

Abstract
TrmA catalyzes S-adenosylmethionine (AdoMet)-dependent methylation of U54 in most tRNAs. We solved the structure of the Escherichia coli 5-methyluridine (m5U) 54 tRNA methyltransferase (MTase) TrmA in a covalent complex with a 19-nt T arm analog to 2.4-Å resolution. Mutation of the TrmA catalytic base Glu-358 to Gln arrested catalysis and allowed isolation of the covalent TrmA–RNA complex for crystallization. The protein–RNA interface includes 6 nt of the T loop and two proximal base pairs of the stem. U54 is flipped out of the loop into the active site. A58 occupies the space of the everted U54 and is part of a collinear base stack G53–A58–G57–C56–U55. The RNA fold is different from T loop conformations in unbound tRNA or T arm analogs, but nearly identical to the fold of the RNA loop bound at the active site of the m5U MTase RumA. In both enzymes, this consensus fold presents the target U and the following two bases to a conserved binding groove on the protein. Outside of this fold, the RumA and TrmA substrates have completely different structures and protein interfaces. Loop residues other than the target U54 make more than half of their hydrogen bonds to the protein via sugar-phosphate moieties, accounting, in part, for the broad consensus sequence for TrmA substrates.
Keywords: RNA modification, substrate specificity, tRNA, x-ray crystallography
Posttranscriptional modification of noncoding RNA occurs in all kingdoms of life. These modifications can alter the physical and/or chemical properties of the nucleotides. Several such modifications are clustered at functionally important sites on tRNA and rRNA and have been shown to contribute to the fidelity and efficiency of mRNA translation (1–3).
S-Adenosylmethionine (AdoMet)-dependent methylation of uridine to 5-methyluridine (m5U) occurs at two sites in the Escherichia coli ribosome and at a single site in E. coli tRNAs. A different enzyme is responsible for modification in each of these three environments. TrmA (formerly known as RUMT) modifies U54 in the T arms of most tRNAs (4). RumA and RumB modify U1939 (5) and U747 (6), respectively, of 23S RNA. TrmA, RumA, and RumB are two- or three-domain proteins that have little overall sequence homology, but contain six conserved sequence motifs that are indicative of a class I AdoMet-dependent MTase fold (7). The chemical mechanism of m5U54 methylation involves Michael addition of a catalytic cysteine to C6 of the uridine (Fig. 1A) (4).
Fig. 1.

Mechanism and substrates of TrmA. (A) The proposed catalytic mechanism of RNA m5U Mtases (4). (B) Consensus sequence of the T arm of tRNA derived from the sequences of E. coli tRNAs known to be methylated (10). Blue indicates conserved and yellow indicates nonconserved bases in the T arm. The target uridine is red. The sequence of the T arm used in this study is indicated next to the base numbering. Circles indicate the additional base pair added to the original T loop sequence. 2′-Se-Me-U is indicated as Use. (C) Side view of TrmA–T arm structure.
The first reported crystal structure of an m5U MTase-RNA complex was that of a ternary complex formed with RumA, AdoMet, and a 37-nt fragment of 23S RNA, (bases 1932–1968) (8). The C5-hydrogen of the target, U1939, in this fragment had been substituted with fluorine, which stalled the methylation reaction at the proton abstraction step, forming a stable covalent complex (presumed to be an analog to intermediate 3 in Fig. 1A). Based on the structure, the invariant Glu-424 was proposed to be the general base in the reaction, and this hypothesis was confirmed by mutagenesis (8). The RNA fragment in the RumA ternary complex binds at the interfaces between three structural domains, an N-terminal OB-fold, a central α-β domain, and the C-terminal catalytic domain, which has the predicted AdoMet MTase fold. The 5′ end of the RNA fragment adopts an intricate conformation that is complementary in shape to the RNA-binding groove formed by the central and catalytic domains but very different from the corresponding nucleotides of the ribosome.
Whereas RumA modifies a single RNA base in the E. coli ribosome, TrmA modifies U54 in the T arms of almost all E. coli tRNAs (9). Systematic mutation of bases surrounding U54 defines a very broad consensus sequence for TrmA substrates (Fig. 1B) (10). Small stem loops with the same sequences as the tRNA T arms are also excellent TrmA substrates with kcat/Kms only ≈10-fold less than those of intact tRNAs (11). To understand the basis for TrmA specificity at the level of atomic structure, we determined the crystal structure of a TrmA–RNA complex. The crystal structure of this complex allows rationalization of the biochemical data on substrate selectivity and suggests mechanisms for substrate binding.
Results and Discussion
The E358Q TrmA–RNA Complex: An Analog of Intermediate 3.
We have solved the structure of a TrmA–RNA complex to 2.43 Å resolution. The TrmA in the complex is the mutant enzyme, E358Q. This residue is equivalent to RumA Glu-424, which is the general base required for the proton abstraction from intermediate 3 (Fig. 1A). Covalent complexes of E358Q TrmA with candidate stem loops were formed by incubation with AdoMet. The RNA substrate for crystallization was designed by tuning the stem length of a 17-nt stem loop with the same sequence as the T arm of tRNAPhe. The RNA fragment that gave the best-ordered crystals was a 19-mer in which the stem of the starting 17-mer was extended by 1 bp (Fig. 1B).
In the structure, C6 of the target uridine is covalently attached to the catalytic sulfhydryl of Cys-324 and a methyl group has been added in trans fashion to C5. The asymmetric unit contains two equivalent TrmA–RNA complexes packed such that their RNA stems are stacked end to end but there are no contacts between the two protein molecules themselves. It is unlikely that the RNA packing interactions affect the binding mode of the RNA stem loop to TrmA because the RNA had proceeded through the first steps of the enzyme reaction. All of the nucleotides in the RNA–protein interface are within the substructure comprising the T loop and the 2 bp at the base of loop, which corresponds to the 11-nt minimal substrate of TrmA (11) (Fig. 1C). The fact that the reaction has stalled after the addition of the methyl group to C5 on the target uridine but before resolution of the covalent bond between uridine C5 and the sulfhydryl of Cys-324 confirms the hypothesis that Glu-358 is the general base in TrmA and that this complex is equivalent to intermediate 3 in Fig. 1A.
Although the methyl transferred to U54 from AdoMet is clearly visible, there is no continuous density for the demethylated AdoMet product, S-adenosylhomocysteine (AdoHcy) in the density maps, suggesting it is disordered or bound at low occupancy. Adding additional AdoMet to the Trma–RNA crystallization solution, as was done for RumA, prevented crystallization. In many AdoMet-dependent MTases, the adenine ring of the cofactor interdigitates between two aromatic side chains. In RumA an RNA base from the substrate flips into the AdoMet site and substitutes for one of these residues, thus forming part of the AdoMet binding site (8). Tyr-218 in TrmA could pack against one side of the adenine ring of the bound cofactor. However, there are no aromatic residues in the TrmA–RNA complex that could pack against the other side of the ring and no substrate bases that could flip into the AdoMet-binding site without disrupting the RNA stem structure. The AdoMet site is exposed to solvent even when whole tRNA is docked to the protein, which may explain why AdoHcy is not bound with high occupancy in the crystal structure.
TrmA and RumA Have the Same Folds but Only the Catalytic Core Structures Are Highly Conserved.
TrmA has two domains with the same folds and topologies as the central RNA binding and catalytic domains of RumA (7) (Fig. 2). As in RumA, the AdoMet MTase fold of the TrmA catalytic domain is extended by the so-called cross-over segment, an α-helix and β-strand formed by a sequence immediately preceding the RNA binding domain (α1 and β1, purple segment, in Figs. 2 and 3) (7). This covalent connection between RNA binding and catalytic domains may help stabilize the RNA binding groove, which spans the two domains. TrmA has a 12-residue insert in the loop between two of the β-strands in the catalytic domain (β13 and β14), and the loop adopts a different conformation than in RumA. Although the AdoMet–MTase fold of the TrmA C-terminal domain was expected, the close structural similarity between the RNA binding domains of TrmA and RumA is more surprising because the sequence conservation within this domain is only 13% and the RNA binding domain of RumA hosts a [Fe4S4] cluster, which is not present in TrmA.
Fig. 2.

Sequence alignment of TrmA and RumA. Conserved residues are white on a red background, and conservative substitutions are red on a white background. The secondary structures of TrmA and RumA are shown above and below their sequences, respectively (coil = helix, arrow = β-strand, T = turn) and are colored purple, yellow, and blue for the cross-over segment, RNA binding, and catalytic domains, respectively. Conserved motifs are marked with blue dashes under the sequence alignment.
Fig. 3.
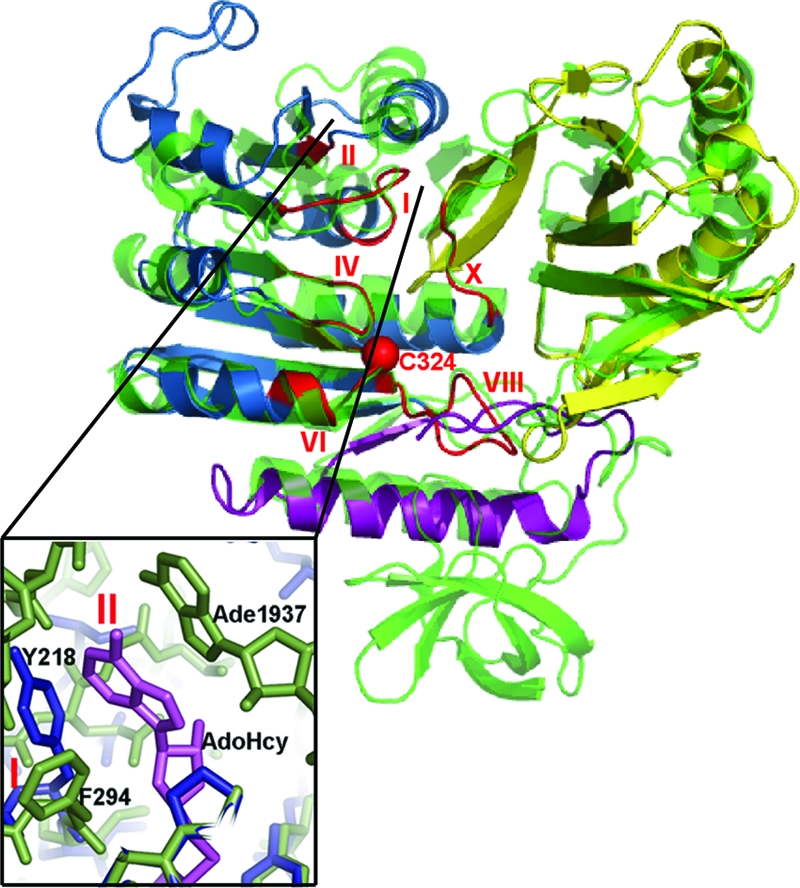
Cartoon of superimposed TrmA and RumA structures. RumA is the background structure (green). The TrmA color scheme is as in Fig. 2. The m5U MTase motifs are red. The Cα of the catalytic sulfhydryl is shown as a red sphere. (Inset) A close-up of the AdoMet binding site, with AdoHcy from RumA complex in violet.
Except for the loop between β13 and β14 in TrmA (Fig. 2), all secondary structure elements in the RNA binding and catalytic domains of TrmA and RumA are conserved, but their relative positions are shifted considerably. We identified the conserved structural core of the enzymes (the largest set of spatially contiguous residues whose Cα positions are in the same relative positions ± 0.5 Å in the two complexes) by a difference-distance method (12). The core comprises 31 residues that cluster around the protein–RNA interface, particularly around the binding site for the target uridine; therefore, we refer to this core as the “catalytic core.” It contains residues from both domains, including residues from four of the six m5U MTase sequence motifs (motifs X, IV, VI, and XIII in Figs. 2 and 3) (7). (There are small structural differences in motifs I and II because of the absence of AdoMet in the TrmA complex; Fig. 3 Inset.) The Cαs of the catalytic cores of the TrmA and RumA RNA complexes align with an rmsd of 0.45 Å, but outside of the core the structures diverge: after alignment of the cores the rmsd for all Cαs is 4.1 Å.
A Consensus RNA Fold: The Fold of the Bound T Loop Resembles the Fold of the Bound RumA Substrate in the Catalytic Core.
As seen for RumA, the TrmA substrate refolds as it binds to the active site (Fig. 4). The T loop is bound to the catalytic site of TrmA in an unstrained conformation that is radically different from both the conformation of the T loop of tRNAPhe (13) and the conformation in solution of 17-nt T arm analogs (14, 15). In mature tRNA, U54 is buried inside the loop, where it forms a reverse-Hoogsteen base pair with A58. In the TrmA–RNA structure, the tip of the loop is folded away from the stem and U54 is flipped out of the loop into the enzyme catalytic site. The bases of G57 and A58 are rotated into the loop to form a nonsequential collinear stack with the bases of G53, U55 and U56 (Figs. 4C and 5A). The base of A58 in this new conformation occupies the space vacated by the everted U54 and the A58 amino group donates a hydrogen bond to the 3′ hydroxyl of G53. C60 and U59 are excluded from the stack of bases and form a separate substructure of the loop that is stabilized in part by an intra-RNA hydrogen bond from C60 4-NH2 to a U59 phosphate oxygen. The C59 and C60 bases are folded into the major groove of the stem, much as they are in the unbound T loop.
Fig. 4.

Substrate binding in TrmA and RumA. (A) Comparison of loop conformations of the T-arm analog from the TrmA–RNA structure (pink and red), with the T loop of unbound tRNAPhe (orange) (13). U54 is shown in stick representation. (B) Comparison of the 5′-loop conformation of the 37-mer in the RumA–RNA cocrystal structure (blue) with the conformation this fragment has in the mature ribosome (orange) (8). U1939 is shown as sticks. (C) Overlap of the RNA loops bound to the active sites of TrmA (pink, bases 53–58) and RumA (blue, bases 1938–1942). (D) (Left) Surface representation of TrmA bound to its 19-mer substrate (magenta), with the RumA 37-mer substrate superposed in the yellow cartoon. (Right) Surface representation of RumA bound to its substrate (yellow), with the TrmA substrate superposed (magenta). RNA binding, catalytic, and OB-fold domains are colored blue, gray, and green, respectively.
Fig. 5.

RNA–protein interactions. (A) Diagram of the secondary structure and interactions of the bound RNA. (B) Stereoview of a stick representation of T-loop bases U54, U55, C56, and G57 bound to the TrmA active site. Putative hydrogen bonds between the target U and the protein are shown with red dotted lines, and hydrogen bonds between the other bases and the protein are shown with green dotted lines. (C) Close-up of the interactions between the target uridine and the amino acid side chains overlaid with Fo − Fc map (4.5 σ) calculated with U54 and surrounding side chains omitted.
The strategy of using an RNA base rather than an amino acid to stabilize the RNA core after base flipping is identical to the strategy used in the RumA–AdoHcy–RNA ternary complex (8). Further, the nonsequential stack (53–58-57–56-55) is surprisingly similar to the stack formed by the RumA substrate bases 1938–1942-1941–1940 (Fig. 4C). The refolding of the TrmA and RumA loops into the stacked arrangement is important for substrate selectivity because it exposes the target uridine and the following two bases for hydrogen bonding with protein side chains and backbone groups in the active-site cavity. We propose that the main requirement for substrates of TrmA and its homologues is the ability to adopt this consensus fold at the substrate-binding site.
Contacts with the Consensus Fold Are Highly Conserved Between TrmA and RumA.
Despite low overall sequence homology in the RNA binding domain, TrmA and RumA display conserved interactions, not mediated by solvent or ions, between protein residues and the consensus RNA fold. In both enzymes the target uridine and the following three nucleotides bind to a deep groove on the protein surface where they form several hydrogen bonds with conserved residues (Fig. 5). Van der Waals, aromatic stacking, and hydrogen-bond interactions stabilize the target base, U54, in a productive orientation. All of these interactions are conserved in RumA (8). There is an edge-to-face aromatic interaction between one side of the uracil ring and Phe-188 (Phe-263/RumA), and there are van der Waals contacts between the opposite side of the uracil ring and Phe-351 (Phe-417/RumA). The guanidinium group of Arg-302 (Arg-366/RumA) forms hydrogen bonds to O2′ and O3′ of U54. The U54 base is hydrogen-bonded to the side chains of three conserved residues with likely roles in catalysis: Gln-190 (Gln-265/RumA), Asp-299 (Asp-363/RumA), and Gln-358 (Glu-358 in wild-type TrmA, Glu-424/RumA) (8, 16).
Most of the interactions between TrmA and nucleotides 55–58 also have conserved counterparts in the RumA substrate complex. The guanidinium group of Arg-45 (Arg-128/RumA) stacks on the pyrimidine ring of U55 and donates hydrogen bonds to its sugar and phosphate moieties. Conserved residues Arg-47 (Arg-130/RumA) and His-356 (His-422/RumA) donate hydrogen bonds to phosphate or ribose moieties in the loop. Amino acids Glu-49 (Arg-132/RumA) and Phe-64 (Arg-149/RumA) are not conserved, but these residues make side-chain and backbone hydrogen bonds with the substrate loop that are homologous to hydrogen bonds in RumA. Only a few of the interactions do not have counterparts in the RumA complex. The conserved protein interactions are possible only when the RNA at the active site is folded into the observed consensus conformation; the interactions are likely involved in guiding the refolding of the consensus fold.
Except for the nucleotides in the consensus fold (nucleotides 53–58 in TrmA), the protein-bound substrates of TrmA and RumA have completely different structures, each of which is complementary in shape to the surface topology of its respective RNA-binding domain (Fig. 4D). The TrmA–RNA “nonconsensus” interface involves only the last two bases of the RNA stem together with nucleotides U59 and C60, which fold into the major groove of the stem. The first four base pairs of the stem of the 19-mer do not interact with TrmA and thus are not determinants in recognition or catalysis. A key surface feature for recognition of the stem is a groove formed by residues 155–158 at the end of the β-strand pair β7 and β8. Residues 155–158 form a “clip” that locks the RNA stem loop to the protein catalytic core.
Proposed TrmA Interactions with tRNA U17, G18 and G19.
We manually docked tRNAPhe [Protein Data Bank IC code 1EHZ (13)] to TrmA by aligning its T stem with the stem of the RNA in the TrmA–RNA structure. In this model, TrmA makes contacts with three D loop nucleotides (U17, G18, and G19) that interact closely with the T loop. Refolding the T loop into the conformation seen in the crystal structure and adjusting side-chain conformations can alleviate the few clashes between tRNA and protein. Thus major conformational changes to tRNA would not be required for binding to TrmA.
The side-chain clashes, Phe-64 and Ile-62 with C56 and Arg-51 with G57 and G18, may serve to destabilize the D loop–T loop interface and help drive the refolding of the T loop into the catalytically productive conformation (Fig. 6). In mature tRNA, ψ55 forms a base pair with G18, and C56 forms a base pair with G19. G57 intercalates between G18 and G19, and the G18 base is intercalated between G57 and A58, forming the base stack A58–G18–G57–G19 (Fig. 6). These interactions must be broken for ψ55, C56, and G57 to form the collinear base stack seen in the crystal structure, and tRNA mutants that can not form the ψ55–G18 or C56–G19 base pairs have ≈10-fold higher kcat/Kms than wild-type tRNA (4).
Fig. 6.

Crossed-eyes stereoview of D loop–T loop interface of a TrmA–tRNA model made by aligning the tRNA T stem with the 19-mer stem in the crystal structure. Protein residues are blue, and tRNA bases are red, except for U17, G18, and G19, which are yellow.
There are several favorable interactions between protein side chains and D-loop nucleotides in the TrmA–tRNA docked complex, which might account for the 6-fold lower Km for TrmA methylation of tRNA compared with methylation of a 17-mer T arm analog (11). Arg-51 is well positioned to intercalate between G18 and G19 after refolding of the T loop, thus substituting for G57 in the interface base stack. His-54, Phe-106, and Trp-53 make hydrophobic or aromatic stacking interactions with U17, G18, and G19, respectively. The His-125 imidazole ring is closely packed against the G18 phosphate. The protein residues involved in these interactions may contribute to TrmA substrate selectivity and none of them is conserved in RumA, whose substrate does not contain a component analogous to the D-loop.
Role of T Loop Bases in Substrate Selectivity.
The seven-base T loops in tRNA substrates of TrmA have three bases besides U54 that are completely conserved (U55, C56, and A58), and two that are semiconserved as Pu57 and Py60 (Fig. 1B) (10). Yet Gu et al. (10) showed that each of these positions tolerate substitutions that are not found in any natural tRNAs. Catalytic efficiency was sensitive to the mutations to various degrees but only the C56G substitution abolished activity. This promiscuity can be examined in light of the structure of the TrmA–RNA interface.
T-loop mutations introduced in a 17-mer T arm analog have a somewhat larger impact on activity than the equivalent mutations in tRNAPhe. Although the proportional decreases from wild-type activity are generally comparable (within a factor of 5 of each other) for the two types of substrates, several mutations that are tolerated in tRNA have no detectable activity in the T-arm analogs. The apparent differences in sensitivity may be a consequence of the different binding interfaces for the two substrates. Our modeled TrmA–tRNAPhe complex shows that TrmA may make several favorable contacts with D-loop residues that would not be present in complexes with T-arm analogs. The D-loop interactions with the protein could anchor the tRNA to the active site, making protein hydrogen bonds with T-loop bases less critical for binding and orientation. Another factor could be that the conformations and mobilities of the T loop in tRNA and the T loops of 17-mer T-arm analogs are different (13–15), thus mutations may affect ground-state energies differently.
We used computer graphics to manually replace T-loop bases in our TrmA–RNA structure with each of the three other canonical RNA bases to see how they could be accommodated in the RNA binding site [supporting information (SI) Fig. S1]. Each modeled substitution that conserves favorable hydrogen bonds with the protein, or that conserves the intra-RNA hydrogen bonds seen in the refolded RNA, is an efficient substrate (10). Reduced levels of activity are seen with substitutions that make less favorable hydrogen bonds or that create steric clashes with the protein. Modeling the single totally inactive substrate, C56G, shows that the guanine 2-NH2 would clash with the backbone of Ile-63 when the T loop is in the conformation observed in the structure.
Decreasing the size of the T loop by deletion of U59 or increasing the size of the T loop from 7 to 8 nt by insertion of U between 60 and 61 results in T-arm analogs that are not methylated by TrmA (10). Thus the seven-base size of the T loop appears to be necessary for binding and catalysis. Substitution of a T-arm analog with m1ψ55 or m1m3ψ55 also abrogates activity (17). Methylated ψ55 could not participate in base stacking seen in the consensus fold of the T loop because the methyl groups are out of the plane of the ψ55 base.
TrmA interacts with the Pu52·Py62 and G53·C61 base pairs only through their sugar and phosphate backbones. The lack of interactions with their bases is in agreement with the published mutational and biochemical data indicating that although a stem-loop structure is required for catalysis, the base composition of the stem is not important (10). The data suggest the stem is important mainly for maintaining the T-loop conformation and orienting the T loop on the TrmA surface.
In general, the only substitutions that completely abolish TrmA activity in both tRNAPhe and T-arm analogs are those that sterically hinder the T loop from binding to the active site with the fold seen in the crystal structure. These results strongly support the contention that the primary requirement for catalysis is the ability to adopt the consensus fold.
Conclusion
tRNA U54 methylation by TrmA requires refolding of the T loop into a conformation that features a nonsequential collinear stack of bases, similar to that formed by the RNA bound to the active site of the m5U MTase RumA (8). In both enzymes, refolding of a target-containing loop segment into this base-stacked conformation displays a U–U–C sequence (where the first U is the target) to the active-site cavity. The U–U–C interactions with the protein are highly conserved between the two enzymes. We propose that the interactions of the three bases with active-site residues and the requirement for bases that can stabilize the consensus base-stacked fold via intra-RNA contacts are common determinants of sequence specificity for TrmA and RumA. The disparate determinants of specificity, which allow RumA and TrmA to uniquely recognize their own substrates, are the highly variable surface features of the RNA binding domain that form the interface to nucleotides outside of the consensus fold. At least for TrmA, refolding of the T loop likely occurs by an induced-fit mechanism: the ensemble of NMR structures of isolated T-arm analogs does not include any conformations resembling the conformation seen in the crystal structure (14, 15). Further experiments are required to determine whether any particular protein residues have a major role in refolding.
Materials and Methods
Preparation and Crystallization of TrmA–RNA Complex.
The TrmA gene was cloned into pET28b (Novagen) with a cleavable N-terminal His6 tag and mutated to TrmA E358Q by using the QuikChange kit (Stratagene). Cell pellet was suspended in buffer A containing 50 mM potassium phosphate (pH 7.5), 300 mM NaCl, 2 mM β-mercaptoethanol, 10% glycerol, and protease inhibitor mixture Complete-EDTA-free (Roche Diagnostics). After lysis by an Emulsiflex-C5 homogenizer (Avestin), the His6 tag protein was purified by using a Talon column (Clontech). The His tag was cleaved by thrombin (Novagen), and thrombin was removed by passage over a benzamidine column (GE Healthcare Bio-Sciences). Protein was dialyzed into a final solution consisting of 50 mM NaCl, 2 mM DTT, and 20 mM Tris·HCl, pH 7.0.
Native and 2′-Se-Me-U (18) modified T-arm analogs were purchased from NedKen. Enzyme–RNA covalent intermediates were formed by incubating 75 μM E358Q TrmA with equimolar RNA in 50 mM Tris·HCl (pH 7.0) containing 1 mM MgCl2, 5 mM DTT, and 1.5 mM AdoMet (Sigma) at room temperature for 1 h. Enzyme–RNA complexes were purified with a HiTrap Q column (Amersham Pharmacia) as described (8).
Crystals were grown by using the hanging drop vapor diffusion method. A solution containing 6 mg/ml of purified complex was mixed with an equal volume of well buffer [100 mM sodium cacodylate (pH 7.4), 200 mM sodium sulfate, and 22% PEG-8000]. Crystals belonged to space group C2 with two complexes in the asymmetric unit, and a = 184.4 Å, b = 70.1 Å, c = 107.9 Å, and β = 120.9° (native crystals) or a = 182.4 Å, b = 63.8 Å, c = 108.4 Å, and β = 120.5° (Se-labeled crystals).
Data Collection and Structure Determination.
Crystals were transferred to the well buffer solution containing 20% (vol/vol) ethylene glycol shortly before immersion in liquid nitrogen. Diffraction data from crystals of native and Se-labeled complexes were collected to 2.4 and 2.9 Å, respectively, at −170°C on a Quantum 315r image plate at the Advance Light Source (Lawrence Berkeley Laboratory, Berkeley, CA). Data were processed and scaled by using the HKL-2000 program suite (19) (Table 1).
Table 1.
Statistics for data collection and refinement
| Statistic | Native | Se-RNA |
|
|---|---|---|---|
| Average* | Remote | ||
| Data collection statistics | |||
| Resolution (last shell), Å | 92.46–2.43 (2.56–2.43) | 40.6–2.93 (4.0–2.93) | |
| Wavelength, Å | 1.11 | 0.9796 | 0.9569 |
| No. of reflections | 42,023 | 23,277 | 23,282 |
| Redundancy (last shell) | 2.2 (2.0) | 7.0 (6.5) | 6.9 (6.5) |
| Completeness (last shell), % | 94.1 (83.7) | 100.0 (99.3) | 100.0 (99.4) |
| Rmerge (last shell) | 0.078 (0.27) | 0.115 (1.15) | 0.120 (1.21) |
| I/σ (last shell) | 5.7 (1.7) | 12.2 (1.5) | 11.7 (1.4) |
| Refinement statistics | |||
| Resolution, Å | 35.0–2.43 | ||
| Reflections in working set | 38,764 | ||
| Rcrystal (last shell), % | 23.2 (35.4) | ||
| Rfree (last shell), % | 28.9 (41.9) | ||
| rmsd bonds, Å | 0.013 | ||
| rmsd angels, ° | 1.61 | ||
| Average B factor, Å2 | |||
| Protein | 58.3 | ||
| RNA | 51.5 | ||
| Water atoms | 294 | ||
| Ramachandran distribution, % | |||
| Most favorable | 88.7 | ||
| Allowed | 11.0 | ||
| Generously allowed | 0.3 | ||
*Average of inflection and peak wavelengths
Phases were determined by multiple-wavelength anomalous dispersion (MAD) methods by using SOLVE (20) and improved by density modification in DM (21). An initial model built to MAD-phased maps was positioned in the native crystal unit cell and refined by manual building using Coot (22) alternated with positional and isotropic B-factor refinement by using REFMAC5 (23) (Table 1). Tight noncrystallographic symmetry (NCS) restraints (σ = 0.05 Å for NCS-related atoms) were used throughout refinement for all but six of the residues.
Sequence alignment was done with CLUSTAL W (24) followed by manual editing. Fig. 2A was made by using ESPript (25). Figs. 1C, 2B, 3, 4B, and 4C were made with PyMOL (DeLano Scientific).
Supplementary Material
Acknowledgments.
We thank Pat Greene for critical reading and editing of the manuscript and Tom James for providing his coordinates for the ensemble of T-stem loop NMR structures. This work was supported by National Institutes of Health Grant GM51232.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3BT7).
This article contains supporting information online at www.pnas.org/cgi/content/full/0802247105/DCSupplemental.
References
- 1.Agris PF. Decoding the genome: A modified view. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore PB, Steitz TA. The involvement of RNA in ribosome function. Nature. 2002;418:229–235. doi: 10.1038/418229a. [DOI] [PubMed] [Google Scholar]
- 3.Ofengand J. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett. 2002;514:17–25. doi: 10.1016/s0014-5793(02)02305-0. [DOI] [PubMed] [Google Scholar]
- 4.Kealey JT, Gu X, Santi DV. Enzymatic mechanism of tRNA m5U54 methyltransferase. Biochimie. 1994;76:1133–1142. doi: 10.1016/0300-9084(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 5.Agarwalla S, Kealey JT, Santi DV, Stroud RM. Characterization of the 23S ribosomal RNA m5U1939 methyltransferase from Escherichia coli. J Biol Chem. 2002;277:8835–8840. doi: 10.1074/jbc.M111825200. [DOI] [PubMed] [Google Scholar]
- 6.Madsen CT, Mengel-Jorgensen J, Kirpekar F, Douthwaite S. Identifying the methyltransferases for m5U747 and m5U1939 in 23S rRNA using MALDI mass spectrometry. Nucleic Acids Res. 2003;31:4738–4746. doi: 10.1093/nar/gkg657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee TT, Agarwalla S, Stroud RM. Crystal structure of RumA, an iron-sulfur cluster containing E. coli ribosomal RNA 5-methyluridine methyltransferase. Structure (London) 2004;12:397–407. doi: 10.1016/j.str.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Lee TT, Agarwalla S, Stroud RM. A unique RNA fold in the RumA–RNA–cofactor ternary complex contributes to substrate selectivity and enzymatic function. Cell. 2005;120:599–611. doi: 10.1016/j.cell.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 9.Sprinzl M, Vassilenko KS. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–D140. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu X, Ivanetich KM, Santi DV. Recognition of the T-arm of tRNA by tRNA m5U54-methyltransferase is not sequence specific. Biochemistry. 1996;35:11652–11659. doi: 10.1021/bi9612125. [DOI] [PubMed] [Google Scholar]
- 11.Gu XR, Santi DV. The T-arm of tRNA is a substrate for tRNA m5U54-methyltransferase. Biochemistry. 1991;30:2999–3002. doi: 10.1021/bi00226a003. [DOI] [PubMed] [Google Scholar]
- 12.Montfort WR, et al. Structure, multiple site binding, and segmental accommodation in thymidylate synthase on binding dUMP and an antifolate. Biochemistry. 1990;29:6964–6977. doi: 10.1021/bi00482a004. [DOI] [PubMed] [Google Scholar]
- 13.Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93-Å resolution: A classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshlap KM, Guenther R, Sochacka E, Malkiewicz A, Agris PF. A distinctive RNA fold: The solution structure of an analogue of the yeast tRNAPhe T Psi C domain. Biochemistry. 1999;38:8647–8656. doi: 10.1021/bi990118w. [DOI] [PubMed] [Google Scholar]
- 15.Schmitz U, Donati A, James TL, Ulyanov NB, Yao L. Small structural ensembles for a 17-nucleotide mimic of the tRNA T psi C-loop via fitting dipolar relaxation rates with the quadratic programming algorithm. Biopolymers. 1998;46:329–342. doi: 10.1002/(SICI)1097-0282(19981015)46:5<329::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Urbonavicius J, Jager G, Bjork GR. Amino acid residues of the Escherichia coli tRNA(m5U54)methyltransferase (TrmA) critical for stability, covalent binding of tRNA, and enzymatic activity. Nucleic Acids Res. 2007;35:3297–3305. doi: 10.1093/nar/gkm205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengupta R, et al. Modified constructs of the tRNA TPsiC domain to probe substrate conformational requirements of m1A58 and m5U54 tRNA methyltransferases. Nucleic Acids Res. 2000;28:1374–1380. doi: 10.1093/nar/28.6.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du Q, et al. Internal derivatization of oligonucleotides with selenium for x-ray crystallography using MAD. J Am Chem Soc. 2002;124:24–25. doi: 10.1021/ja0171097. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowtan K. Joint CCP4 ESF-EACBM Newsl Protein Crystallogr. 1994;31:34–38. [Google Scholar]
- 22.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.