

Abstract
Oxidative DNA damage is generated by reactive oxygen species. The mutagenic base, 8-oxoguanine, formed by this process, is removed from oxidatively damaged DNA by base excision repair. Genes coding for DNA repair enzymes that recognize 8-oxoguanine have been reported in bacteria and yeast. We have identified and characterized mouse and human cDNAs encoding homologs of the 8-oxoguanine DNA glycosylase (ogg1) gene of Saccharomyces cerevisiae. Escherichia coli doubly mutant for mutM and mutY have a mutator phenotype and are deficient in 8-oxoguanine repair. The recombinant mouse gene (mOgg1) suppresses the mutator phenotype of mutY/mutM E. coli. Extracts prepared from mutY/mutM E. coli expressing mOgg1 contain an activity that excises 8-oxoguanine from DNA and a β-lyase activity that nicks DNA 3′ to the lesion. The mouse ogg1 gene product acts efficiently on DNA duplexes in which 7,8-dihydroxy-8-oxo-2′-deoxyguanosine (8-oxodG) is paired with dC, acts weakly on duplexes in which 8-oxodG is paired with dT or dG, and is inactive against duplexes in which 8-oxodG is paired with dA. Mouse and human ogg1 genes contain a helix–hairpin–helix structural motif with conserved residues characteristic of a recently defined family of DNA glycosylases. Ogg1 mRNA is expressed in several mouse tissues; highest levels were detected in testes. Isolation of the mouse ogg1 gene makes it possible to modulate its expression in mice and to explore the involvement of oxidative DNA damage and associated repair processes in aging and cancer.
Genomic DNA is susceptible to attack by reactive oxygen species produced by cellular aerobic metabolism and by exogenous agents, including certain chemical carcinogens and ionizing radiation (1, 2). A variety of mutagenic and cytotoxic DNA lesions are formed by oxidative DNA damage (3), one of the most abundant being 8-oxoguanine (4, 5). Oxidative DNA damage has been implicated in the pathophysiology of both aging and degenerative diseases such as cancer, heart disease, cataracts, and brain dysfunction (6–8).
In Escherichia coli, 8-oxoguanine is excised from oxidatively damaged DNA by a DNA repair enzyme known variously as formamidopyrimidine DNA glycosylase (9, 10), 8-oxoguanine DNA glycosylase (Ogg) (11), and MutM [formamidopyrimidine/Ogg (Fpg)] protein (12). DNA repair activities that recognize 7,8-dihydroxy-8-oxo-2′-deoxyguanosine (8-oxodG) have also been identified in Saccharomyces cerevisiae (13–15). These activities differ with respect to their substrate specificity. Yeast Ogg1 excises 8-oxodG paired with dC but has little or no effect when the lesion is paired with other DNA bases, or on DNA substrates containing N7-methylformamidopyrimidine (14, 15). At least one related activity found in yeast acts preferentially on DNA containing formamidopyrimidine and weakly on duplexes in which 8-oxodG is paired with dG or dA (13, 15, 16).
8-oxoguanine-DNA repair activities have been identified in extracts prepared from human leukocytes (17), HeLa cells (18), and animal tissues (19). Two HeLa cell activities were separated by column chromatography; one of these excises 8-oxoguanine from DNA, the other nicks DNA substrates 3′ and 5′ to the oxidized base (18).
We have used the yeast ogg1 sequence to search for mammalian homologs of this gene and describe here the cloning of mouse and human cognate genes. The mouse ogg1 gene product was identified as a DNA 8-oxoguanine glycosylase. The substrate specificity of this enzyme resembles closely that of yeast Ogg1 (14, 15) and is readily distinguished from Fpg protein of E. coli (10, 20), Ogg2 of S. cerevisiae (14), and functionally related DNA repair activities in yeast (13, 16). Isolation of the human and mouse ogg1 genes should make it possible to create transgenic animals and/or cell lines deficient in the repair of oxidative DNA damage and to investigate the role of this process in aging and cancer.
MATERIALS AND METHODS
DNA Substrates and Enzymes.
Unmodified and modified oligodeoxynucleotides were prepared by solid state synthesis using an Applied Biosystems model 394 automated DNA synthesizer and purified by denaturing PAGE and HPLC, as described (20). Sequences of oligodeoxynucleotide substrates and mobility markers employed in this study are listed in Table 1. An oligodeoxynucleotide mobility marker (M3) containing an abasic site at the 3′ end was prepared by acid depurination of M4 (21). A substrate containing a single thymine glycol residue (C6) was prepared by treatment of an oligodeoxynucleotide with osmium tetroxide (22) and purified by PAGE. When the modified oligomer was annealed to a complementary strand, the duplex was efficiently cleaved by E. coli endonuclease III (gift of R. Cunningham, State University of New York, Albany). Oligodeoxynucleotides were labeled at the 5′ end using [γ-32P]ATP (Amersham) and bacteriophage T4 polynucleotide kinase (New England Biolabs). A 14C-labeled substrate containing 7-methylformamidopyrimidine was prepared by treating poly(dG⋅dC) (Boehringer Mannheim) with [14C]dimethyl sulfate (Sigma) and ammonia (11). Fpg protein was purified from an overproducing strain of E. coli (23).
Table 1.
Oligonucleotides used in this study
| Mobility markers | ||
| M1 | pCpTpCpTpCpCpCpTpTpC | |
| M2 | pCpTpCpTpCpCpCpTpTpCp | |
| M3 | pCpTpCpTpCpCpCpTpTpCpX | (X = natural abasic site) |
| M4 | pCpTpCpTpCpCpCpTpTpCpG | |
| M5 | pCpTpCpTpCpCpCpTpTpCpXpCpTpCpCpTpTpTpCpCpTpCpT | X = F |
| M6 | pCpTpCpTpCpCpCpTpTpCpXpCpTpCpCpTpTpTpCpCpTpCpT | X = 8-oxodG |
| Substrates | ||
| S1 | CTC TCC CTT CAC TCC TTT CCT CT | |
| S2 | CTC TCC CTT CGC TCC TTT CCT CT | |
| S3 | CTC TCC CTT CXC TCC TTT CCT CT | (X = 8-oxodG) |
| S4 | CTC TCC CTT CXC TCC TTT CCT CT | (X = 8-oxodN) |
| S5 | CTC TCC CTT CXC TCC TTT CCT CT | (X = F) |
| C1 | AGA GGA AAG GAG AGA AGG GAG AG | |
| C2 | AGA GGA AAG GAG CGA AGG GAG AG | |
| C3 | AGA GGA AAG GAG GGA AGG GAG AG | |
| C4 | AGA GGA AAG GAG TGA AGG GAG AG | |
| C5 | AGA GGA AAG GAG XGA AGG GAG AG | (X = 8-oxodG) |
| C6 | AGA GGA AAG GAG XGA AGG GAG AG | (X = dTg) |
F, tetrahydrofuran; Tg, thymine glycol.
Bacterial Strains and Plasmids.
The strain of E. coli K12 used in these experiments, CC104 mutM mutY [ara, Δ (gpt-lac)5, rpsL/F′ (lacI378, lacZ461, pro A+B+), fpg::tetR (fpg), micA (mutY)::kanR] (24), was obtained from Jeffrey Miller (University of California, Los Angeles). The cloning vector and plasmid expressing Fpg have been described (12). Mouse cDNA clone 388707 and human cDNA clones 283432, 284190, 416941, and 298625 were obtained from the American Type Culture Collection and renamed pmogg1, phogg1, phogg2, phogg3, and phogg4, respectively. The expression plasmid pLacZmogg1 was prepared by digesting pmogg1 with XhoI and NheI, filling in ends with the Klenow fragment of DNA pol I and ligating with T4 ligase. The LacZ promoter drives expression of a fusion protein containing the first 12 amino acids of LacZ linked to the full 345 amino acid sequence of mouse Ogg1.
DNA Sequencing.
Terminal sequences of cDNA clones were obtained through GenBank and confirmed by DNA sequencing conducted in the Stony Brook Sequencing Facility. The design of synthetic primers, used to determine internal sequences, was based on previous sequencing runs. Clones were sequenced on both strands. Sequences were compiled and analyzed using the dnasis v2.0 software package (Hitachi, Tokyo).
Mutation Assays.
Approximately 1,000 cells were inoculated into Luria–Bertani (LB) media containing 25 mg/ml kanamycin, 25 mg/ml tetracycline, and 50 mg/ml ampicillin; the culture was grown overnight with shaking at 37°C. Serial dilutions were plated onto kanamycin–LB plates with and without rifampicin; the number of colonies was determined after overnight incubation at 37°C.
Northern Blot Analysis.
Total RNA was isolated from tissues of an adult male FVB/N mouse using Trizol reagent (Life Technologies, Grand Island, NY) following the manufacturer’s protocol. RNA (20 μg) was subjected to electrophoresis in a formaldehyde denaturing gel, then transferred to a Duralon (Stratagene) membrane. The blot was hybridized to a 0.9-kb 32P-labeled probe (synthesized with a Life Technologies random priming kit) corresponding to the 5′ untranslated region and amino acids 1–224 of the mouse Ogg1 coding sequence.
Preparation of Bacterial Extracts and Partial Purification of Enzyme Activities.
Bacteria were grown at 37°C in 50 ml of yeast tryptone broth (YTx2) containing 100 μg/ml ampicillin and grown to A600 = 0.9. Isopropyl β-d-thiogalactoside was added (final concentration, 0.5 mM), after which the culture was incubated for 3 additional hours. Cells were harvested by centrifugation at 10,000 × g for 10 min at 4°C, then resuspended in 0.5 ml of lysis buffer (25 mM Tris⋅HCl, pH 7.5/250 mM NaCl/1 mM EDTA/1 mM DTT/1 mM phenylmethylsulfonyl fluoride). Lysozyme was added to a final concentration of 0.1 mg/ml. Cells were incubated on a rotating platform for 1 hr at 4°C then disrupted by two 7-sec pulses with a Fisher Model 550 Sonic Dismembrator operated at maximum power and equipped with an one-eighth inch microtip. Cell extracts were clarified by centrifugation at 10,000 × g for 30 min at 4°C. The supernatant was divided into aliquots and stored at −20°C.
Partially purified extracts containing mouse mOgg1 were prepared from a 4 liter culture. After cell lysis, the protein concentration was adjusted to 8 mg/ml and NaCl was added to a final concentration of 300 mM. This extract was loaded onto a Q Sepharose Fast Flow column (Pharmacia, 80 ml bed volume) equilibrated with Buffer A (10 mM Hepes-KOH, pH 7.5/1 mM EDTA/1 mM DTT) containing 300 mM NaCl. The column was washed with 2 vol of this solution; eluate and flow-through were pooled and loaded onto an S Sepharose Fast Flow column (Pharmacia, 80 ml bed volume) equilibrated with Buffer A. A gradient of 300–600 mM NaCl in Buffer A was used to develop the column. Nicking of oligodeoxynucleotide duplexes containing a single 8-oxodG residue was used to detect DNA glycosylase activity and strand breaks, as described below. Active fractions were pooled, precipitated with 80% (NH4)2SO4, dialyzed overnight against Buffer A containing 50% glycerol and stored at −20°C.
Activity Assays.
The standard reaction mixture contained 52 mM Hepes-KOH (pH 7.5), 60 mM NaCl, 5 mM EDTA, 0.2 mM DTT, 50 nM (100 nM for competition experiments) duplex oligodeoxynucleotide substrate (32P-labeled at the 5′ end of the strand containing the modified base), varying amounts of cell extract (added last), and where indicated, KCN or unlabeled oligonucleotide. Reactions were incubated at 25°C for periods of 15 sec to 1 hr and terminated by adding 0.5 vol formamide dye loading buffer, then heating for 5 min at 95°C. Aliquots (5 μl) were loaded onto denaturing gels and subjected to 20% PAGE. Bands on the gel were quantified using a Molecular Dynamics PhosphorImager system.
RESULTS
Identification of Potential Mammalian ogg1 cDNAs.
The National Center for Biotechnology Information tblastn program (25) was used to search the GenBank expressed sequence tag database for potential homologs of S. cerevisiae ogg1 (14, 15). Candidate sequences were used to search GenBank and other DNA databases for homologous sequences. A group of overlapping human expressed sequence tags and a corresponding set of overlapping mouse expressed sequence tags were identified, each showing strong homology to the N-terminal sequence of yeast ogg1. One mouse clone and three human clones were sequenced. The mouse clone (1.5 kb) contains an open reading frame encoding a 345 amino acid protein with extensive homology to yeast ogg1 (Fig. 1). The three human cDNA clones were incomplete; one of them contains an 18 bp deletion relative to the other two sequences, possibly reflecting an alternate splice site. Being >99% identical in their overlapping regions, the partial clones were assumed to be isolates from the same gene and a composite human ogg1 protein sequence with extensive homology to the mouse protein was assembled.
Figure 1.

Lineup of yeast ogg1 with putative human and mouse ogg1 proteins. The helix–hairpin–helix (HhH)–PVD loop motif, conserved in the DNA endonuclease III (endo III) family of DNA glycosylases, is underlined. Asterisks indicate conserved residues. The boxed lysine residue is found in DNA glycosylases with AP lyase activity; the boxed aspartic acid residue is conserved throughout this family. Underlined residues (W23 and S31) near the amino termini of the mouse and human proteins indicate potential mitochondrial cleavage sites. The underlined sequence near the COOH terminus represents a potential nuclear localization sequence.
Fig. 1 shows an alignment of the predicted mouse and human ogg1 proteins with the yeast ogg1 sequence. Underlined in this figure are previously noted motifs conserved between yeast ogg1 and the DNA endo III/MutY/AlkA family of DNA glycosylases (15, 26, 27). These motifs include a HhH DNA-binding domain followed by a proline/valine-rich loop (labeled PVD loop in Fig. 1). Both mammalian proteins contain conserved lysine and aspartic acid residues that have counterparts in yeast Ogg1 (Lys-241 and Asp-260) and E. coli endo III (Lys-120 and Asp-138) but lack glycine residues in the loop region. In addition to a HhH/PVD motif, four regions of homology were identified in yeast and mammalian proteins (Fig. 1). Putative mitochondrial targeting sequences are located near the amino-terminal end of both mammalian proteins; a potential nuclear localization signal is located near the COOH terminus of mouse and human Ogg1 (Fig. 1).
Mouse ogg1 Can Replace E. coli fpg.
Fpg protein, encoded by mutM (fpg), efficiently excises 8-oxoguanine from oxidatively damaged DNA (11); mutY protein is involved in postreplication repair of this lesion (24). The mutM/mutY double mutant has a 25- to 75-fold higher mutation rate than either mutator alone, reflected by an increase in rifampicin-resistant colonies (24). We tested the ability of mouse ogg1 to suppress the mutator phenotype of mutM/mutY bacteria. A plasmid was constructed that expresses a fusion protein containing the first 12 amino acids of the LacZ α fragment linked to the product of the mouse ogg1 gene. Following transformation, this plasmid reduces the spontaneous mutation rate of mutM/mutY E. coli by 19-fold, a reduction similar to that conferred by transformation with a plasmid expressing Fpg protein (Table 2).
Table 2.
Suppression of spontaneous rifr by mouse ogg1
| Strain | rifr/109 cells |
|---|---|
| fpg, mutY + pKK223-3 | 1,700 ± 800 (n = 5) |
| fpg, mutY + pFpg | 47 ± 21 (n = 5) |
| fpg, mutY + pLacZmOgg1 | 88 ± 56 (n = 5) |
Expression of mOgg1 in E. coli.
mutM/mutY double mutants of E. coli were transformed with plasmids expressing Fpg or mouse Ogg1. Extracts prepared from these cells were tested for their ability to nick a 23-mer duplex oligodeoxynucleotide substrate containing a single 8-oxodG residue. Extracts of cells transformed by the vector alone do not produce detectable cleavage of this substrate (Fig. 2, lane 1). Extracts of cells transformed with plasmids expressing Fpg or mouse Ogg1 generate a labeled product with a mobility reflecting cleavage at the lesion site (Fig. 2, lanes 2 and 3).
Figure 2.
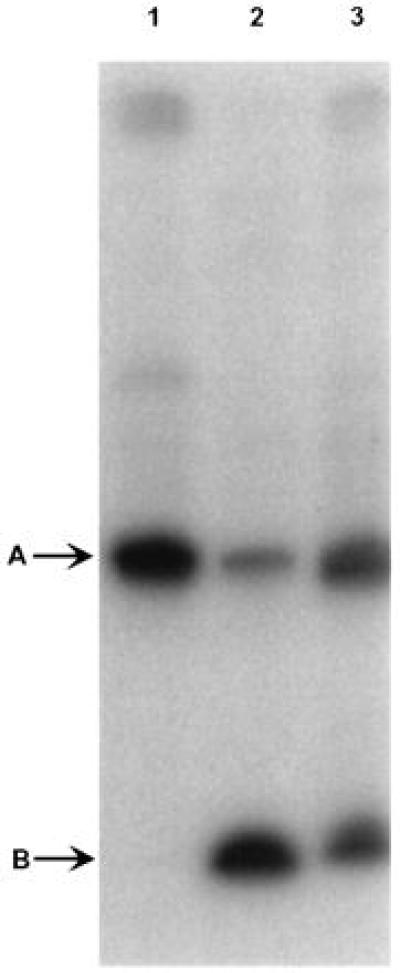
Nicking of substrates containing 8-oxodG by extracts prepared from E. coli CC104 mutM/mutY harboring different plasmids. A 32P-labeled oligonucleotide containing a single 8-oxodG residue (S3) was annealed to a complementary strand (C2) containing dC positioned opposite the lesion. Reaction mixtures containing 30 μg protein were incubated for 10 min at 25°C, as described. The arrow A indicates the position of unmodified 23-mer duplex (M6); the arrow B indicates the position of the Fpg cleavage product (M2) (see Table 1 for a description of mobility markers). Lane 1, extract prepared from E. coli harboring pKK223–3. Lane 2, extract prepared from E. coli harboring pFpg. Lane 3, extract prepared from E. coli harboring pLacZmogg1.
Nature of Products Generated by mogg1.
Several DNA glycosylases, including E. coli endo III, contain an AP lyase activity that catalyzes a β elimination reaction at the abasic site produced by excision of the damaged base, thereby generating an α,β-unsaturated sugar moiety at the 3′ terminus (28). Some glycosylases, including Fpg protein, further process the 3′ terminus by δ elimination, removing the sugar residue and leaving 3′ and 5′ phosphates flanking a one-base gap (29, 30). We examined the mechanism of cleavage by mOgg1 under various experimental conditions. Partially purified extracts containing mOgg1 were incubated with a 23-mer duplex substrate containing 8-oxoguanine (S3/C2). Products of this reaction (Fig. 3, lanes 4 and 9) include Band A, reflecting loss of a base, and a rapidly migrating double band (C and D), resolved by 20% denaturing PAGE. The double band was observed even without heating (data not shown). Band D corresponds to oligodeoxynucleotide marker M3 (lane 3) containing an abasic site at the 3′ terminus. The ratio of Band C to Band D decreases slowly with time (data not shown). Neither band corresponds to the marker M2 for δ elimination (lane 2). Band B was not reproducibly observed.
Figure 3.
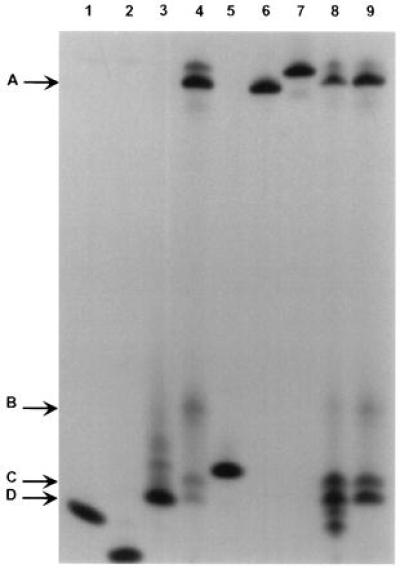
Cleavage by mOgg1 generates a product with an unsaturated sugar residue at the 3′ end. The standard reaction mixture (see Materials and Methods) contained 0.5 μg protein of the active fraction recovered following S Sepharose chromatography and a 32P-labeled duplex oligonucleotide substrate containing 8-oxodG:dC (S3/C2). (See Table 1 for a description of mobility markers.) Lanes: 1, M1; 2, M2; 3, M3; 4, standard reaction mixture incubated for 10 min at 25°C; 5, M4; 6, M5; 7, M6; 8, standard reaction mixture containing 10 mM 3-MPA incubated for 30 min at 37°C; 9, standard reaction mixture containing 100 mM 2-mercaptoethanol incubated for 30 min at 37°C.
To confirm the presence of an unsaturated sugar moiety in the partially cleaved substrate, the reaction mixture was incubated in the presence of 10 mM 3-mercaptopropionic acid (3-MPA). 3-MPA adds to the double bond of α,β-unsaturated aldehydes, introducing a negative charge and increasing the electrophoretic mobility of the addition product. Two new bands, migrating more rapidly than Bands C and D, were observed in the presence of 3-MPA (Fig. 3, lane 8), suggesting that both cleavage products contain a 3′-terminal unsaturated sugar. The band shift was not observed when 3-MPA was replaced by 2-mercaptoethanol, a neutral molecule, (Fig. 3, lane 9).
The principal reaction product formed after 10 min incubation (Band A in Fig. 3, lane 4) comigrates with an oligodeoxynucleotide (M3) containing tetrahydrofuran, a synthetic abasic site (31). The time course of this reaction was determined (data not shown). Band A forms within 10 min while the nicked product accumulates significantly only after 30 min (Fig. 3, lane 9).
Inhibition by KCN.
Cyanide ion inhibits DNA glycosylases containing AP lyase activity by reacting with imino intermediates, as shown for bacteriophage T4 endonuclease V (32) and E. coli Fpg protein (D.O.Z. unpublished observations). KCN inhibits abasic site formation and the strand-nicking reactions catalyzed by extracts of mouse Ogg1. I50 = 16 mM for both reactions (data not shown). Pre-incubation of extracts with 0.1 M KCN for 30 min does not lead to a decrease in activity of the enzyme if the reaction mixture contains 0.1 M KCl.
Substrate Specificity of mogg1.
Initial rates of cleavage (vo) were established for substrates containing various base pairs or mispairs, using extracts containing mOgg1 (Table 3 and Fig. 4). A duplex containing 8-oxodG:dC was cleaved efficiently; duplexes containing 8-oxodG:dT, 8-oxodG:dG, 8-oxodNeb:dC, and 7,8-dihydroxy-8-oxo-2′-deoxynebularine (8-oxodNeb):dG were weak substrates, and duplexes containing 8-oxodG:dA, 8-oxodNeb:dT, 8-oxodNeb:dA, and dG:dC were not cleaved. Extracts containing mOgg1 did not excise adenine from dA:8-oxodG or dA:dG, the preferred substrates for MutY protein (24). The rate of excision of 7-methylformamidopyrimidine from methylated, alkali-treated poly(dG-dC) by mOgg1 was less than 10% the rate observed with Fpg (data not shown).
Table 3.
Rate of cleavage of DNA substrates by extracts of E. coli harboring pLacZmogg1
| Labeled strand | Complementary strand | vo, fmol·min−1 |
|---|---|---|
| 8-oxodG | dA | 0.15 ± 0.04 |
| dC | 14.3 ± 3.8 | |
| dG | 1.02 ± 0.07 | |
| dT | 2.7 ± 0.03 | |
| dG | dA | 0.11 ± 0.08 |
| dC | 0.11 ± 0.1 | |
| dG | 0.12 ± 0.09 | |
| dT | 0.11 ± 0.1 | |
| 8-oxodNeb | dA | 0.15 ± 0.06 |
| dC | 1.51 ± 0.1 | |
| dG | 1.52 ± 0.04 | |
| dT | 0.25 ± 0.08 | |
| dA | dG | 0.15 ± 0.07 |
| 8-oxodG | 0.12 ± 0.05 |
Figure 4.
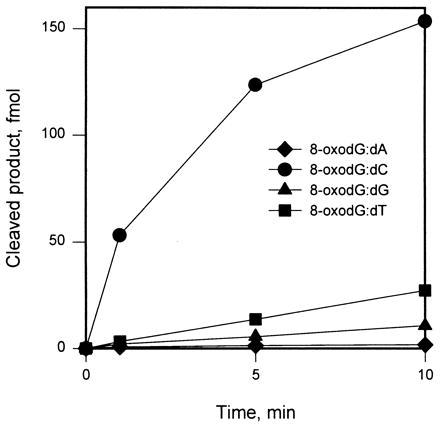
mOgg1 efficiently cleaves substrates containing the 8-oxodG:dC base pair. A 32P-labeled oligonucleotide containing a single 8-oxodG residue (S3) was annealed to a complementary strand (C2) containing the indicated dN positioned opposite the lesion. Reactions with extracts of E. coli harboring pLacZmogg1 (10 μg protein) were performed at 25°C.
Our experiments were conducted with partially purified extracts of E. coli; thus, nicking of the DNA phosphodiester backbone by contaminating AP endonucleases or AP lyases could occur after base excision. However, none of the reaction products comigrated with M1 (Fig. 3, lane 1), a marker for products of cleavage by Class II AP endonucleases. Selected oligodeoxynucleotides were tested for their ability to compete with natural substrate in base excision and strand-nicking reactions. Unlabeled duplexes containing 8-oxodG or tetrahydrofuran (model abasic site) positioned opposite dC competed efficiently in both reactions, yielding 50% inhibition when added in 2-fold excess over substrate. Duplexes containing dA:dTg, dG:dC, or dA:dG, added in 10-fold excess, inhibited base excision and cleavage by only 20%. Results of this experiment are not conclusive, but suggest that mOgg1 can carry out both base excision and strand-scission reactions.
Expression of Mammalian ogg1.
Total RNA from adult mouse tissues was analyzed for expression of mOgg1 RNA. Low level expression of RNA was reproducibly detected in several tissues (Fig. 5). The size of the band (1.5 kb) was similar to that of the mOgg1 cDNA clone. Highest levels of expression were observed in testes.
Figure 5.
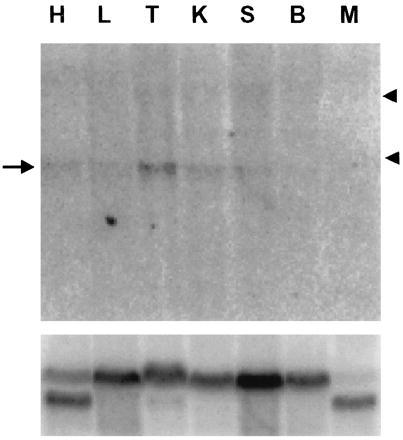
Expression of mouse ogg1 in adult tissues. Total RNA (20 μg) was isolated from tissues, subjected to electrophoresis in a formaldehyde–agarose gel, blotted, hybridized to a mouse ogg1 cDNA probe (Upper) and reprobed with a β-actin sequence (Lower). H, heart; L, lung; T, testes; K, kidney; S, spleen; B, brain; M, skeletal muscle. Arrow indicates mOgg1 RNA. Arrowheads indicate the positions of 28S and 18S rRNAs.
DISCUSSION
We report here the cloning of human and mouse cDNAs that encode homologs of the yeast ogg1 gene, together with partial characterization of the product of mouse ogg1, a repair activity that selectively excises 8-oxoguanine from oxidatively damaged DNA. The human and mouse genes contain a conserved HhH region, which identifies them as members of a recently defined family of DNA glycosylases. Other members of this family include endo III, MutY, Micrococcus luteus UV endonuclease, and 3-methyladenine DNA glycosylase II of E. coli (15, 26, 27). Notably, the human and mouse Ogg1 sequences include lysine and aspartic acid residues in a region corresponding to the active site of these proteins. Mutation of the counterpart residues in endo III (Lys-120 and Asp-138) and 3-methyladenine DNA glycosylase II (Asp-238) markedly reduces the catalytic activity of these enzymes (15, 26, 27). In addition to the HhH/PVD motif, four regions of unknown function in yeast ogg1 and mouse ogg1 are conserved.
Base excision repair is initiated by recognition of a damaged DNA base. In excising this lesion, DNA glycosylases generate, at least transiently, an abasic site. Steps in the repair process may differ; for example, Fpg protein and endo III form Schiff base intermediates, catalyzing β-elimination of the 3′ phosphate. Monofunctional DNA glycosylases such as MutY have a kinetically distinct AP lyase activity; in this case, attack at C1′ of deoxyribose appears to be mediated through a water molecule (33, 34). Mouse Ogg1 excises 8-oxoguanine, generating an abasic site. The phosphodiester backbone is then nicked; the two steps are distinguished by the time course and by showing that duplexes containing an abasic site analog are competitive inhibitors of this reaction. The product of the nicking reaction appears to contain an unsaturated residue, indicated by formation of an addition product with 3-MPA. The DNA glycosylase/AP lyase activity of mouse Ogg1 may involve a Schiff base intermediate, implied by the presence of Lys-240 in the putative active site (Fig. 1) and demonstrated by showing that both reactions are inhibited by KCN (35). Definitive interpretation of the mechanism of action of mOgg1 awaits studies with purified enzyme.
Northern blot analysis reveals that mouse ogg1 RNA is present in low abundance in several mammalian tissues; highest expression was observed in testes. Elevated expression in testes has been observed for other base excision repair proteins, including DNA polymerase β (36) and XRCC1 (37). The relatively high level of expression in this tissue may reflect a need to minimize germ-line mutations. Three of the mammalian ogg1 expressed sequence tag clones were isolated from embryonic tissues suggesting that ogg1 protects against oxidative DNA damage during fetal development.
Mouse ogg1 was not detected in skeletal muscle or brain; however, low levels of RNA in these tissues might not have been detected. Alternatively, the product(s) of other mammalian genes may repair 8-oxoguanine, as suggested by the identification of two 8-oxoguanine repair activities in HeLa cells (18). One of these activities, a DNA glycosylase, resembles mouse Ogg1; a similar activity was detected in human placenta (D.O.Z., unpublished observation). The other activity, an endonuclease that recognizes 8-oxodG and cleaves the phosphodiester backbone 5′ to the lesion, is mechanistically different from Ogg1 and may be encoded by a different gene. A mammalian gene coding for N-methylpurine DNA glycosylase also has been shown to have 8-oxoguanine glycosylase activity and to complement fpg deficient bacteria (38).
In addition to Ogg1, yeast have an activity that excises 8-oxodG but acts preferentially on substrates containing methylated formamidopyrimidines and on duplexes in which 8-oxoguanine is positioned opposite dA or dG (13, 15). These properties resemble those of a yeast protein cloned through its sequence similarity to endo III (16).
Between 104 and 105 8-oxoguanine residues are produced per cell each day in mammals (7). DNA replication past 8-oxoguanine promotes incorporation of adenine opposite the lesion and leads to G:C → T:A mutations in E. coli (39). 8-oxoguanine glycosylase plays a central role in protecting cells from the effects of this mutagenic lesion. Mouse Ogg1 does not act on substrates containing oxodG:dA; this mispair presumably is repaired by the mammalian homolog of MutY (40). The human homolog of MutT, a nucleotide triphosphotase that removes 8-oxodGTP from the dNTP pool, also has been described (41). Thus, functional homologs for each of the three activities required to effect repair of 8-oxoguanine in bacteria (42) have now been identified in mammalian cells and cloned.
It is difficult to evaluate the contribution of oxidative DNA damage and its repair to the pathophysiology of human diseases. Cloning of mouse ogg1 makes it possible to knock out or otherwise modulate expression of this gene in experimental animals. Transgenic mice that over- or underexpress 8-oxoguanine DNA glycosylase should prove useful in determining whether oxidative DNA damage contributes to aging and cancer and in assessing effects of antioxidants in counteracting this process.
Acknowledgments
Robert Rieger synthesized the modified and unmodified oligodeoxynucleotides used in this study. Nikolay Bulychev prepared the substrate containing thymine glycol. We thank Richard Cunningham (State University of New York, Albany) for a gift of endonuclease III, Francis Johnson and Michael Frohman for helpful discussion, and Sidney Strickland and Daniel Bogenhagen for critically reviewing this manuscript. We thank Susumu Nishimura (Banyu Tsukuba Research Institute) for communicating pre-publication results. This research was supported by National Institutes of Health Grants CA17395 and ES04068.
ABBREVIATIONS
- 8-oxodG
7,8-dihydroxy-8-oxo-2′-deoxyguanosine
- 8-oxodNeb
7,8-dihydroxy-8-oxo-2′-deoxynebularine
- endo III
DNA endonuclease III
- Ogg
8-oxoguanine-DNA glycosylase
- Fpg
formamidopyrimidine/8-oxoguanine DNA glycosylase
- 3-MPA
3-mercaptopropionic acid
- HhH
helix–hairpin–helix
Footnotes
References
- 1.Wiseman H, Halliwell B. Biochem J. 1996;313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedberg E C, Walker G C, Seide W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 1–58. [Google Scholar]
- 3.Dizdaroglu M. In: DNA and Free Radicals. Halliwell B, Arouma O I, editors. Chichester, U.K.: Horwood; 1993. pp. 19–39. [Google Scholar]
- 4.Kasai H N, Nishimura S. In: Oxidative Stress and Antioxidants. Sies H, editor. London: Academic; 1991. pp. 99–116. [Google Scholar]
- 5.Grollman A P, Moriya M. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 6.Totter J R. Proc Natl Acad Sci USA. 1980;77:1763–1767. doi: 10.1073/pnas.77.4.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ames B N, Shigenaga M K, Hagen T M. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames B N, Gold L S, Willett W C. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chetsanga C J, Lindahl T. Nucleic Acids Res. 1979;6:3673–3684. doi: 10.1093/nar/6.11.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boiteux S, O’Connor T R, Lederer F, Gouyette A, Laval J. J Biol Chem. 1990;265:3916–3922. [PubMed] [Google Scholar]
- 11.Tchou J, Kasai H, Shibutani S, Chung M-H, Laval J, Grollman A P, Nishimura S. Proc Natl Acad Sci USA. 1991;88:4690–4694. doi: 10.1073/pnas.88.11.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michaels M L, Pham L, Cruz C, Miller J H. Nucleic Acids Res. 1991;19:3629–3632. doi: 10.1093/nar/19.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Oliveira R, van der Kemp P A, Thomas D, Geiger A, Nehls P, Boiteux S. Nucleic Acids Res. 1994;22:1760–1764. doi: 10.1093/nar/22.18.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Kemp P A, Thomas D, Barbey R, de Oliveira R, Boiteux S. Proc Natl Acad Sci USA. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nash H M, Bruner S D, Schärer O D, Kawate T, Addona T A, Spooner E, Lane W S, Verdine G L. Curr Biol. 1996;6:968–980. doi: 10.1016/s0960-9822(02)00641-3. [DOI] [PubMed] [Google Scholar]
- 16.Eide L, Bjørås M, Pirovano M, Alseth I, Berdal K G, Seeberg E. Proc Natl Acad Sci USA. 1996;93:10735–10740. doi: 10.1073/pnas.93.20.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung M-H, Kim H-S, Ohtsuka E, Kasai H, Yamamoto F, Nishimura S. Biochem Biophys Res Commun. 1991;178:1472–1478. doi: 10.1016/0006-291x(91)91059-l. [DOI] [PubMed] [Google Scholar]
- 18.Bessho T, Tano K, Kasai H, Ohtsuka E, Nishimura S. J Biol Chem. 1993;268:19416–19421. [PubMed] [Google Scholar]
- 19.Yamamoto F, Kasai H, Bessho T, Chung M-H, Inoue H, Ohtsuka E, Hori T, Nishimura S. Jpn J Cancer Res. 1992;83:351–357. doi: 10.1111/j.1349-7006.1992.tb00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tchou J, Bodepudi V, Shibutani S, Antoshechkin I, Miller J, Grollman A P, Johnson F. J Biol Chem. 1994;269:15318–15324. [PubMed] [Google Scholar]
- 21.Castaing B, Boiteux S, Zelwer C. Nucleic Acids Res. 1992;20:389–394. doi: 10.1093/nar/20.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedmann T, Brown D M. Nucleic Acids Res. 1978;5:615–622. doi: 10.1093/nar/5.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zharkov D O, Rieger R A, Iden C R, Grollman A P. J Biol Chem. 1997;272:5335–5341. doi: 10.1074/jbc.272.8.5335. [DOI] [PubMed] [Google Scholar]
- 24.Michaels M L, Cruz C, Grollman A P, Miller J H. Proc Natl Acad Sci USA. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Tayer M M, Ahern H, Xing D, Cunningham R P, Tainer J A. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamagata Y, Kato M, Odawara K, Tokuno Y, Nakashima Y, Matsushima N, Yasumura K, Tomita K, Ihara K, Fujii Y, Nakabeppu Y, Sekiguchi M, Fujii S. Cell. 1996;86:311–319. doi: 10.1016/s0092-8674(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 28.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 29.Bailly V, Verly W G, O’Connor T R, Laval J. Biochem J. 1989;262:581–589. doi: 10.1042/bj2620581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhagwat M, Gerlt J A. Biochemistry. 1996;35:659–665. doi: 10.1021/bi9522662. [DOI] [PubMed] [Google Scholar]
- 31.Giloni L, Takeshita M, Johnson F, Iden C, Grollman A P. J Biol Chem. 1981;256:8606–8615. [PubMed] [Google Scholar]
- 32.Dodson M L, Schrock R D, III, Lloyd R S. Biochemistry. 1993;32:8284–8290. doi: 10.1021/bi00083a032. [DOI] [PubMed] [Google Scholar]
- 33.Dodson M L, Michaels M L, Lloyd R S. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- 34.Bulychev N V, Varaprasad C V, Dorman G, Miller J H, Eisenberg M, Grollman A P, Johnson F. Biochemistry. 1996;35:13147–13156. doi: 10.1021/bi960694h. [DOI] [PubMed] [Google Scholar]
- 35.Tennant G. In: Comprehensive Organic Chemistry: The Synthesis and Reactions of Organic Compounds. Vol. 2: Nitrogen Compounds, Carboxylic Acids, Phosphorus Compounds. Sutherland I O, editor. New York: Pergamon; 1979. p. 409. [Google Scholar]
- 36.Hirose F, Hotta Y, Yamaguchi M, Matsukage A. Exp Cell Res. 1989;181:169–180. doi: 10.1016/0014-4827(89)90191-2. [DOI] [PubMed] [Google Scholar]
- 37.Walter C A, Lu J, Bhakta M, Zhou Z-Q, Thompson L H, McCarrey J R. Somatic Cell Mol Genet. 1994;20:451–461. doi: 10.1007/BF02255837. [DOI] [PubMed] [Google Scholar]
- 38.Bessho T, Roy R, Yamamoto K, Kasai H, Nishimura S, Tano K, Mitra S. Proc Natl Acad Sci USA. 1993;90:8901–8904. doi: 10.1073/pnas.90.19.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibutani S, Takeshita M, Grollman A P. Nature (London) 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 40.McGoldrick J P, Yeh Y-C, Solomon M, Essigmann J M, Lu A-L. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M. J Biol Chem. 1993;268:23524–23530. [PubMed] [Google Scholar]
- 42.Tchou J, Grollman A P. Mutat Res. 1993;299:277–287. doi: 10.1016/0165-1218(93)90104-l. [DOI] [PubMed] [Google Scholar]