

Abstract
A human and a mouse gene have been isolated based on homology to a recombinational repair gene from the corn smut Ustilago maydis. The new human (h) gene, termed hREC2, bears striking resemblance to several others, including hRAD51 and hLIM15. hREC2 is located on human chromosome 14 at q23–24. The overall amino acid sequence reveals characteristic elements of a RECA-like gene yet harbors an src-like phosphorylation site curiously absent from hRAD51 and hLIM15. Unlike these two relatives, hREC2 is expressed in a wide range of tissues including lung, liver, placenta, pancreas, leukocytes, colon, small intestine, brain, and heart, as well as thymus, prostate, spleen, and uterus. Of greatest interest is that hREC2 is undetectable by reverse transcription-coupled PCR in tissue culture unless the cells are treated by ionizing radiation.
The RecA protein of Escherichia coli provides the enzymatic function for recognition of DNA sequence homology and DNA strand exchange. These are essential steps in the molecular systems necessary for genetic recombination and recombinational repair. recA-like genes are ubiquitous in prokaryotes, and the pairing mechanism derived from studies on the E. coli RecA protein is the prototype for this class of enzymes. The discovery that the Rad51 protein of Saccharomyces cerevisiae is a RecA structural homologue and is required for recombination and repair proficiency has provided strong evidence in favor of the universality of the RecA paradigm (1–3). This view has been strengthened by the discovery of Rad51 homologues in organisms representing numerous groups of eukaryotes including human.
One puzzling aspect of the genetic control of recombination and repair in eukaryotes is the multiplicity of genes encoding RecA-like proteins. In S. cerevisiae a total of four genes encoding proteins with structural homology to RecA have been discovered. In addition to RAD51 these include RAD55 (4), RAD57 (5), and DMC1 (6), the latter of which is expressed only during meiosis. RAD51, RAD55, and RAD57 are expressed mitotically, as well as meiotically, but are not redundant in function. Inactivation of any one of these genes leads to loss of proficiency in DNA repair and to defective meiosis.
Two mitotically expressed genes encoding proteins with RecA homology have been found in the fungus Ustilago maydis. One (UmRAD51) encodes a protein very close in structure to the human Rad51 protein (7), and the other (REC2) encodes a protein more than twice as large as HsRad51 but nonhomologous over most of the 200-amino acid region corresponding to the “homologous core” residues of RecA protein, except for a 50-amino acid stretch of conserved residues that is essential for activity in recombination and repair (8). Nonetheless, biochemical studies have shown that Rec2 protein, the product of this structurally divergent gene, is active in promoting RecA-like DNA pairing reactions in vitro (9). The relationship between the U. maydis REC2 and RAD51 genes is by no means clear, but genetic analysis has indicated some overlap in function in recombination and repair (7).
PCR methodology has provided a successful strategy in cloning RecA homologues from mammalian species as the isolation of two genes has been reported. The product of one, HsRad51 (10), is highly similar to Rad51 of S. cerevisiae, and the product of the other, HsLim15 (11), is closer to the meiotically expressed DMC1 homologue of Lilium longiflorum (12). Herein we report the cloning of RecA homologues from human and mouse libraries by a PCR screening strategy based on the U. maydis REC2 gene.
MATERIALS AND METHODS
Cloning of Human (h) REC2.
To maintain consistency among the genes from various species described in this paper, we designate the human REC2 gene as hREC2, the mouse (m) REC2 gene as mREC2, and the Ustilago maydis REC2 gene as REC2. The human analogs of the LIM15 and RAD51 genes are labeled hLIM15 and hRAD51. Finally all proteins from humans are designated HsRec2, HsRad51, HsLim15, etc.
Degenerate primers were used for PCR amplification. Based on sequences in the conserved and divergent regions of the U. maydis REC2 gene, a set of 10 primers were designed with codon degeneracy. This was accomplished by using human codon usage tables and incorporating the inosine nucleotide into DNA primers on an Applied Biosystems DNA synthesizer. These primers were used in the PCR using the Expand system (Boehringer), and after two rounds of amplification, the bands were purified on a 3% SeaPlaque (FMC) low-melting-temperature agarose TAE gel. Approximately 60 gene fragments were cloned into the pCRII plasmid (Invitrogen) by T-A cloning and sequenced with a fluorescent PCR-based Sanger dideoxynucleotide chain-termination procedure. One clone amplified from human kidney cDNA with GKTQ-f [5′-gcggiaa(g/a)acica(g/a)atg-3′] and STIY-r [5′-cciccg(c/g)(t/a)igtiat(a/g)ta-3′] produced a 110-bp fragment that contained significant homology to Rec2. This sequence and the Rec2 sequence containing the RecA core were used to query the database of expressed sequence tags by the blastx program. The query revealed an IMAGE clone (no. 153195) that is a cDNA sequence derived from human breast tissue cloned into pT3T7 vector (Pharmacia) and bearing significant homology to REC2 (P = 0.90). Therefore, we obtained this clone from the IMAGE consortium at Lawrence Livermore National Laboratory and it was sequenced by the Sanger dideoxynucleotide chain-termination procedure in a stepwise fashion by primer walking. Data were collected, assembled, and edited on a PowerMac using inherit software (Applied Biosystems). The sequence was translated with the macvector program (Kodak), and a restriction map was generated using the GCG software package on a UNIX work station. Next, the 5′ end of the cDNA was restored because the IMAGE sample was only a partial clone. A testis SuperScript (pCMVSPORT) cDNA library (BRL) was then screened by high-fidelity PCR amplification using a set of nested primers (16r, 5′-acctgtaatctctgtgag-3′; 18r, 5′-gcagactctgtgtcaatg-3′) to amplify reverse from the 5′ end of the partial cDNA to the SP6 promoter in the pCMVSPORT plasmid. Four specific bands (150, 200, 450, and 500 bp) were obtained, reamplified, and cloned into pCRII vector using T-A cloning (Invitrogen). The products were sequenced and found to have exact homology with an overlap of ∼100 bp with the 5′ end of clone 153195. The longest clone was selected and analyzed for suitable cloning sites within this 100-bp overlap. A single unique restriction site (BsrGI) was identified. Therefore, a 3′ end fragment of clone 153195 from BsrGI to XbaI was ligated to the synthesized 5′ fragment in pCRII to create a complete cDNA.
Cloning of mREC2.
The hREC2 gene, which contained the entire coding region and 160 bp of 3′ untranslated region, was excised from plasmid, hsREC2 2, by endonuclease digestion of the KpnI and XbaI restriction sites. The resultant 1.2-kb fragment was isolated from a 1% agarose gel by electroelution, purified, and randomly labeled (Prime-It Kit, Stratagene) with [α-32P]dCTP. The labeled fragment was first used as a heterologous probe in a Southern blot analysis of genomic murine DNA digested with EcoRI, XbaI, or HindIII (WEHI and NP3 strains); a specific banding pattern on film was observed with each enzyme. The probe was then used to screen the 129SVJ Mouse Genomic Library in the λ Fix II vector (Stratagene).
Positive phage DNA clones were mapped by digestion with several different restriction endonucleases and Southern blot analysis showed two clones (5D2A and 7B1A) containing restriction fragments that hybridized strongly to the hREC2 probe. Subclone 5D2A was sequenced in a stepwise fashion by primer walking with an automated fluorescent PCR-based Sanger dideoxynucleotide chain-termination procedure. Contigs were assembled and edited using inherit software and macvector was used for sequence translation and restriction mapping. Sequence translation analysis of 5D2A revealed two homologous exons (separated by a 2-kb intron sequence) to the first 200 bp of the 5′ coding region of hREC2 cDNA. In addition, part of the 5′ untranslated sequence was also revealed.
Two primers were designed from mouse REC2 coding sequence to prime mREC2 cDNA sequences to amplify in opposite directions. The primer set EXFor7 (5′-gtgccccgcagatgcaaa-3′) and EXRev5 (5′-ggccagtcactttcataag-3′) was used in the PCR amplification to inversely amplify mREC2 cDNA from a Superscript Mouse Liver Library (GIBCO/Life Technologies). The inverse PCR amplification was designed to produce a fragment that would contain the entire muRec2 cDNA (devoid of approximately 50 bp of sequence between the priming regions for the oligonucleotides) and cloning vector. The inverse amplification generated a fragment that was made blunt ended in a T4 DNA polymerase reaction. The ends were subsequently joined in a self-ligation reaction and transformed into competent αDH5 E. coli cells. After restriction analysis, several clones were sequenced as previously described.
The Chromosomal Position of hREC2.
To determine the chromosomal localization of the hREC2 gene, oligonucleotide primers were used to screen a panel of rodent–human hybrids by PCR amplification. Only hybrids retaining human chromosome 14 exhibited a human specific product for hREC2. In addition, the Stanford Radiation Hybrid mapping panel (Research Genetics, Huntsville, AL) was screened by PCR with the same oligonucleotide primers to refine the localization of the Rec2 gene. The scoring data was submitted to the Stanford Radiation Hybrid server (rhserver@shgc.stanford.edu). Radiation Hybrid server analysis showed linkage of hREC2 to the D14S258 marker with a logarithm of odds score greater than 6 at a distance of 35.88 centiRays 8000, confirming rodent–human hybrid mapping data. The Centre Européan pour l’Etude du Polymorphism Humain (CEPH) places D14S258 at 0.65 morgans (M) on chromosome 14 and at 14q24.1 on the physical map (13). When we combined our data with published CEPH data (13), we therefore placed the hREC2 gene at 14q24.1. Fluorescence in situ hybridization (FISH) analysis with a REC2-positive yeast artificial chromosome (YAC) clone was used to confirm physical mapping. The CEPH B YAC library (Research Genetics) was screened for YAC clones positive for hREC2, and YAC clone 958C2 was found positive for the hREC2 gene. A search through the Whitehead Institute database and CEPH contigs (13, 14) showed that clone 958C2 is part of Whitehead Institute contig 14.9 and of the CEPH contig located at 0.61 morgans on chromosome 14, immediately centromeric to the contig anchored at D14S528. Clone 958C2 DNA was used as probe for FISH analysis on normal human metaphase chromosomes and was found to be chimeric and to hybridize to chromosomes 5q13–14 and to chromosome 14q23.3–24.1. FISH was performed with total yeast DNA on normal blood metaphase chromosomes as described (15).
Induction and RNA Analysis.
Apparently normal primary human foreskin fibroblasts were obtained from Coriell Institute for Medical Research (GM03468A). Cells were grown to near confluency in MEM with Earle’s balanced salt solution and 20% fetal bovine serum in a T-175 flask. Prepared cells were γ-irradiated at a 2-, 10-, or 20-Gy dose from a Gammacell 40 137Cs linear accelerator at a rate of 1.1 cGy/min. The cells were then harvested at several time points after irradiation and frozen as cell pellets for RNA extraction.
Total RNA was purified from the samples by lysis in guanidinium isothiocyanate and ultracentifugation over a cesium chloride gradient (16). The RNA samples were carefully quantitated by spectrophotometry and ethidium bromide staining after electrophoresing on a denaturing formaldehyde/1% agarose gel. One microgram of total RNA from each sample was analyzed by a semiquantitative reverse transcription–PCR (RT-PCR) technique to compare levels of hREC2 transcript. The cDNA was synthesized by SuperScript reverse transcriptase (BRL) with a poly(T) primer (18-mer) followed by 35 cycles of expand PCR (Boehringer) using primers 1f (5′-gttatcttgacgaatcag-3′) and 3r (tgtcagtgccatggtatc-3′) to produce a 335-bp product. Primers designed to actin cDNA (βAf, 5′-ccaaggccaaccgcgagaagatga-3′; βAr, 5′-agggtaggtgccgccagac-3′) that produce a 550-bp product. This product was used as a standard to ensure proper quantitation of RNA. The samples were electrophoresed on a 1.5% agarose gel, stained with CyberGreen (FMC), and visualized and quantitated on a FluorImager (Molecular Dynamics).
Northern Blot Analysis.
A membrane containing 2 μg of poly(A+) mRNA isolated from various tissues was obtained from CLONTECH. The mRNA samples were electrophoresed in a 1.2% agarose gel under denaturing conditions, then transferred to a charge-modified nylon membrane, and fixed by UV cross-linking. The Northern blot contained RNA from human heart, brain, placenta, lung, liver, skeletal muscle, kidney, pancreas, spleen, thymus, prostate, testis, uterus, small intestine, colon, and peripheral leukocytes. The Northern blot was then hybridized with a randomly primed radioactive 1.2-kb hREC2 cDNA fragment in ExpressHyb hybridization solution (CLONTECH) at 60°C for 1 h. The intensity of hybridization was normalized by comparing hREC2 hybridization intensity to the intensity of a control hybridization with a G3PDH probe. Washes were performed in two steps. Three low-stringency [2× standard saline citrate (SSC)/0.1% SDS, for 10 min at 22°C] washes were performed, followed by three high-stringency (0.1× SSC/0.1% SDS, for 20 min at 50°C) washes. Specific bands indicating hybridization to RNA samples were visualized by autoradiography.
Southern Blot Analysis.
A positively charged nylon membrane to which 4 μg of digested genomic DNA samples was transferred by Southern blotting, was obtained from CLONTECH. The blot contains EcoRI-digested DNA from nine eukaryotic species: human, monkey (Rhesus), rat (Sprague–Dawley), mouse (BALB/c), dog, cow, rabbit, chicken, and yeast (S. cerevisiae). The blot was then hybridized with a randomly primed radioactive 1.2-kb hREC2 cDNA fragment in ExpressHyb hybridization solution (CLONTECH) at 60°C for 1 h. Washes were performed in two steps. Three low-stringency (2× SSC/0.1%SDS, for 10 min at 22°C) washes were performed, followed by three high-stringency washes (0.1× SSC/0.1%SDS, for 20 min at 50°C). Specific bands indicating hybridization to genomic DNA samples were visualized by autoradiography.
Dot Blot Analysis.
Total cellular RNA was isolated as above, of which 20 μg was transferred to Hybond N+ nylon membrane under denaturing conditions with a Bio-Dot SF Microfiltration Apparatus (Bio-Rad). The RNA was fixed by UV cross-linking and hybridized with the hREC2 1.2-kb cDNA probe in ExpressHyb hybridization solution (CLONTECH) at 60°C for 1 h. The intensity of hybridization was normalized by comparing hREC2 hybridization intensity to the intensity of a control hybridization with a G3PDH probe. Washes were performed in two steps, three low-stringency (2× SSC/0.1% SDS, for 10 min at 22°C) washes followed by three high-stringency (0.1× SSC/0.1% SDS, for 20 min at 50°C) washes. Intensities of each dot were visualized and quantitated on a PhosphorImager (Molecular Dynamics).
RESULTS
Cloning of hREC2.
Primers for PCR amplification were designed from the U. maydis Rec2 protein sequence. One primer was based on the nucleotide binding P-loop, which is highly conserved in the RecA family of proteins. The second primer was based on a tract of residues between the Walker A and B boxes, a region that is unique to the U. maydis Rec2 protein. These primers were used in PCR amplification of DNA fragments from a human kidney cDNA library. Approximately 60 DNA fragments in the size range of 60 to 120 bp were obtained. After DNA sequence determination, one fragment was identified that contained significant homology to REC2. This sequence was used to query the database of Expressed Sequence Tags and an IMAGE clone corresponding to a cDNA derived from human breast tissue was identified. This clone was obtained from the IMAGE consortium at the Lawrence Livermore National Laboratory and was sequenced in stepwise fashion by primer walking. Analysis revealed that the IMAGE clone contained only a partial sequence that was lacking the 5′ end of the open reading frame. A complete sequence was obtained from a testis cDNA library by PCR amplification with primers to reverse-amplify from the 5′ end of the partial clone to a unique sequence in the vector. An open reading frame was identified in which an ATG start codon lay within a suitable context for initiation of translation (17).
The hREC2 gene encodes a protein of 350 amino acids with a molecular mass of 3.8 × 104 (Fig. 1A). The homologous core central region of about 200 amino acid residues that is highly conserved among almost all the RecA family members is evident in HsRec2 beginning at residue 83. However, there is less homology in the N-terminal region that is conserved in many of the eukaryotic RecA homologues but not prokaryotic homologues.
Figure 1.

Sequence of the human and mouse REC2 genes and amino acid alignments. (A) The complete cDNA and predicted amino acid sequence is presented along with upstream and downstream nucleotide sequence. (B) The predicted amino acid sequence of the mouse REC2 gene and its divergence from the human homologue. (C) Homology of hREC2 with other human RECA family members. Aligned sequences were generated with a clustular method viewed with msashade on the National Center for Supercomputing Applications molecular biology workbench (http://biology.NCSA.uiuc.edu). Identical residues are box-shaded.
By using hREC2 cDNA as a probe, exons 1 and 2 of a closely related mouse gene were identified and cloned. The cDNA of the mouse homologue of REC2 (mREC2) has been identified and cloned by an inverse PCR technique designed from mREC2 exon 2 sequence. Sequence analyses revealed a nucleotide homology of 84.8% within the open reading frame of the gene, and sequence translation revealed an 83.7% identity and a 94.3% similarity at the amino acid level (Fig. 1B).
The coding region of hREC2 translates into a protein of 38.3 kDa and has significant homology to the U. maydis Rec2 protein (P = 2.8 e−5) as well as homology to other prokaryotic and eukaryotic RecA-like proteins. Amino acid comparison with the human RecA homologues HsLim15 and HsRad51 provides additional clues to the type of category to which HsRec2 might belong (Fig. 1C). A significant level of identity and putative consensus is conserved for all three genes, with several sites of particular interest. The highest level of homology among the three genes exists in the central region (from approximately amino acids 80 through 213 in the HsRec2 sequence). Within this region, the so-called “A box” consensus sequence is matched perfectly between HsLim15 and HsRad51 but only partially matched within HsRec2. There is a potential src phosphorylation site in HsRec2 (amino acids 160 through 165), which is partially deleted in HsLim15 and HsRad51. The “B box”, amino acids 205 through 211 (in HsRec2 sequence), is highly conserved among all three genes. In the other sections of the HsRec2 protein, the large gap present near the N-terminal end of HsRec2 is compensated near the C-terminal end. Since HsRad51 (and theoretically HsLim15) has been shown to participate in DNA repair/recombination pathways, it is likely that HsRec2 plays a role in one or both of these processes. As described in Materials and Methods, the hREC2 gene was placed at 14q23.3–24.1 within a 5-centimorgan region flanked by markers D14S98 and D14S258(13) using the Stanford Radiation Hybrid mapping panel screen and FISH (Fig. 2).
Figure 2.
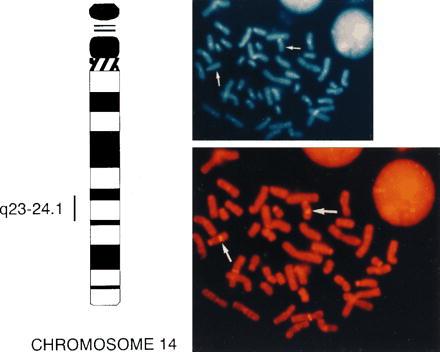
Regional chromosomal localization of hREC2. The position of hREC2 is indicated by a vertical bar to the left of the idiogram for human chromosome 14, as determined by FISH analysis. (Upper Right) shows the 4,6-diamidino-2-phenylindole image. (Lower Right) Corresponding propidium iodide image of FISH analysis of a normal lymphocyte metaphase squash with an hREC2-positive YAC (no. 958C2). Arrows indicate the position of the hREC2 gene.
hREC2 Is Conserved Among Mammalian Species.
The degree of conservation of hREC2 was determined by Southern blot hybridization to restriction enzyme-digested genomic DNA. Positive hybridization in increasingly stringent conditions indicates the amount of selective pressure to maintain REC2 homologues in the genomes of diverse species. A so-called “Zoo blot” (CLONTECH) containing EcoRI-digested DNA from nine eukaryotic species: human, monkey (Rhesus), rat (Sprague–Dawley), mouse (BALB/c), dog, cow, rabbit, chicken, and yeast (S. cerevisiae) was employed to ascertain evolutionary conservation. The blot was hybridized with a randomly primed radioactive 1.2-kb hREC2 cDNA fragment, first with multiple low-stringency washes and then with multiple high-stringency washes. Specific bands, indicating hybridization to genomic DNA samples, were visualized by autoradiography. A low-stringency wash showed banding in all lanes including yeast (data not shown), whereas a high-stringent wash revealed high conservation of the gene among higher eukaryotic species (Fig. 3). Strong specific banding appeared in all lanes containing mammalian species and in a representative from avian tissue. Yeast may be too far removed to be detected under high stringency on a nucleotide level.
Figure 3.

Conservation of hREC2 among eukaryotes. A high-stringency (0.1× SSC/0.1% SDS for 20 min at 50°C) hybridization detected specific bands in all higher eukaryotic species tested with 4 μg of digested genomic DNA from the indicated species. The probe was a randomly primed radioactive 1.2-kb hREC2 cDNA fragment. Marker bands to the left indicate standard size fragment.
Expression of the Human REC2 Gene.
Expression of hREC2 in various human tissues was examined by Northern blot hybridization. A 2.0-kb RNA was detected with a probe prepared from the entire hREC2 coding region. The results presented in Fig. 4A reveal that hREC2 is expressed in a variety of tissues of mitotic and meiotic origin. The range of tissues found to be positive for hREC2 differs from the tissue screens using hRAD51 and hLIM15. hLIM15 is found only in meiotic tissue (11) and hRAD51 is found primarily in spleen, thymus, testes, and ovaries (10). Initial observations suggest that hREC2 is also expressed in spleen, thymus, heart, placenta, and prostate. After the level of G3PDH expression (provided as a control for RNA amounts) was taken into consideration, we found that hREC2 was expressed most abundantly in the following tissues (Fig. 4B): placenta, lung, liver, pancreas, spleen, thymus, uterus, prostate, small intestine, and colon. The level of hREC2 RNA was detected to be lowest in skeletal muscle and highest in spleen. Other highly expressing tissues that exhibit a 20- to 40-fold increase over skeletal muscle include placenta, lung, thymus, prostate, uterus, and peripheral leukocytes. Lower expressing tissues at a 2- to 20-fold level include heart, brain, kidney, pancreas, intestine, colon, and testis. Alternate transcripts were detected in some tissues, e.g., a major 1.6-kb band hybridized in heart and uterus. Minor transcripts larger in size were detected in lymphoid and highly vascularized tissues and placenta, but the significance of all of these related transcripts is not yet known.
Figure 4.

Expression of hREC2 in various tissues. (A) Northern blot analysis of hREC2 mRNA. Each lane contains 2 μg of poly(A+) RNA from the indicated tissues. The membrane was probed with the full-length cDNA, then stripped, and reprobed with G3PDH DNA radioactive probe as a control. Positions of RNA size markers are indicated. Arrows indicate potential secondary transcripts. (B) Bar graph displaying levels of expression after correcting for the presence of G3PDH mRNA, a transcript used as a control.
Induction of hREC2 by γ-Irradiation.
The hREC2 gene product conserves functional domains from other eukaryotes that have been shown to be involved in recombination or repair. Expression of these proteins may be responsible for the detection and repair of damaged DNA caused by chemical and physical insults. There is evidence that such genes can be induced by the damaging effects of ionizing radiation and radiomimetic drugs (18, 19). hREC2 may be a human counterpart of these inducible genes regulating DNA repair; therefore, we analyzed mRNA levels after exposure to DNA damaging agents.
Normal primary human foreskin fibroblasts were grown to near confluency in MEM. Prepared cells were irradiated with 2, 10, or 20 Gy from a 137Cs irradiator. The cells were then harvested at several time points after irradiation, and total cellular RNA was extracted and normalized. Analysis of the RNA samples (through RT-PCR) for the control transcript β-actin revealed constant levels throughout (Fig. 5A Right). HsRec2 mRNA was present at nearly undetectable levels in untreated cells, but after treatment of cells with a sublethal (2 Gy) dose of radiation, hREC2 was rapidly induced and maintained over an 8-h period before beginning to taper off (Fig. 5A Left). A 20-Gy lethal dose showed an immediate response of hREC2 at an even higher transcript level but began to drop after 1 h possibly due to catastrophic DNA damage (data not shown).
Figure 5.

Induction of hREC2 in γ-irradiated cultured cells. (A) The 524-bp RT-PCR product shown in the left section represents relative amounts of hREC2 transcript in RNA samples isolated from cells harvested at time points after irradiation. The hREC2 level in untreated cells was near the limit of detection by this technique but was rapidly induced after irradiation in increasing levels to a peak around 4 h after induction. RNA was previously quantitated and normalized to β-actin levels as shown in the right section. The control lanes (O Gy) represent the RT-PCR product after 8 h (8) of culture with irradiation and a reaction (−) in which no RNA was present. (B) Each bar represents comparative hREC2 transcript levels detected in 137Cs-irradiated cultured cells. Cells irradiated at three different doses (2, 10, and 20 Gy) were harvested at three time points (0, 2, and 4 h). Loading of RNA was normalized by comparing to G3PDH transcript levels in each dot. Fold increase of hREC2 transcript level over the lowest represented tissue, skeletal muscle, is shown.
The response to γ-irradiation was also measured by a Northern dot blot. As shown in Fig. 5B, after treatment with 2 or 10 Gy of irradiation, greater than a 4-fold increase in RNA levels is observed. At a higher dosage (20 Gy), the response is greatly reduced. Since this approaches the lethal dose level for these cells, it is possible that the high range of irradiation is simply killing the majority of cells. But even under these conditions, and after 4 h of recovery, a detectable amount of hREC2 message is present. Hence, the Northern dot-blot analysis of RNA isolated from γ-irradiated cells supports the RT-PCR data.
DISCUSSION
These results show that an additional member of the RecA family of proteins distinct from the HsRad51 and HsLim15 proteins is present in human cells. The mouse REC2 gene has also been isolated and cloned. The strategy used in identification and isolation of both genes was a PCR-based approach using oligonucleotide primers designed from sequences in the Rec2 protein. The primers included a sequence taken from a region conserved among RecA family members and a region unique to Rec2. The gene isolated has similarity to REC2 as well as to the other members of the RECA family, but the overall size and sequence similarity of hREC2 is actually closer to hRAD51 and hLIM15. Nevertheless, the tissue-specific expression patterns of hREC2 indicate that its functional role in recombination and repair could be more general than either RAD51 or LIM15. Unlike hLIM15 gene expression, confined to germ cells, or hRAD51 expression, which is strongly expressed in thymus, spleen and germ cells (3, 20), there appears to be detectable expression of HsRec2 in almost all organs and tissues examined. There is a significant difference in levels of expression with more than a 40-fold variation between the weakest expressing tissue and the strongest.
As seen in several tissue samples, most notably heart and placenta, multiple transcripts hybridize to the hREC2 probe. The importance of secondary transcripts and the significance of these particular tissues is unknown, but the existence of alternative mRNAs is not unprecedented for DNA recombination/repair genes. For example, mouse RAD51 contains multiple transcripts in the thymus, spleen, and testes, whereas mouse also expresses two transcripts for DMC1 in testis through alternative splicing (10, 11). The latter group indicates that the expression of hRAD51 is particularly low in lung and liver but hREC2 transcripts are prevalent in these two tissues. Perhaps hREC2 and hRAD51 expression is different among the body’s tissues and although transcriptional activity overlaps in several tissues, a conservation of function is maintained by such regulation. Ferguson et al. (7) demonstrated that REC2 and RAD51 from Ustilago act in similar pathways of recombination and repair and in some cases could be considered epistatic. Perhaps in humans, hREC2 and hRAD51 can regulate the same processes and so are used in different tissues in a multicellular organism for similar purposes.
The presence in human of at least two somatically expressed RecA family member genes is not surprising given that in S. cerevisiae there are four known RecA family members and three of these are expressed in mitotic cells. Genetic analyses in S. cerevisiae of these three mitotically expressed genes indicate that they do not have exclusively redundant functions. All three are necessary for repair and recombination proficiency, and mounting evidence indicates that the three genes work in concert. Epistasis analyses of their relationships in radiation repair and intrachromosomal recombination have established that the three genes act in the same functional pathway (21, 22) and yeast two-hybrid analysis has provided evidence for physical interaction between combinations of the proteins (23, 24). Nevertheless, it is clear from molecular analysis of modes of recombination that the individual contributions of the three genes to the overall process are different (22, 25).
Expression of the hREC2 gene is inducible by treatment with ionizing radiation. Based on control samples, the corrected induction approaches 4- to 5-fold, clearly aligning with the response levels of the previously identified DNA damage inducible genes GADD45 (26) and p21 (27). Interestingly, hREC2 is also induced by UV damage to the same level (P.H. and E.B.K., unpublished results). The significance of this bilateral response is unknown, but the Ustilago REC2 gene is also inducible by UV and ionizing irradiation (28). Other experiments have demonstrated that hREC2 is associated with proliferating cell nuclear antigen (PCNA) and p53 in vitro and upon overexpression contributes to a delay in the G1- to S-phase transition of the cell cycle (P.H. and E.B.K., unpublished results). These observations, coupled with the results presented in this study, provide evidence that hREC2 modulates the rate of DNA synthesis (through association with PCNA) or extends the time frame allotted for DNA repair (by association with p53 or on its own) as an inducible response to DNA damage. Hence, we suspect that hREC2 plays a role in genome surveillance, occupying a position in the feedback loop regulating the rate of DNA synthesis and cell division.
One feature of the amino acid sequence of HsRec2 is that the consensus src-like phosphorylation site is present, whereas in HsLim15 and HsRad51, this site is partially deleted. Perhaps, this site contributes to activity/regulation of the protein. Since site-specific alterations at that position have been constructed, experimentation is now underway to test this hypothesis.
Acknowledgments
We are grateful to members of the Kmiec laboratory for insightful comments throughout the course of the work and to Kimeragen, Inc., for financial support. We are indebted to Mr. Anthony Rice for manuscript preparation and graphics design.
ABBREVIATIONS
- h
human
- m
mouse
- CEPH
Centre Européan pour l’Etude du Polymorphism Humain
- FISH
fluorescence in situ hybridization
- YAC
yeast artificial chromosome
- RT-PCR
reverse transcription–PCR
Footnotes
References
- 1.Aboussekhra A, Chanet R, Adjiri A, Fabre F. Mol Cell Biol. 1992;12:3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basile G, Aker M, Mortimer R K. Mol Cell Biol. 1992;12:3235–3246. doi: 10.1128/mcb.12.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shinohara A, Ogawa H, Ogawa T. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 4.Lovett S T. Gene. 1994;142:103–106. doi: 10.1016/0378-1119(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 5.Kans J A, Mortimer R K. Gene. 1991;105:139–140. doi: 10.1016/0378-1119(91)90527-i. [DOI] [PubMed] [Google Scholar]
- 6.Bishop D K, Park D, Xu L, Kleckner N. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson D O, Rice M C, Rendi M H, Kotani H, Kmiec E B, Holloman W K. Genetics. 1997;145:243–252. doi: 10.1093/genetics/145.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubin B P, Ferguson D O, Holloman W K. Mol Cell Biol. 1994;14:6287–6296. doi: 10.1128/mcb.14.9.6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kmiec E B, Cole A, Holloman W K. Mol Cell Biol. 1994;14:7163–7172. doi: 10.1128/mcb.14.11.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinohara A, Ogawa H, Matsuda Y, Ushio N, Ikeo K, Ogawa T. Nat Genet. 1993;4:239–243. doi: 10.1038/ng0793-239. [DOI] [PubMed] [Google Scholar]
- 11.Sato S, Seki N, Hotta Y, Tabata S. DNA Res. 1995;2:183–186. doi: 10.1093/dnares/2.4.183. [DOI] [PubMed] [Google Scholar]
- 12.Terasawa M, Shinohara A, Hotta Y, Ogawa H, Ogawa T. Genes Dev. 1993;9:239–243. doi: 10.1101/gad.9.8.925. [DOI] [PubMed] [Google Scholar]
- 13.Chumakov I M, Rigault P, Legall I, Bellannechantelot C, Billault A, et al. Nature (London) 1995;377:175. [Google Scholar]
- 14.Whitehead Institute/MIT Center for Genome Research. Human Genetic Mapping Project. Cambridge, MA: MIT; 1995. , Data Release 9. [Google Scholar]
- 15.Veronese M L, Ohta M, Finan J, Nowell P C, Croce C M. Blood. 1995;85:2132–2138. [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 17.Kozak M. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fornace A J, Jr, Nebert D W, Hollander M C, Luethy J D, Papathanasiou M, Fargnoli J, Holbrook N J. Mol Cell Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith M L, Chen I T, Zhan Q, O’Connor P M, Fornace A J., Jr Oncogene. 1995;10:1053–1059. [PubMed] [Google Scholar]
- 20.Haaf T, Golub E I, Reddy G, Radding C M, Ward D C. Proc Natl Acad Sci USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Game J C. Semin Cancer Biol. 1993;4:73–83. [PubMed] [Google Scholar]
- 22.Rattray A J, Symington L S. Genetics. 1995;139:45–56. doi: 10.1093/genetics/139.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hays S L, Firmenich A A, Berg P. Proc Natl Acad Sci USA. 1995;92:6925–6929. doi: 10.1073/pnas.92.15.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson R D, Symington L S. Mol Cell Biol. 1995;15:4843–4850. doi: 10.1128/mcb.15.9.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rattray A J, Symington L S. Genetics. 1994;138:587–595. doi: 10.1093/genetics/138.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papathanasiou M A, Kerr N C, Robbins J H, McBride O W, Alamo I, Jr, Barrett S F, Hickson I D, Fornace A J., Jr Mol Cell Biol. 1991;11:1009–1016. doi: 10.1128/mcb.11.2.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagano M, Theodoras A M, Tam S W, Draetta G F. Genes Dev. 1994;8:1627–1639. doi: 10.1101/gad.8.14.1627. [DOI] [PubMed] [Google Scholar]
- 28.Rubin B P, Li D, Holloman W K. Gene. 1994;140:131–135. doi: 10.1016/0378-1119(94)90743-9. [DOI] [PubMed] [Google Scholar]