this increase was less than that in H/R-stimulated WT aortic rings (n = 4−5, data are means ± SD, *P < 0.05 compared with H/R; #P < 0.05 compared with baseline).
Fig. 8.
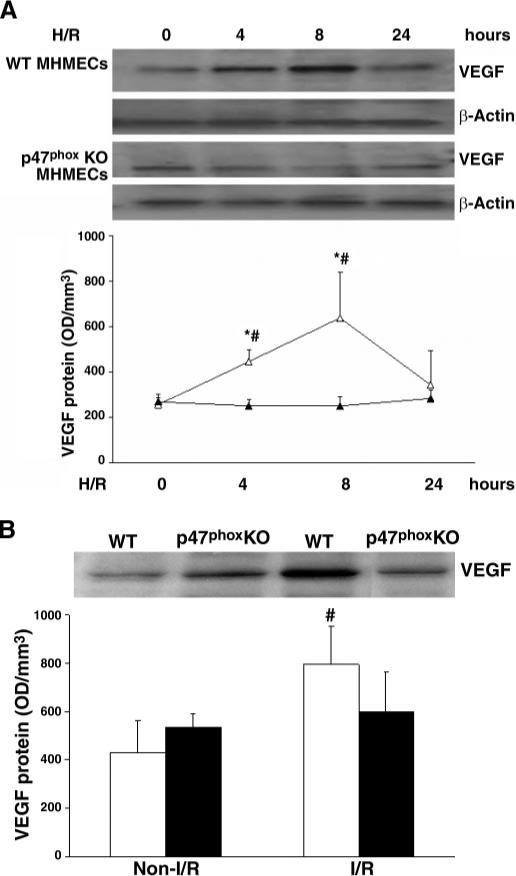
A: Western blot analysis showing time course and patterns of H/R-induced VEGF expression in MHMECs isolated from WT or p47phox−/− mice. Exposure of WT MHMECs (△) to H/R increased VEGF expression at 4 and 8 h, whereas exposure of p47phox−/− MHMECs (▲) to H/R failed to induce VEGF expression (n = 3 cell lines, data are means ± SD, *P < 0.05 compared with H/R; #P < 0.05 compared with baseline). B: Western blot analysis reveals that exposure of I/R increases myocardial VEGF expression in WT (white bars) but not in p47phox−/− mice (black bars, n = 4−5 mice, data are means ± SD, #P < 0.05 compared with control).
DISCUSSION
The novel findings in this study are that exposure of myocardial endothelial cells to hypoxia for 2 h followed by various periods of oxygenation increases NADPH oxidase-derived ROS formation. This is accompanied by a gradual increase in Akt and ERK1/2 phosphorylation, increases in VEGF expression, and increased angiogenesis. Pharmacological inhibition of NADPH oxidase activity largely prevents H/R-induced ROS formation, Akt and ERK1/2 activation, and angiogenesis. Using mice with a genetic deletion of the NADPH oxidase subunit p47phox in an I/R in vivo model further demonstrated that the NADPH oxidase and its p47phox subunit are required for the activation of myocardial Akt and ERK1/2 and for the stimulation of angiogenic growth factor expression and angiogenesis in the setting of I/R.
MAPKs constitute an essential signal transduction cascade that plays a central role in cell proliferation, differentiation, apoptosis, and stress signaling. ERK1/2 activation is induced by ischemia in vivo and has been implicated as a modulator of I/R. In addition, accumulating data suggest that ERK1/2 is an important regulator of endothelial cell proliferation and is a major signaling mediator in angiogenesis (23, 41, 47). Recent studies reveal that ERK1/2 regulates cell survival and death during I/R (4, 13, 24). I/R may induce the rapid and brief activation of survival signals as an early response, followed by activation of the death signals. Within the heart, the MAPKs pathways have been implicated to play an antiapoptotic role in cardiomyocytes after I/R. For instance, genetic manipulation of activated ERK1/2 in the mouse myocardium has been shown to diminish I/R-induced apoptosis and injury, suggesting a prerequisite role for activated ERK1/2 in protecting cardiomyocytes against I/R injury (24). The intracellular molecular mechanism by which ERK1/2 was activated in the myocardium during I/R is not clear. Oxidative stress is increased in cellular and experimental models of H/R and I/R. In porcine aortic endothelial cells, ROS generated by hypoxic mitochondria have been shown to contribute to the activation of the ERK1/2 pathway and to be required for cell proliferative responses to hypoxia (39). NADPH oxidase is a major enzymatic source of ROS produced in both myocardial endothelial cells and cardiomyocytes (10, 11, 30, 50). Our finding that inhibition of NADPH oxidase activity or deletion of its p47phox subunit attenuates H/R and I/R-induced ERK1/2 phosphorylation suggests that activation of NADPH oxidase and its p47phox subunit are important regulators of ERK1/2 in the setting of H/R and I/R myocardium. In addition, our data also demonstrate that H/R significantly increases intracellular ROS formation and superoxide production in an endothelial NADPH oxidase-dependent manner. Our finding that pretreatment with 200 μM Apo significantly inhibited ROS production but had no effect on ERK1/2 phosphorylation following H/R was somewhat surprising and suggests that ROS generated from sources other than NADPH oxidase, such as from hypoxic mitochondria, may also contribute to the H/R-induced ERK1/2 activation.
The PI3K/Akt pathway is activated by a number of growth factors and modulates many important aspects of cellular function, such as cell survival, cell motility, and endothelial migration. Akt is also a central player in the regulation of cardiac hypertrophy, angiogenesis, and apoptosis (5, 29, 40, 42, 43). The PI3K/Akt pathway has also been shown to be activated by I/R in various organs, including the heart. Activation of Akt in the heart protects against cardiomyocyte apoptosis after I/R (12, 17, 25), but little is known regarding the signaling pathway by which I/R regulates activation of Akt in the myocardium. To our knowledge, our data are the first to demonstrate that H/R and I/R-induced Akt phosphorylation is significantly blunted by pretreatment with NADPH oxidase inhibitors or by deletion of its p47phox subunit. Our data implicate that myocardial NADPH oxidase is required for the activation of antiapoptotic Akt signaling after the myocardium is subjected to H/R and I/R. Our present data also indicate that activation of the Akt and ERK1/2 pathways are critical in the regulation of myocardial angiogenesis in response to H/R and I/R.
Our results may shed light on the previously reported findings by Hoffmeyer et al. (19) who investigated the role of NADPH oxidase in I/R-induced myocardial infarction in p47phox−/− mice. These authors reported that myocardial infarction size following 30 min of ischemia and 24 h of reperfusion was similar in p47phox−/− and WT mice. Thus, in the Hoffmeyer et al. study, deficiency of the NADPH oxidase subunit p47phox failed to protect the myocardium against I/R-induced myocardial infarction. Our in vitro and in vivo findings suggest that failure to activate ERK1/2 and Akt, an antiapoptotic pathway, and a failure to stimulate angiogenic growth factor expression and angiogenesis in p47phox−/− mouse myocardial endothelial cells and the myocardium in response to H/R and I/R may contribute to the diminished protection against I/R-induced myocardial infarction seen in the p47phox−/− mouse.
NADPH oxidase-derived ROS or superoxide () are important mediators in the regulation of expression of angiogenic factors, such as VEGF, PDGF, and hypoxia-inducible factor-1[H9251].] Several studies have shown that NOX2, the gp91phox homo-logue of NADPH oxidase, is a critical component for super-oxide generation in endothelial cells and for VEGF and ischemia-mediated angiogenesis (1, 46–48). Furthermore, in vitro and in vivo genetic approaches to overexpress NOX1 lead to increased ROS formation and upregulation of VEGF expression and matrix metalloproteinase activity. The effect of over-expression of NOX1 on VEGF expression is eliminated by catalase (2). Consistent with these findings, our data reveal that NADPH oxidase-derived ROS are also required for the H/R and I/R-induced myocardial VEGF expression, highlighting the fundamental role of NADPH oxidase in H/R and I/R-induced myocardial angiogenesis.
Although endothelial NADPH oxidases have been shown to contribute to angiogenesis and vascular remodeling, the relative contribution of each of these NADPH oxidase components is incompletely understood. In addition, there are many cellular sources of ROS other than NADPH oxidases, including cyclooxygenase, uncoupled NOS, lipoxygenase, xanthine oxidase, and mitochondria, which may also modulate angiogenesis and vascular remodeling. It is possible that ROS from other sources may also play a role in the regulation of angiogenic growth factor expression and angiogenesis in response to H/R and I/R. To overcome this obstacle, in our present study, we used p47phox-deficient mice to determine whether the p47phox subunit of NADPH oxidase is necessary and required for the H/R and I/R-induced Akt and ERK1/2 activation, VEGF expression, and angiogenesis. Our data clearly demonstrate that deficiency of p47phox significantly suppresses H/R and I/R-induced angiogenic growth factor expression and vessel outgrowth, strongly associating this cytosolic subunit with angio-genesis. Our data also indicate that the cytosolic p47phox activation is a key step in the initiation of Akt and MAPKs activation in response to H/R and I/R in the heart. Using the NADPH oxidase subunit knockout mouse models represents a particularly promising approach to elucidate the relative contributions of these multiple ROS-generating enzymes in vascular remodeling and angiogenesis.
In summary, the present study demonstrates that exposure of myocardial endothelial cells to hypoxia followed by reoxygenation causes a significant increase in NADPH oxidase-derived intracellular ROS and superoxide formation and induces NADPH oxidase-dependent Akt and ERK1/2 activation. With the use of both in vitro and ex vivo models, our data show that inhibition of NADPH oxidase activity suppresses H/R-induced VEGF expression and angiogenesis. Additional studies in p47phox−/− mice subjected to I/R confirm the essential role of NADPH oxidase in H/R and I/R-induced angiogenic responses. We conclude that NADPH oxidase plays a critical role in the regulation of angiogenic signaling and angiogenesis in the H/R and I/R heart.
ACKNOWLEDGMENTS
We thank Natasha Strauss for editorial assistance.
GRANTS
This work was supported by grants from the American Heart Association, 0565196B (to J. X. Chen) and the National Institute of Health DK074995 (to J. X. Chen) and HL075511 (to J. L. Aschner).
REFERENCES
- 1.Abid MR, Kachra Z, Spokes KC, Aird WC. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000;486:252–256. doi: 10.1016/s0014-5793(00)02305-x. [DOI] [PubMed] [Google Scholar]
- 2.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci USA. 2002;99:715–720. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell RM, Cave AC, Johar S, Hearse DJ, Shah AM, Shattock MJ. Pivotal role of NOX-2-containing NADPH oxidase in early ischemic preconditioning. FASEB J. 2005;19:2037–2039. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
- 4.Brar BK, Jonassen AK, Stephanou A, Santilli G, Railson J, Knight RA, Yellon DM, Latchman DS. Urocortin protects against ischemic and reperfusion injury via a MAPK-dependent pathway. J Biol Chem. 2000;275:8508–8514. doi: 10.1074/jbc.275.12.8508. [DOI] [PubMed] [Google Scholar]
- 5.Chen JX, Lawrence ML, Cunningham G, Christman BW, Meyrick B. HSP90 and Akt modulate Ang-1-induced angiogenesis via NO in coronary artery endothelium. J Appl Physiol. 2004;96:612–620. doi: 10.1152/japplphysiol.00728.2003. [DOI] [PubMed] [Google Scholar]
- 6.Chen JX, Meyrick B. Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab Invest. 2004;84:182–190. doi: 10.1038/labinvest.3700027. [DOI] [PubMed] [Google Scholar]
- 7.Chen JX, Zeng H, Lawrence ML, Blackwell TS, Meyrick B. Angiopoietin-1-induced angiogenesis is modulated by endothelial NADPH oxidase. Am J Physiol Heart Circ Physiol. 2006;291:H1563–H1572. doi: 10.1152/ajpheart.01081.2005. [DOI] [PubMed] [Google Scholar]
- 8.Das S, Engelman RM, Maulik N, Das DK. Angiotensin preconditioning of the heart: evidence for redox signaling. Cell Biochem Biophys. 2006;44:103–110. doi: 10.1385/CBB:44:1:103. [DOI] [PubMed] [Google Scholar]
- 9.Das S, Otani H, Maulik N, Das DK. Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling. J Mol Cell Cardiol. 2006;41:248–255. doi: 10.1016/j.yjmcc.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280:H2649–H2657. doi: 10.1152/ajpheart.2001.280.6.H2649. [DOI] [PubMed] [Google Scholar]
- 11.El Jamali A, Freund C, Rechner C, Scheidereit C, Dietz R, Bergmann MW. Reoxygenation after severe hypoxia induces cardiomyocyte hyper-trophy in vitro: activation of CREB downstream of GSK3beta. FASEB J. 2004;18:1096–1098. doi: 10.1096/fj.03-1054fje. [DOI] [PubMed] [Google Scholar]
- 12.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germack R, Dickenson JM. Adenosine triggers preconditioning through MEK/ERK1/2 signalling pathway during hypoxia/reoxygenation in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:429–442. doi: 10.1016/j.yjmcc.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res. 2000;87:26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 15.Gorlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, Kietzmann T, Busse R. Thrombin activates the hypoxiainducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22phox-containing NADPH oxidase. Circ Res. 2001;89:47–54. doi: 10.1161/hh1301.092678. [DOI] [PubMed] [Google Scholar]
- 16.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 17.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 18.Hoefer IE, Piek JJ, Pasterkamp G. Pharmaceutical interventions to influence arteriogenesis: new concepts to treat ischemic heart disease. Curr Med Chem. 2006;13:979–987. doi: 10.2174/092986706776360996. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmeyer MR, Jones SP, Ross CR, Sharp B, Grisham MB, Laroux FS, Stalker TJ, Scalia R, Lefer DJ. Myocardial ischemia/reperfusion injury in NADPH oxidase-deficient mice. Circ Res. 2000;87:812–817. doi: 10.1161/01.res.87.9.812. [DOI] [PubMed] [Google Scholar]
- 20.Kim YM, Kim KE, Koh GY, Ho YS, Lee KJ. Hydrogen peroxide produced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006;66:6167–6174. doi: 10.1158/0008-5472.CAN-05-3640. [DOI] [PubMed] [Google Scholar]
- 21.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 22.Lelkes PI, Hahn KL, Sukovich DA, Karmiol S, Schmidt DH. On the possible role of reactive oxygen species in angiogenesis. Adv Exp Med Biol. 1998;454:295–310. doi: 10.1007/978-1-4615-4863-8_35. [DOI] [PubMed] [Google Scholar]
- 23.Li ZD, Bork JP, Krueger B, Patsenker E, Schulze-Krebs A, Hahn EG, Schuppan D. VEGF induces proliferation, migration, and TGF-beta1 expression in mouse glomerular endothelial cells via mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Biochem Biophys Res Commun. 2005;334:1049–1060. doi: 10.1016/j.bbrc.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba-El-Leil MK, Meloche S, Pouyssegur J, Pages G, De Windt LJ, Doevendans PA, Molkentin JD. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109:1938–1941. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 25.Matsui T, Tao J, del MF, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 26.Maulik N. Redox signaling of angiogenesis. Antioxid Redox Signal. 2002;4:805–815. doi: 10.1089/152308602760598963. [DOI] [PubMed] [Google Scholar]
- 27.Maulik N. Ischemic preconditioning mediated angiogenic response in the heart. Antioxid Redox Signal. 2004;6:413–421. doi: 10.1089/152308604322899486. [DOI] [PubMed] [Google Scholar]
- 28.Maulik N, Das DK. Redox signaling in vascular angiogenesis. Free Radic Biol Med. 2002;33:1047–1060. doi: 10.1016/s0891-5849(02)01005-5. [DOI] [PubMed] [Google Scholar]
- 29.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del MF, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takihara K. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 31.Nordlie MA, Wold LE, Simkhovich BZ, Sesti C, Kloner RA. Molecular aspects of ischemic heart disease: ischemia/reperfusion-induced genetic changes and potential applications of gene and RNA interference therapy. J Cardiovasc Pharmacol Ther. 2006;11:17–30. doi: 10.1177/107424840601100102. [DOI] [PubMed] [Google Scholar]
- 32.Oudot A, Vergely C, Ecarnot-Laubriet A, Rochette L. Angiotensin II activates NADPH oxidase in isolated rat hearts subjected to ischaemia-reperfusion. Eur J Pharmacol. 2003;462:145–154. doi: 10.1016/s0014-2999(03)01315-3. [DOI] [PubMed] [Google Scholar]
- 33.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47phox may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 34.Ray PS, Estrada-Hernandez T, Sasaki H, Zhu L, Maulik N. Early effects of hypoxia/reoxygenation on VEGF, ang-1, ang-2 and their receptors in the rat myocardium: implications for myocardial angiogenesis. Mol Cell Biochem. 2000;213:145–153. doi: 10.1023/a:1007180518474. [DOI] [PubMed] [Google Scholar]
- 35.Ray PS, Sasaki H, Estrada-Hernandez T, Zu L, Maulik N. Effects of hypoxia/reoxygenation on angiogenic factors and their tyrosine kinase receptors in the rat myocardium. Antioxid Redox Signal. 2001;3:89–102. doi: 10.1089/152308601750100560. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki H, Fukuda S, Otani H, Zhu L, Yamaura G, Engelman RM, Das DK, Maulik N. Hypoxic preconditioning triggers myocardial angiogenesis: a novel approach to enhance contractile functional reserve in rat with myocardial infarction. J Mol Cell Cardiol. 2002;34:335–348. doi: 10.1006/jmcc.2001.1516. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki H, Ray PS, Zhu L, Galang N, Maulik N. Oxidative stress due to hypoxia/reoxygenation induces angiogenic factor VEGF in adult rat myocardium: possible role of NFkappaB. Toxicology. 2000;155:27–35. doi: 10.1016/s0300-483x(00)00274-2. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki H, Ray PS, Zhu L, Otani H, Asahara T, Maulik N. Hypoxia/reoxygenation promotes myocardial angiogenesis via an NF kappa B-dependent mechanism in a rat model of chronic myocardial infarction. J Mol Cell Cardiol. 2001;33:283–294. doi: 10.1006/jmcc.2000.1299. [DOI] [PubMed] [Google Scholar]
- 39.Schafer M, Schafer C, Ewald N, Piper HM, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. [DOI] [PubMed] [Google Scholar]
- 40.Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, Namchuk M, Salituro F, Yao YM, Hou WM, Chen X, Aronovitz M, Tsichlis PN, Bhattacharya S, Force T, Kilter H. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res. 2006;98:111–118. doi: 10.1161/01.RES.0000197781.20524.b9. [DOI] [PubMed] [Google Scholar]
- 41.Shin EY, Lee JY, Park MK, Chin YH, Jeong GB, Kim SY, Kim SR, Kim EG. Overexpressed alpha3beta1 and constitutively activated extracellular signal-regulated kinase modulate the angiogenic properties of ECV304 cells. Mol Cell. 1999;9:138–145. [PubMed] [Google Scholar]
- 42.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 44.Shono T, Ono M, Izumi H, Jimi SI, Matsushima K, Okamoto T, Kohno K, Kuwano M. Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol Cell Biol. 1996;16:4231–4239. doi: 10.1128/mcb.16.8.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shyu KG, Chang CC, Wang BW, Kuan P, Chang H. Increased expression of angiopoietin-2 and Tie2 receptor in a rat model of myocardial ischaemia/reperfusion. Clin Sci (Lond) 2003;105:287–294. doi: 10.1042/CS20030025. [DOI] [PubMed] [Google Scholar]
- 46.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. 2005;111:2347–2355. doi: 10.1161/01.CIR.0000164261.62586.14. [DOI] [PubMed] [Google Scholar]
- 47.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 48.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91phox-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Gabrielsen A, Lawler PR, Paulsson-Berne G, Steinbruchel DA, Hansson GK, Kastrup J. Myocardial gene expression of angiogenic factors in human chronic ischemic myocardium: influence of acute ischemia/cardioplegia and reperfusion. Microcirculation. 2006;13:187–197. doi: 10.1080/10739680600556811. [DOI] [PubMed] [Google Scholar]
- 50.Woo AY, Cheng CH, Waye MM. Baicalein protects rat cardiomyocytes from hypoxia/reoxygenation damage via a prooxidant mechanism. Cardiovasc Res. 2005;65:244–253. doi: 10.1016/j.cardiores.2004.09.027. [DOI] [PubMed] [Google Scholar]