

Abstract
A sensitive and precise in vitro technique for detecting DNA strand discontinuities produced in vivo has been developed. The procedure, a form of runoff DNA synthesis on molecules released from lysed bacterial cells, mapped precisely the position of cleavage of the plasmid pMV158 leading strand origin in Streptococcus pneumoniae and the site of strand scission, nic, at the transfer origins of F and the F-like plasmid R1 in Escherichia coli. When high frequency of recombination strains of E. coli were examined, DNA strand discontinuities at the nic positions of the chromosomally integrated fertility factors were also observed. Detection of DNA strand scission at the nic position of F DNA in the high frequency of recombination strains, as well as in the episomal factors, was dependent on sexual expression from the transmissable element, but was independent of mating. These results imply that not only the transfer origins of extrachromosomal F and F-like fertility factors, but also the origins of stably integrated copies of these plasmids, are subject to an equilibrium of cleavage and ligation in vivo in the absence of DNA transfer.
Keywords: conjugational DNA transfer, rolling-circle DNA replication, relaxosome, nicking-closing activity
We are interested in understanding the events leading to the cleavage and initiation of DNA strand transfer of the self-transmissible, F-like, antibiotic resistance factor R1, and in clarifying the molecular mechanisms governing the initiation of DNA replication of the pMV158 family of promiscuous, rolling-circle (RC)-replicating plasmids. These systems have the common biological objective of mobilizing single-stranded (ss) DNA. Current models of plasmid leading strand replication and conjugational transfer of F-like plasmid DNA resemble each other in several mechanistic aspects (refs. 1–3 and references therein).
Plasmid pMV158 is a 5.5-kb multicopy natural promiscuous replicon that is the prototype of a family of RC-replicating plasmids isolated from several eubacteria (2). Initiation of its replication is mediated by RepB protein, which cleaves the phosphodiester bond between G-448 and A-449 of the double-strand origin (dso; ref. 4 and references therein). The nicking-closing activity of the purified RepB protein on various substrates in vitro has been examined (4, 5).
For conjugational transfer of plasmid DNA to occur, a specific plasmid strand is cleaved at a unique nick site (nic) within the origin of transfer (oriT). In keeping with present models of this process in the F system, nicking of oriT DNA of the closely related plasmid R1 should be catalyzed by a multienzyme complex, the relaxosome, that includes the Escherichia coli integration host factor protein, and plasmid-encoded TraY and TraI proteins (ref. 3 and references therein). In the R1 system, at least one additional plasmid protein, TraM, acts as an accessory DNA binding protein at oriT (E.L.Z., unpublished data). The site-specific DNA strand transferase activity of the relaxosome is provided by the TraI protein (6, 7).
Elegant biochemical studies have characterized the activities of relaxosomes, reconstituted in vitro from purified proteins, of plasmids of the F, P, and Q groups (8–11). However, the properties and regulation of these complexes at their respective transfer origins in vivo are less well understood. Notably, the conditions that induce initiation of conjugational DNA transfer remain obscure as does the nature of the activation. Recently, precedence was found in the R6K system for two functional oriTs on one plasmid molecule (12). Because transfer can initiate from only one origin, this finding reveals an additional complexity in the regulation of the start of transfer, namely, relaxosome choice.
To analyze the performance of nickases and enzyme complexes such as the R1 relaxosome in vivo, we have developed a methodology that permits detection of intracellular cleavage of DNA strands. This report characterizes the experimental system and presents evidence that DNA strand interruptions can be readily detected with nucleotide resolution in large (≈90 kb), very low copy number Gram-negative plasmids and in small (5 kb), RC plasmids in Gram-positive bacteria. Remarkably, nic-specific DNA strand discontinuities at the sites of integration of F plasmids in the chromosomes of E. coli high frequency recombination (Hfr) strains were also observed. From this finding, it is inferred that the transfer proteins expressed by chromosomally integrated fertility factors form functional relaxosomes that maintain an equilibrium of cleavage and joining of nic DNA at the point of origin of an Hfr strain even in the absence of bacterial mating.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Media.
All E. coli strains were K12 derivatives. J5 and MC1061 were used for the maintenance and transfer of R1 and F plasmid derivatives. M1174 sfrA+ and M1164 sfrA5 (13) were kindly provided by P. Silverman (Oklahoma Medical Research Foundation, Oklahoma City). The Hfr donor strains were Hfr3000 (14), KL96 (15), 61-1 deoB-serBΔ, and 122-1 deoB-arcAΔ (16). 61-1 and 122-1 were the gift of A. Dijkstra (Hoffmann–LaRoche, Basel). W3110, LE392, and JC3272 were used for (F−) controls.
Plasmids R1drd19 (R1-19) and R1drd16 (R1-16) are derepressed derivatives of plasmid R1. pOX38-Km (17) was the gift of L. Frost (University of Alberta, Edmonton, Canada). pGZ119HE (18), employed for the expression of E. coli arcA and the R1 traI gene, was kindly provided by E. Lanka (Max-Planck-Institut für Molekulare Genetik, Berlin). pUAA-1 (19) was the gift of G. Sawers (University of Sussex, Brighton, U.K.). pCK217 (20) contains positions 1238–2297 of R1 DNA (numbering as in ref. 21) in pBluescript SK+ (Stratagene). pLS1 is a deletion variant of pMV158 that lacks mobilization sequences (22–24). pCGA12 (24) contains the entire dso of pLS1 inserted into the HindIII site of pC194 (25). pLS86 contains a 5.3-kb insertion of Streptococcus pneumoniae DNA including the sulA gene (26). The pLS1cop7 derivative of pLS1 (27) has undergone a spontaneous C to A transversion at position 743 (22). As a result, the copy number of pLS1cop7 is elevated 5-fold relative to pLS1. pCGA12, pLS86, and pLS1cop7 were maintained in S. pneumoniae MP551 [R6 end-1 hexo-3 noz-19 polAΩ(1052::cat)] (28).
The medium for E. coli was 2xTY (29). Media and growth of S. pneumoniae have been described (30). When required, antibiotics were added at the indicated concentrations (in μg ml−1): kanamycin, 40; streptomycin, 25; dihydroampicillin, 50; chloramphenicol, 10 (E. coli) or 3 (S. pneumoniae); tetracycline-HCl, 1.
Oligonucleotides, Enzymes, and Reagents.
The sequences of the oligonucleotides in the nicking assay and for nucleotide sequence standards were (5′ to 3′): no. 6, complementary to F factors, CCCGGCCTGCAAGATCAAAGC [R1 positions 1865–1885] and primer pMV158, used with the Gram(+) plasmids, TTCATGCGTCACCTCTC [pMV158 positions 659–643]. No. 6*, AATTGCTTTGATCTTGCAGGCCGGGAGCT was used for the control template. For the molecular cloning of traI, TRAI-1, TCTCCCATGCTGCCCAGCAC and TRAI-2, GACCTGCAGAAAGAGAAAACCCTGGG were used.
Restriction endonucleases, calf intestinal phosphatase, T4 DNA ligase, and T4 polynucleotide kinase were purchased from Boehringer Mannheim. Klenow enzyme was purchased from New England Biolabs. The Thermus brockianus thermostable DNA polymerase, DyNAoyme, was obtained from Finnzymes (Espoo). Radiochemicals were purchased from NEN.
Preparation of Expression Constructions for the E. coli arcA Gene and the R1 traI Gene.
Standard recombinant DNA techniques (31) or those suggested by the manufacturers were used.
Molecular Cloning of the traI Gene of Plasmid R1-16.
An 11.3-kb Nsi fragment from R1-16 DNA, identified by hybridization to 5′ and 3′ traI probes, was ligated to linear pBluescript SK+ DNA. Partial DNA sequence of the region upstream of traI of positive clone pHP1 was obtained using primer TRAI-1 and the T7 sequencing kit (Pharmacia). These data were sufficient to identify an AsnI site 51 nt upstream of the traI start codon. A 6.1-kb AsnI fragment of pHP1, containing traI, was made blunt and subcloned to SmaI-digested pGZ119HE DNA (Ptac-lacIq). Clone pHP2 was used for complementation of nicking in vivo.
The arcA gene of E. coli was excised from pUAA-1 (EcoRI/HindIII) and introduced to pGZ119HE DNA. Positive clones were identified by PCR amplification of the arcA gene and complementation of conjugal donor ability of E. coli 122-1 arcAΔ.
Preparation of the Standard DNA Template.
Oligonucleotide 6* was complementary to primer 6 and had additionally four nt at the 5′ and 3′ ends compatible with EcoRI/SacI digested vector. Hybridized oligonucleotides 6* and 6 were ligated to linear pBluescript DNA, and transformed XL1 cells were selected with dihydroampicillin and 5-bromo-4-chloro-3-indolyl β-d-galactoside. One positive clone, pHP7, was purified, linearized, and reisolated for use as a standard DNA template in the nicking assay.
Conditions for PCR Amplification and for the Runoff DNA Synthesis Assay.
E. coli donor strains for conjugation were cultured to stationary phase, then diluted 1:25 in fresh medium at 37°C. They were then grown at 37°C without shaking for one doubling (final OD600 0.2–0.3). A 100-fold excess of recipients over donor cells was added directly from overnight culture (ONC) to the growing donor culture. Mating cultures were incubated at 37°C without shaking for 15–30 min. Transfer was stopped by vigorous mixing for 1 min and rapid cooling to 0°C. Plasmid-plus and plasmid-free strains not subjected to mating were cultivated as described above, and harvested at a final OD600 0.1–0.6. In some experiments, ONCs were used directly. For PCR amplification of DNA, 1–5 μl of an ONC or a single colony resuspended in reaction buffer were used as template. For the runoff DNA synthesis assay, viable cell counts were obtained at harvest.
Aliquots of cultures containing 0.4–20 × 106 colony forming units (cfu) were centrifuged for 4–10 min at 12,000 × g. The medium was thoroughly removed. Cell pellets kept at 0°C were used directly (E. coli) or frozen on dry ice and stored at −70°C for later use (S. pneumoniae). For the reaction mixture, bacteria were resuspended in 25 μl ice-cold buffer containing 10 mM Tris⋅HCl (pH 8.8), 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, 100 ng oligonucleotide, 100 μM dNTPs, and 5–10 μCi [α-32P]dATP, 3,000 Ci/mmol (1 Ci = 37 GBq). For assays including F-factors, 0.5–1.0 ng standard DNA template was also present as indicated. DyNAoyme polymerase (2.5 units) was added to each mixture. The reaction was started by incubation. For experiments with F-factors, either the cycle program 95°C for 90 s/60°C for 2 min/72°C for 3 min (see Figs. 2, 3, 4) or 95°C for 90 s/60°C for 1 min/72°C for 1 min (see Figs. 5 and 6) was applied for 35 cycles. RC plasmids were treated 95°C for 90 s/60°C for 40 s/72°C for 40 s for 35 cycles. These conditions were chosen empirically by first optimizing the yield and specificity of DNA amplification from plasmid-carrying bacteria with the nicking oligonucleotide plus a second primer.
Figure 2.
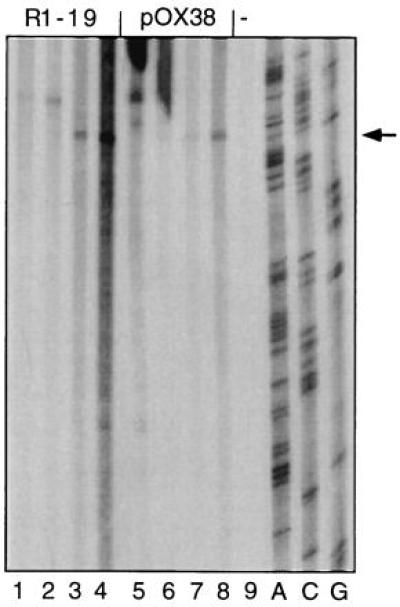
The 3′ end of the oriT-specific reaction product maps to the same nucleotide position in plasmids R1 and F. In two separate experiments, 100, 200, 400, and 800 μl of mating cultures containing either J5[R1-19] (lanes 1–4, respectively); 50, 100, 200, and 400 μl of J5[pOX38-Km] (lanes 5–8, respectively); or 400 μl of J5 without plasmid (lane 9) were centrifuged and the cell pellets treated as described. Autoradiograms of the gels are shown. Arrow indicates reaction products terminated specifically at nic. Correcting for culture density, comparable numbers of cells of both strains were present in the reaction mixtures.
Figure 3.
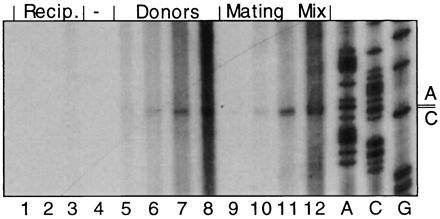
The R1-19 plasmid is nicked at oriT in the absence of recipient cells. Culture medium containing no cells (lane 4); 60, 120, or 240 μl of recipients alone (lanes 1–3, respectively); 60, 120, 240, and 480 μl of J5[R1-19] alone (lanes 5–8); or a mating mixture of J5[R1-19] and a recipient following 15 min of incubation at 37°C (lanes 9–12) was processed for runoff DNA sequencing as described. The number of plasmid-carrying cells in reaction mixtures containing either donor alone or the mating mixture was the same. The position of oriT-terminated reaction products is indicated by the R1 nucleotides C-2040 and A-2041 (numbering as in ref. 21), which complement the nick site within the consensus 3′-TCGTGG↓TGTGG-5′ (3).
Figure 4.
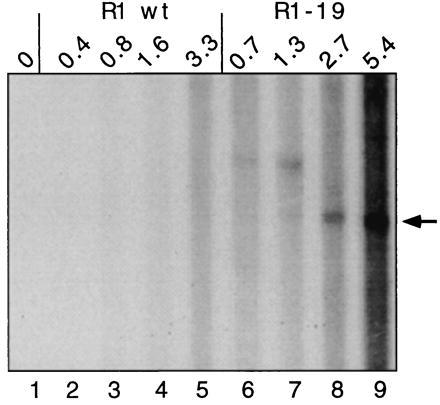
Synthesis of nic-terminated DNA in vitro was observed from R1-19 (finO−) but not the fertility-inhibited R1 plasmid. Medium (lane 1), increasing amounts of cultures containing J5[R1] (lanes 2–5), or J5[R1-19] (lanes 6–9) were prepared for the DNA synthesis assay. Arrow indicates position of nic-specific reaction products. The number of viable cells present in each reaction mixture is indicated above the lanes (cfu × 106).
Figure 5.
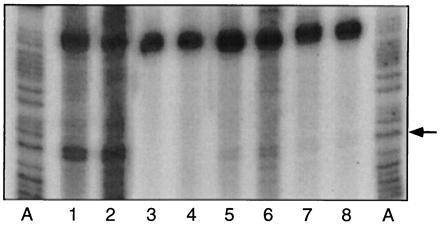
Synthesis of nic-terminated DNA in vitro requires transfer gene expression from the R1 plasmid in vivo. Reaction mixtures containing M1174[R1-16+pGZ119] (lanes 1 and 2), M1164[R1-16+pGZ119] (lanes 3 and 4), M1164[R1-16+pGZarcA] (lanes 5 and 6), or M1164[R1-16+pHP2] (lanes 7 and 8) were subjected to runoff nucleotide synthesis. The numbers of viable cells analyzed in lanes 1–8 were, respectively, 2.3, 4.6, 7.8, 15.6, 8.8, 17.6, 5.6, and 11.2 (× 106 cfu).
Figure 6.
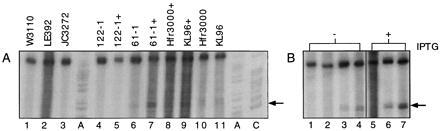
Detection of transfer-independent cleavage activity at the oriTs of Hfr strains requires synthesis of F transfer proteins in vivo. (A) Forty microliters from ONCs of three F(−) strains (lanes 1–3) and four Hfr strains without (lanes 4, 6, 10, and 11) and with (+) the R1-16 plasmid (lanes 5, 7, 8, and 9) were prepared for a runoff nucleotide sequencing assay. (B) Forty and 80 μl of ONCs of 122-1[pGZ119] (lanes 1 and 2) and 122-1[pGZarcA] (lanes 3 and 4), or aliquots of cultures of 122-1[pGZ119] (lane 5) or 122-1[pGZarcA] (lanes 6 and 7), which were diluted first in fresh medium containing 10 mM isopropyl β-d-thiogalactoside and incubated with shaking at 37°C for 2.5 h, were prepared for the nicking assay. The numbers of viable cells analyzed in the reaction mixtures in lanes 1–7 were, respectively, 0.65, 1.3, 30.8, 61.5, 22.1, 0.09, and 0.15 (× 106 cfu). The nic-specific reaction products are indicated by arrows.
DNA products were deproteinized, recovered by ethanol precipitation, dissolved in formamide, and analyzed by electrophoresis at 40 W for 2.5–3.5 h through 6% polyacrylamide (19:1, acrylamide:bisacrylamide) gels (19.5 × 45 × 0.04 cm) containing 7 M urea. The electrophoresis buffer was 89 mM Tris borate (pH 8.3) and 2.5 mM EDTA. Gels were autoradiographed. The autoradiograms were scanned using a Umax PowerLook II system. Figures shown are the scanned images analyzed with photoshop software and printed with an Apple LaserWriter.
Preparation of Polynucleotide Size Markers.
The same primers used in the nicking assay were used to form primed DNA templates with plasmids containing the R1 oriT (pCK217) or the pMV158 dso (pLS1). The conditions for annealing and for the in vitro DNA sequencing reactions in the presence of 5 μCi [α-32P]dATP, 3,000 Ci/mmol were as specified in the Sequenase kit for double-stranded DNA (United States Biochemical).
RESULTS
oriT DNA of Derepressed Plasmids Is Nicked in Vivo in the Absence of Conjugative Transfer.
Increasing amounts of liquid cultures containing mating E. coli, where the conjugative plasmid was either R1-19, which is derepressed for transfer gene expression (see Fig. 2, lanes 1–4), or pOX38-Km, a transfer-proficient derivative of the naturally derepressed F plasmid (see Fig. 2, lanes 5–8) were harvested and used in the runoff DNA synthesis assay as described (Fig. 1). An autoradiogram of the gel indicated that, when sufficient numbers of plasmid-carrying bacteria were present in the reaction mixture, a single band of specifically terminated reaction products was apparent (Fig. 2). The DNA sequence of the size standard and of the reaction products were identical. In accordance with the identity of the DNA sequence of both the R1 and F plasmids between the site of complementation of the primer and the expected nic site (32–34), specifically terminated reaction products corresponding in size to the distance separating the primer from nic were generated from both conjugative plasmids. This places the site of intracellular cleavage at the R1 oriT between position G 2040 and T 2041 (numbering as in ref. 21).
Figure 1.
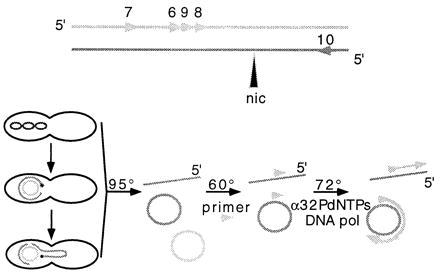
Diagram of the nicking assay. The reaction is a runoff DNA synthesis with a thermostable DNA polymerase using as template ss linear or ss circular molecules, released from lysed bacteria, which have been hybridized to a single primer (triangle) 100–250 bases 3′ to the 5′ end of a suspected nick site. DNA synthesis proceeds (Upper, top strand) until the polymerase encounters an interruption (nic) in the DNA template (nicked substrates) or reaches the limits of its processivity (unnicked molecules). These unspecifically terminated reaction products can be distinguished by size from the much smaller specific termination products using a sequencing gel. The source of the specific DNA cleavage event in vivo could be conjugative transfer of a plasmid, as depicted (Lower). Cell lysis and denaturation of the released double-stranded DNA molecules (95°C), hybridization of one specific complementary DNA primer (60°C), and synthesis on primed DNA templates (72°C) are achieved in three successive incubations of the reaction mixture for 35 cycles in a thermocycler. The reaction products are labeled with [α-32P]dNTP precursors. (Upper) Primers 6–9, complementary to the sequences of the T strand and primer 10, complementary to the retained strand, within the oriT of F-like plasmids.
Specific termination of DNA synthesis at this site within the R1 oriT was not dependent on the oligonucleotide used as primer. DNA synthesis initiated from three additional oligonucleotides (Fig. 1, nos. 7, 8, and 9), which anneal to the transferred strand upstream from the expected nic site, led to site-specific termination at this position (not shown). In agreement with the strand-specificity of the nicking reaction in vivo, synthesis from a primer complementary to the opposite strand (Fig. 1, no. 10) did not terminate at this position (not shown).¶
To determine whether formation of the specific nic-terminated reaction product was dependent on conjugal transfer of the plasmid, samples of separate cultures of plasmid-free E. coli MC1061 and E. coli J5[R1-19] were compared in the nicking assay with aliquots of a mixed culture of these two strains undergoing mating (Fig. 3). No specific reaction product was formed in reaction mixtures without bacteria (lane 4) or in those containing plasmid-free bacteria (lanes 1–3). Reaction products terminated specifically at nic were observed when the culture of mating cells was tested (lanes 9–12), as well as when E. coli J5[R1-19], which was grown in the absence of a recipient strain, was sampled (lanes 5–8). Formation of this unique product was dependent on the presence of plasmid but was independent of conjugal transfer. The specific in vitro DNA reaction products observed here and in all subsequent experiments appear to migrate as two bands that differ in length by one nt. We expect that formation of two specific products was due to the terminal transferase activity of the DNA polymerase and not to the presence of two independent cleavage sites in the plasmid DNA.
Synthesis of nic-Terminated DNA in Vitro Is Dependent on Transfer Gene Expression from the R1 Plasmid in Vivo.
The enzyme that relaxes the transfer origin of F factors, TraI, is plasmid encoded. To ascertain whether formation of the nic-specific in vitro reaction product was dependent on transfer gene expression, separate cultures of E. coli J5 carrying two R1 plasmid derivatives that exhibit different levels of transfer gene expression were compared in the nicking assay (Fig. 4). The wild-type R1 plasmid is subject to fertility inhibition (fin+) and, as a result, transfers 1000-fold less frequently than the fin− R1-19 derivative. Although comparable numbers of plasmid-carrying bacteria were assayed, nic-terminated reaction products were not observed with the fin+ R1 plasmid (lanes 2–5), but were readily detected when reaction mixtures contained at least 2.7 × 106 viable cells carrying the R1-19 plasmid, which produces its transfer proteins constitutively (lanes 6–9).
To be able to monitor the efficiency of the DNA polymerization, as well as the recovery from additional handling of the reaction products, a DNA template was created for use as an internal control. Briefly, the sequence of primer 6, specific for the oriT region of F-like plasmids, was inserted in an unrelated plasmid vector. This DNA was isolated and cleaved once with a restriction endonuclease. When introduced as purified DNA to the reaction mixture of the nicking assay, together with the same oligonucleotide primer, this DNA template yields a second reaction product of defined length (194 nt) that is distinguishable in size from those produced on nicked molecules of the conjugative plasmids (176 nt). Following optimization, 0.5–1 ng of purified standard DNA template was chosen for routine use in the assay (not shown). Presence of the second DNA template in the reaction mixture did not inhibit synthesis from the R1-16 plasmid DNA. However, aberrant products were formed when excessive amounts of the standard DNA were used.
The results shown in Fig. 4 suggested strongly that formation of nic-terminated reaction products in vitro were dependent on transfer gene expression from the plasmid in vivo. Despite the fact that the transfer origins of plasmids R1 and R1-19 are identical, the comparison is still one of two distinct plasmids. Therefore, we sought to corroborate this evidence with another approach and additionally include the internal control in the assay. For this experiment, the same R1 plasmid derivative was now compared in two different host strains that individually support different levels of transfer gene expression from the plasmid. E. coli M1174 and M1164 are isogenic strains that differ by a V203M mutation in the arcA gene of M1164 (P. Silverman, unpublished data). E. coli arcA, also known as sfrA (sex factor regulator A) and fex (F expression) encodes the regulator element of a two-component signal transduction system, which is known to positively regulate sexual expression from F factors (13, 35). Ten-fold lower levels of expression from the main transfer operon promoter of F were found in M1164 sfrA5 than in the wild-type M1174 strain (36). To determine whether the specific in vitro reaction product we observed was dependent on the presence of arcA and thus required sexual expression in vivo, overnight cultures of R1-16-carrying M1174 and M1164 were compared in the nicking assay (Fig. 5, lanes 1–4). Synthetic products terminated at the position of nic were apparent only in reaction mixtures when the R1-16 plasmid was maintained in the wild-type arcA strain. Formation of this product in vitro was complemented by the coresidency in M1164 sfrA5[R1-16] of a second plasmid capable of expressing in trans the E. coli arcA gene (lanes 5 and 6), or the traI gene of plasmid R1 (lanes 7 and 8). Coresidency of the expression vector alone was not responsible for this effect. Overexpression from the LacI-regulated tac promoter was not induced chemically in this experiment. Complementation of the nicking activity mediated by traI in trans was notably weaker than when arcA was present. This difference results from the ability of arcA to complement transfer gene expression to wild-type levels under these conditions (G. Koraimann and E.L.Z., unpublished data), whereas coresidency of a traI-carrying plasmid provides TraI alone. In this case, the reduced abundance of the accessory DNA binding proteins seems to limit the efficiency of the nicking reaction in vivo.
As presence of either the arcA gene or the traI gene of R1 in trans to the R1-16 plasmid in the M1164 strain complemented formation of the nic-specific reaction product in vitro, we conclude that formation of this product was dependent on transfer gene expression in vivo. These results, taken together, established that the nic-specific reaction product was generated from plasmid DNA, which was nicked in vivo at the transfer origin.
The F Transfer Origins in Hfr Strains Are Subject to Transfer-Independent Cleaving-Joining Activity in Vivo.
Hfr donor strains emerge from the stable integration of a transfer-proficient plasmid into the chromosome. This study has shown that nicked T strands of R1 derivatives were readily detectable in E. coli if their transfer proteins were produced constitutively (Fig. 4). Tra gene expression from the F plasmid is naturally derepressed (finO−). Therefore, we wondered whether the chromosomally integrated copies of the F plasmids in Hfr strains would similarly experience transfer-independent cleaving-joining activity at their transfer origins. To investigate this possibility two Hfr strains, Hfr3000 and KL96, and three female (F−) strains were analyzed in the nicking assay using oriT-specific primer 6 for F factors (Fig. 6A). Two additional HfrH strains, 61-1 deoB-serBΔ and 122-1 deoB-arcAΔ, were also examined to assess simultaneously the dependence of the reaction products on gene expression from the F factors in vivo. In vitro DNA synthesis from the standard DNA template was essentially equivalent in all reaction mixtures (upper band). A second band, migrating at the position expected for nic-terminated reaction products from the F plasmid (arrow), was observed in each reaction including an Hfr strain (lanes 6, 10, and 11), except for ΔarcA strain 122-1 (lane 4), and was absent from those containing an F(−) strain (lanes 1–3). When the Hfr strains contained additionally R1-16, a band with the same apparent mobility and increased intensity was observed (lanes 7–9), suggesting that both the R1 and F oriTs served as a DNA template for the in vitro DNA synthesis. A band of this size was not generated from the HfrH ΔarcA strain without (lane 4) or with plasmid R1-16 (lane 5).
If initiation of synthesis of this reaction product took place on the primed origin DNA of F plasmids linked to the bacterial chromosome and, if termination of the in vitro reaction occurred at the 5′ ends of the T strands cleaved in vivo by F-determined relaxase, then it should be possible to complement formation of this DNA product in the 122-1 HfrH ΔarcA strain by complementing positive regulation of gene expression from F. An expression construction, pGZarcA, with the E. coli arcA gene under Ptac control and vector alone, were introduced to 122-1. Reaction products from noninduced and isopropyl β-d-thiogalactoside-induced cultures of these strains were compared in the nicking assay (Fig. 6B). A reaction product corresponding in size to nic-specific termination in vitro was obtained in 122-1 when the arcA gene was provided on a plasmid. Induction of arcA expression resulted in a higher yield of the unique nic product despite a more than 300-fold reduction in the number of cells analyzed under conditions of overexpression. Given this result, we infer that the DNA strand scissions detected in these experiments with Hfr donor strains reflect the cleaving-joining activity of relaxosomes positioned at the oriTs of the integrated F factors.
Identification of the Site of Initiation of Plasmid pMV158 Leading Strand Replication in S. pneumoniae.
To determine whether the cleavage ability exhibited by the purified replication initiator protein, RepB, on dso-containing DNA substrates in vitro reflects properly the performance of the enzyme in initiating DNA replication in vivo, linear replication intermediates of pMV158 derivatives were released from S. pneumoniae and analyzed in the nicking assay (Fig. 7). A unique band corresponding in size to the distance separating the primer and the site of in vitro initiation, nic, was observed when S. pneumoniae carried a pLS1 derivative encoding wild-type replication elements (lane 1) or a copy-up mutant (lane 2), but not when the pLS1 dso alone was present heterologously on a different RC replicon (lane 3). In agreement with the elevated copy number of pLS1cop7 compared with wild-type pLS1, the abundance of the nic-terminated reaction product generated from the pLS1cop7 mutant was greater than that from pLS86 when equivalent numbers of bacteria were analyzed.
Figure 7.
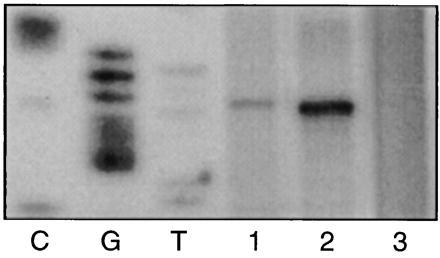
Leading strand replication of pMV158 derivatives initiates at the same site in S. pneumoniae that is cleaved by RepB protein in vitro. Bacteria were cultured to OD600 0.25–0.3. Aliquots containing 4 × 106 cfu of S. pneumoniae strains MP551[pLS86] (lane 1), MP551[pLS1cop7] (lane 2), and MP551[pCGA12] (lane 3) were subjected to runoff DNA synthesis, as described, except that 10 ng of primer pMV158 was present in each reaction mixture.
DISCUSSION
Conjugative plasmids play an important role in genome plasticity by mediating horizontal gene transfer. Dispersal of genetic material occurs via plasmid self-transfer as well as through mobilization of nonconjugative plasmids and chromosomal genes. The identification and characterization of naturally occurring replicons, therefore, will continue to receive attention in bacterial genetics. The localization of Tra, Mob, and replication origins belongs to the basic criteria with which new plasmids and conjugative systems are analyzed. The presented methodology was shown to be well suited to mapping discontinuities in DNA strands generated in vivo, and to locate precisely cleavage sites not previously mapped, such as the nic site of plasmid R1 (Fig. 2). The technique should prove particularly useful for fine mapping in new systems once an origin-containing restriction fragment has been identified. This feature is important, since conventional analyses of an oriT DNA fragment, for example, can reveal several putative origins, any one or more of which may be active in vivo (37). It is an advantage that this procedure is as effective in Gram-positive as in Gram-negative bacteria. In some instances, the characterization of origins from Gram-positive systems must be carried out in surrogate Gram-negative hosts because properties of the natural host preclude isolation of relaxation complexes (38). In this context, it is also significant that the application of this methodology was not limited by low plasmid copy number nor by the size of the DNA substrate. In addition, proposed nick sites within the dso of RC plasmids can now be unambiguously defined. As shown in this study (Fig. 7), the procedure can be used to confirm whether the position of strand scission observed in vitro properly reflects the position of bond breakage catalyzed in vivo.
The formation of DNA–protein relaxation complexes is a general feature of plasmids of E. coli (39), first observed with ColE1 by Clewell and Helinski (40), and subsequently found for a variety of conjugative and mobilizable plasmids (41). Since the initial observation, the issue of whether or not the supercoiled DNA of nucleoprotein relaxation complexes, which is released in a relaxed form following treatment with heat or protein denaturing agents, is actually maintained in an equilibrium of nicking and closing in vivo, or whether cleavage is simply induced by the treatment in vitro, has not been resolved unequivocally. Several lines of evidence exist to support the view that relaxosomes maintain an equilibrium of cleaving and joining activity in vivo. Compelling evidence is available from IncW, IncF, and IncQ plasmids, where enhanced recombination of oriT-containing DNA in vivo has been observed that required additionally the cognate cleaving–joining activity but was independent of conjugative transfer (42–44). Additional evidence stems from the well-characterized in vitro properties of the relaxosomal proteins of the IncP plasmid RP4. The key component of this multienzyme complex, TraI, has been shown to possess cleaving–joining activity, resembling a type I topoisomerase (45). Cleavage of ss oligonucleotides by TraI does not require induction by a protein denaturing agent. A mutant form of TraI (S74A) was also shown to act together with the TraJ protein to convert form I oriT DNA to partially relaxed covalently closed plasmid forms (46). The partial relaxation took place in the absence of denaturing agents, therefore, cleaving–joining of the plasmid DNA must occur continuously and not only when the complex is disrupted. Based on these in vivo and in vitro data, we assume that relaxosomes sustain an equilibrium of nicking and closing of nic DNA in vivo without the stimulus of conjugation. This report has been written based on this premise.
In this study, the specificity of the products of the nicking assay with F factors was demonstrated according to several criteria (Figs. 2, 3, 4, 5). Based on the premise stated above, it follows, therefore, that the transfer protein-dependent formation of the same specific reaction products from the Hfr donor strains (Fig. 6) was due to the cleaving–joining activity exerted by relaxosomes positioned at the oriTs of the integrated fertility factors.
The strength of this approach lies in the ability to observe directly the position and frequency of DNA processing reactions that occur in vivo with substrate and enzyme in their natural context. This technique could, therefore, prove useful for monitoring a variety of DNA transactions producing intermediates with DNA strand discontinuities. This could include initiation, termination, and paused intermediates of DNA replication, as well as intermediates of site-specific recombination, transposition, and perhaps viral integration. Extension of the technique to studies with eukaryotes is feasible but remains to be tested. The next step is to address questions about the concerted mechanisms that affect the replicative and conjugative DNA processing reactions observed here.
Acknowledgments
This work was financed by Fonds zur Förderung der Wissenschaftlichen Forschung Grant 9141, Grant BIO94–1029 from the Comisión Interministerial de Ciencia y Technología, and by the Joint Program between Spain and Austria Grants 13B and 30/96.
ABBREVIATIONS
- Hfr
high frequency recombination
- RC
rolling circle
- ss
single stranded
- dso
double-stranded origin
- cfu
colony forming unit(s)
- ONC
overnight culture
Footnotes
Two types of artefacts were formed routinely in these experiments when too few bacteria were used relative to the amount of primer present in the reaction mixtures (Fig. 2, lanes 1, 2, 5, and 6). The elongated smears were common to several different oligonucleotide primers when used in excess in the assay and may result from primer–primer interactions. The specific band approximately 10 bases longer than the reaction products terminated at nic, which was also present at high primer to cell ratios, was reproducible and unique to the particular primer used (Fig. 2, lanes 1, 2, 5, and 6; Fig. 4, lanes 6 and 7).
References
- 1.Waters V L, Guiney D G. Mol Microbiol. 1993;9:1123–1130. doi: 10.1111/j.1365-2958.1993.tb01242.x. [DOI] [PubMed] [Google Scholar]
- 2.del Solar G, Moscoso M, Espinosa M. Mol Microbiol. 1993;8:789–796. doi: 10.1111/j.1365-2958.1993.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 3.Lanka E, Wilkins B M. Annu Rev Biochem. 1995;64:141–169. doi: 10.1146/annurev.bi.64.070195.001041. [DOI] [PubMed] [Google Scholar]
- 4.Moscoso M, del Solar G, Espinosa M. J Biol Chem. 1995;270:3772–3779. doi: 10.1074/jbc.270.8.3772. [DOI] [PubMed] [Google Scholar]
- 5.Moscoso M, del Solar G, Espinosa M. J Bacteriol. 1995;177:7041–7049. doi: 10.1128/jb.177.24.7041-7049.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matson S W, Morton B S. J Biol Chem. 1991;266:16232–16237. [PubMed] [Google Scholar]
- 7.Reygers U, Wessel R, Müller H, Hoffmann-Berling H. EMBO J. 1991;10:2689–2694. doi: 10.1002/j.1460-2075.1991.tb07812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson W C, Howard M T, Sherman J A, Matson S W. J Biol Chem. 1995;270:28374–28380. [PubMed] [Google Scholar]
- 9.Howard M T, Nelson W C, Matson S W. J Biol Chem. 1995;270:28381–28386. [PubMed] [Google Scholar]
- 10.Pansegrau W, Balzer D, Kruft V, Lurz R, Lanka E. Proc Natl Acad Sci USA. 1990;87:6555–6559. doi: 10.1073/pnas.87.17.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scherzinger E, Lurz R, Otto S, Dobrinski B. Nucleic Acids Res. 1992;20:41–48. doi: 10.1093/nar/20.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avila P, Núnez B, de la Cruz F. J Mol Biol. 1996;261:135–143. doi: 10.1006/jmbi.1996.0447. [DOI] [PubMed] [Google Scholar]
- 13.Beutin L, Achtman M. J Bacteriol. 1979;139:730–737. doi: 10.1128/jb.139.3.730-737.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes W. Cold Spring Harbor Symp Quant Biol. 1953;18:75–93. doi: 10.1101/sqb.1953.018.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Low K B. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Ed. Neidhardt F C, editor. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2402–2405. [Google Scholar]
- 16.Roeder W, Somerville R L. Mol Gen Genet. 1979;176:361–368. doi: 10.1007/BF00333098. [DOI] [PubMed] [Google Scholar]
- 17.Chandler M, Galas D. J Mol Biol. 1983;170:61–91. doi: 10.1016/s0022-2836(83)80227-7. [DOI] [PubMed] [Google Scholar]
- 18.Lessl M, Balzer D, Lurz R, Waters V L, Guiney D G, Lanka E. J Bacteriol. 1992;174:2493–2500. doi: 10.1128/jb.174.8.2493-2500.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drapal N, Sawers G. Mol Microbiol. 1995;16:597–607. doi: 10.1111/j.1365-2958.1995.tb02422.x. [DOI] [PubMed] [Google Scholar]
- 20.Koraimann G, Schroller C, Graus H, Angerer D, Teferle K, Högenauer G. J Bacteriol. 1993;177:4279–4288. doi: 10.1111/j.1365-2958.1993.tb01732.x. [DOI] [PubMed] [Google Scholar]
- 21.Graus H, Hödl A, Wallner P, Högenauer G. Nucleic Acids Res. 1990;18:1046. doi: 10.1093/nar/18.4.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacks S A, López P, Greenberg B, Espinosa M. J Mol Biol. 1986;192:753–765. doi: 10.1016/0022-2836(86)90026-4. [DOI] [PubMed] [Google Scholar]
- 23.Priebe S D, Lacks S A. J Bacteriol. 1989;171:4778–4784. doi: 10.1128/jb.171.9.4778-4784.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Solar G, Moscoso M, Espinosa M. Mol Gen Genet. 1993;237:65–72. doi: 10.1007/BF00282785. [DOI] [PubMed] [Google Scholar]
- 25.Horinouchi S, Weisblum B. J Bacteriol. 1982;150:804–814. doi: 10.1128/jb.150.2.804-814.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López P, Espinosa M, Lacks S A. Mol Gen Genet. 1984;195:402–410. doi: 10.1007/BF00341440. [DOI] [PubMed] [Google Scholar]
- 27.del Solar G, de la Campa A G, Pérez-Martín J, Choli T, Espinosa M. Nucleic Acids Res. 1989;17:2405–2420. doi: 10.1093/nar/17.7.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Díaz A, Lacks S A, López P. Mol Microbiol. 1994;14:773–783. doi: 10.1111/j.1365-2958.1994.tb01314.x. [DOI] [PubMed] [Google Scholar]
- 29.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. pp. 431–433. [Google Scholar]
- 30.Lacks S A. Genetics. 1966;53:207–235. doi: 10.1093/genetics/53.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 32.Everett R, Willetts N. J Mol Biol. 1980;136:129–150. doi: 10.1016/0022-2836(80)90309-5. [DOI] [PubMed] [Google Scholar]
- 33.Thompson R, Taylor L, Kelly K, Everett R, Willetts N. EMBO J. 1984;3:1175–1180. doi: 10.1002/j.1460-2075.1984.tb01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson T L, Centola M B, Deonier R C. J Mol Biol. 1989;207:505–512. doi: 10.1016/0022-2836(89)90460-9. [DOI] [PubMed] [Google Scholar]
- 35.Lerner T J, Zinder N D. J Bacteriol. 1979;137:1063–1065. doi: 10.1128/jb.137.2.1063-1065.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silverman P M, Wickersham E, Harris R. J Mol Biol. 1991;218:119–128. doi: 10.1016/0022-2836(91)90878-a. [DOI] [PubMed] [Google Scholar]
- 37.Jaworski D D, Clewell D B. J Bacteriol. 1995;177:6644–6651. doi: 10.1128/jb.177.22.6644-6651.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang A, Macrina F L. J Bacteriol. 1995;177:4199–4206. doi: 10.1128/jb.177.15.4199-4206.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helinski D R. Annu Rev Microbiol. 1973;27:437–470. doi: 10.1146/annurev.mi.27.100173.002253. [DOI] [PubMed] [Google Scholar]
- 40.Clewell D B, Helinski D R. Proc Natl Acad Sci USA. 1969;62:1159–1166. doi: 10.1073/pnas.62.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willetts N, Wilkins B. Microbiol Rev. 1984;48:24–41. doi: 10.1128/mr.48.1.24-41.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llosa M, Bolland S, Grandoso G, de la Cruz F. J Bacteriol. 1994;176:3210–3217. doi: 10.1128/jb.176.11.3210-3217.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter J R, Porter R D. J Bacteriol. 1991;173:1027–1034. doi: 10.1128/jb.173.3.1027-1034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer R. J Bacteriol. 1989;171:799–806. doi: 10.1128/jb.171.2.799-806.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pansegrau W, Schröder W, Lanka E. Proc Natl Acad Sci USA. 1993;90:2925–2929. doi: 10.1073/pnas.90.7.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pansegrau W, Schröder W, Lanka E. J Biol Chem. 1994;269:2782–2789. [PubMed] [Google Scholar]