

Abstract
Reduced B cell numbers and a mutation in Btk are considered sufficient to make the diagnosis of X-linked agammaglobulinaemia. In the process of conducting family studies, we identified a 58-year-old healthy man with an amino acid substitution, Y418H, in the adenosine-5′-triphosphate binding site of Btk. Immunofluorescence studies showed that this man had 0·85% CD19+ B cells (normal 4–18%) in the peripheral circulation and his monocytes were positive for Btk. He had borderline low serum immunoglobulins but normal titres to tetanus toxoid and multiple pneumococcal serotypes. To determine the functional consequences of the amino acid substitution, a Btk– chicken B cell line, DT40, was transfected with expression vectors producing wild-type Btk or Y418H Btk. The transfected cells were stimulated with anti-IgM and calcium flux and inositol triphosphate (IP3) production were measured. Cells bearing the mutant protein demonstrated consistently a 15–20% decrease in both calcium flux and IP3 production. These findings indicate that even a modest decrease in Btk function can impair B cell proliferation or survival. However, a mutation in Btk and reduced numbers of B cells are not always associated with clinical disease.
Keywords: agammaglobulinaemia, B lymphocytes, immunological deficiency syndromes, penetrance, protein–tyrosine kinases
Introduction
Patients with X-linked agammaglobulinaemia (XLA) are typically recognized to have immunodeficiency in the first 3 years of life because of recurrent or severe bacterial infections [1–4]. On evaluation, these patients generally have severe reductions in all immunoglobulin isotypes and less than 1% CD19+ B cells in the peripheral circulation. However, after Btk was identified as the gene responsible for XLA in 1993 [5,6], it was noted that some patients with proven mutations in Btk do not come to medical attention in the first 10 years of life and some have higher than expected concentrations of serum immunoglobulins [3,7–11]. Recent evidence suggests that there is some genotype/phenotype correlation [3,12,13], but it is clear that the specific mutation in Btk is not the only factor that influences the severity of disease.
Btk is a cytoplasmic tyrosine kinase that is expressed in monocytes and platelets as well as B cells [6,14,15]. In addition to the carboxyterminal kinase domain, Btk has an amino terminal pleckstrin homology domain, followed by a Tec homology domain, an Src homology 3 (SH3) domain and an SH2 domain [5,6]. These domains allow Btk to act as a scaffold protein as well as an enzyme. Until recently, no polymorphic variants changing the amino acid sequence of Btk had been reported. In 2007, Perez et al. described a family in which the proband had an amino acid substitution in the SH3 domain, alanine to valine at codon 230, as well as a known disease causing mutation in the kinase domain, arginine to histidine at codon 641 [16]. A male maternal cousin with the amino acid substitution at codon 230 but no mutation at codon 641 had normal numbers of B cells and no signs of immunodeficiency.
Over 600 different mutations in Btk have been reported in patients with XLA [17] and no single mutation accounts for more than 3% of patients [18]. More than 95% of mutations are single base pair substitutions or the gain or loss of less than 20 base pairs. The remaining mutations are larger deletions or duplications, insertions or inversions that can be detected by Southern blot analysis [19,20]. Amino acid substitutions constitute about one-third of all mutations and the majority of these mutations destabilize the protein, such that no Btk can be detected by immunofluorescence staining of monocytes [18,21]. Amino acid substitutions, particularly amino acid substitutions that are associated with stable Btk protein, tend to be associated with older age at diagnosis, higher concentrations of serum IgM and slightly more B cells in the peripheral circulation [12].
The diagnostic criteria for XLA indicate that a mutation in Btk plus reduced numbers of CD19+ cells are sufficient to make the diagnosis of XLA [22]. However, it is not clear that a mutation in Btk and reduced numbers of B cells are always associated with clinical disease. We have identified a family in which the proband and his brother had a premature stop codon in Btk and an amino acid substitution in the kinase domain. Their mother was heterozygous for both alterations but their healthy maternal grandfather had only the amino acid substitution. The grandfather had markedly reduced numbers of B cells in the peripheral circulation but no clinical signs of immunodeficiency.
Materials and methods
Patients
The subjects included in this study were analysed as part of a research study approved by the St Jude Children's Research Hospital Institutional Review Board. Written informed consent was obtained for each subject.
Mutation detection
Mutation detection was performed by single strand conformation polymorphism (SSCP) screening followed by direct sequencing of relevant polymerase chain reaction (PCR) products, as described previously [23].
Monoclonal antibody production and immunofluorescence staining
To assess Btk expression, a monoclonal antibody to Btk, DFS, was produced by subcutaneous immunization of C57/B × 129 mice with a purified fusion protein consisting of codons 212–275 of Btk fused to a −3′ glutathione S transferase (GST) domain. Two days after the fourth immunization, lymphocytes from draining lymph nodes and the spleen were fused to the X63–AG8·653 fusion partner and hybridomas were grown in HAT media with interleukin-6 (100 units/ml). Supernatants from clones that reacted with the Btk fusion construct but not GST were analysed by indirect immunofluorescence for staining of monocytes from normal controls and patients with premature stop codons in Btk to identify clones useful for immunofluorescence.
Peripheral blood mononuclear cells were isolated by Ficoll gradient separation to examine cytoplasmic staining for Btk and B cell percentage and phenotype. For cytoplasmic Btk staining, 0·5 × 106 cells were incubated with Cytofix (BD Biosciences, San Jose, CA, USA) for 40 min at room temperature. The cells were washed twice and then incubated with 50 μl of hybridoma tissue culture supernatant for 10 min. The cells were washed and then stained with fluorescein isothiocyanate-labelled goat anti-mouse IgG1. Staining for CD19 and surface IgM was performed as described previously [24].
Vector construction and transfection
Site-directed mutagenesis was used to produce the Btk construct with the Y418H alteration. This construct was placed in the pApuro vector and transfected into DT40 chicken B cells by electroporation as described previously [25].
Calcium analysis and inositol triphosphate generation
Wild-type, Btk– and transfected Btk– DT40 cells were loaded with 3 mM Fura-2/AM and stimulated with 2 μg/ml of a monoclonal anti-chicken IgM (M4) and monitored for fluorescence as described previously [25]. To examine inositol triphosphate (IP3) production, the same cells were incubated with 10 μg/ml of the M4 antibody, and the Biotrak IP3 assay system (Amersham, Piscataway, NJ, USA) was used according to the manufacturer's protocol.
Results
A 14-month-old boy (III-2) was hospitalized in the intensive care unit for severe pneumonia requiring intubation. He had had a previous hospitalization at 8 months of age for oral ulcers, fever and dehydration. Immunological evaluation demonstrated profound hypogammaglobulinaemia and less than 1% CD19+ B cells. Genomic DNA from this child was screened by SSCP for alterations in Btk. Analysis of exons 2, 14 and 18 demonstrated altered fragment migration. Therefore, these exons were amplified by PCR and sequenced. Analysis of exon 2 showed a C to T at the +11 position of the splice donor site for intron 2. This previously described but uncommon polymorphism [26] provided a useful marker for haplotype analysis. The alteration in exon 14 was a T to C substitution in codon 418, resulting in the replacement of the wild-type tyrosine with histidine. This histidine occurs in the adenosine-5′-triphosphate (ATP) binding subdomain of the kinase domain and is conserved in murine Btk, but a leucine is seen in this position in other Btk family members including Tec, Itk, Bmx and Rlk/Txk. The change in exon 18 was an A to T substitution in codon 625; this mutation changes the wild-type lysine to a premature stop codon.
DNA samples from other family members were examined for all three alterations (Fig. 1). The asymptomatic older brother of the proband had all three base pair substitutions and the mother of the proband was heterozygous for all three alterations. DNA from the maternal grandfather demonstrated the polymorphism in intron 2 and the amino acid substitution in exon 14 but not the premature stop codon in exon 18. Both of the mother's sisters were heterozygous for the polymorphism in exon 2 and the amino acid substitution in exon 14 but not the premature stop codon. Use of the highly polymorphic marker DXS101 provided additional support for the observation that the mutant allele was inherited from the maternal grandfather; however, the brother of the grandfather (I-2) did not inherit this allele.
Fig. 1.
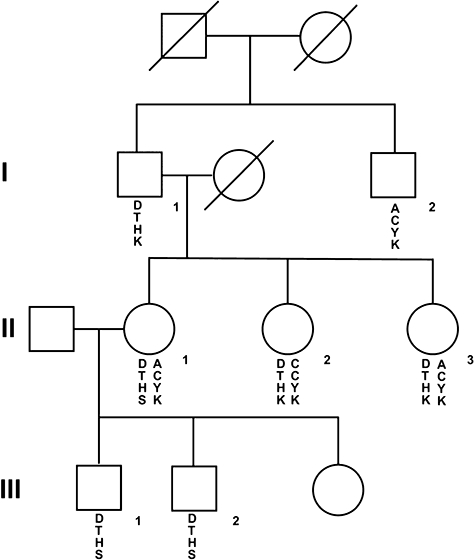
The pedigree for a family with two different alterations in Btk is shown. Squares represent males and circles indicate females. Genetic markers in and near Btk are shown below the symbol for each individual. The top marker is the highly polymorphic marker DXS101, which is 700 kb 5′- to Btk; the second marker is the uncommon polymorphism in intron 2 of Btk, the third marker is the alteration in codon 418 giving rise to a histidine (H) rather than the wild-type tyrosine (Y). The fourth marker is the alteration in codon 625 giving rise to a premature stop codon (S) rather than the wild-type lysine (K).
The tyrosine to histidine substitution at codon 418 has been reported as a disease causing mutation in the XLA data base (http://bioinf.uta.fi/BTKbase/) and the Cardiff mutation data base (http://www.hgmd.cf.ac.uk/ac/index.php); however, I-1 did not have a medical history compatible with immunodeficiency. He had had his tonsils removed at 8 years of age, but he had not been hospitalized for infection and he did not have signs of chronic sinus or pulmonary disease. As shown in Table 1, this 58-year-old man had borderline low serum immunoglobulins, but he made adequate titres of antibody to both T cell-dependent and T cell-independent antigens. He was blood group A with an anti-B titre of 1:4. His titre to tetanus toxoid was high normal, greater than 1:6000, and in the absence of recent immunization he had normal titres to nine of 12 pneumococcal serotypes.
Table 1.
Serum immunoglobulins.*
| I-1 | III-1 | III-2 | Adult controls | Age-matched controls | |
|---|---|---|---|---|---|
| IgG | 690 | 851† | 1002† | 694–1618 | 533–1678 |
| IgM | 36 | <1 | <1 | 48–271 | 26–218 |
| IgA | 85 | <4 | <4 | 81–463 | 24–121 |
As measured in mg/dl.
On intravenous gammaglobulin therapy.
Peripheral blood was obtained from the proband, his brother and his grandfather to evaluate expression of Btk and the B cell phenotype. Although monocytes from the proband and his brother were negative for Btk, the grandfather's monocytes showed normal expression of Btk (Fig. 2). The percentage of B cells was decreased in all three individuals, although more impressively in the proband and his brother (0·03% and 0·14% respectively) than in the grandfather (0·85%). As is typical of patients with XLA, the B cells from III-1 and III-2 demonstrated variable intensity CD19 and high intensity surface IgM. The phenotype of the B cells from the grandfather was more similar to that seen in the normal control, although the CD19 was slightly more variable and a higher percentage of the cells were stained brightly for IgM. Approximately 35% of the CD19+ cells from the grandfather were positive for CD27, a marker of mature memory B cells (Fig. 2).
Fig. 2.
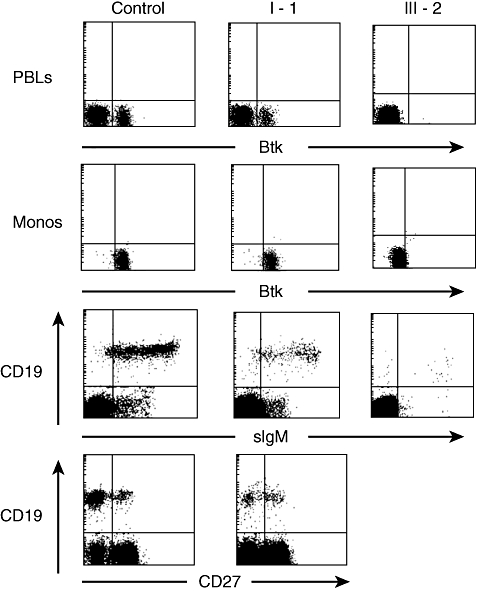
Cytoplasmic staining for Btk and surface staining for B cell markers on peripheral blood mononuclear cells from the grandfather (I-1) and the patient (III-2). The top panel shows indirect cytoplasmic staining of Btk in periperal blood lymphocytes, gated by forward- and side-scatter. The second panel shows Btk staining in the cell population identified as monocytes by forward- and side-scatter. The third and fourth panels show surface staining for CD19 and either IgM or CD27 on peripheral blood lymphocytes. The number of events analysed was 20 000 for the control, 50 000 for I-1 and 110 000 for III-2.
To examine the functional consequences of the tyrosine to histidine substitution, expression vectors producing wild-type or Y418H Btk were transfected into a chicken B cell line (DT40) lacking Btk. Previous studies have shown that Btk– DT40 cells have minimal calcium flux and no IP3 production after cross-linking of the surface B cell receptor [27,28]. Wild-type DT40 cells and cells that had been transfected with normal or Y418H Btk were stimulated with anti-IgM and calcium flux and IP3 production were measured. Cells containing the Y418H Btk vector had consistently a 15–25% decrease in calcium flux and IP3 production at 0·5 min when compared with cells that received the wild-type Btk vector (Fig. 3).
Fig. 3.
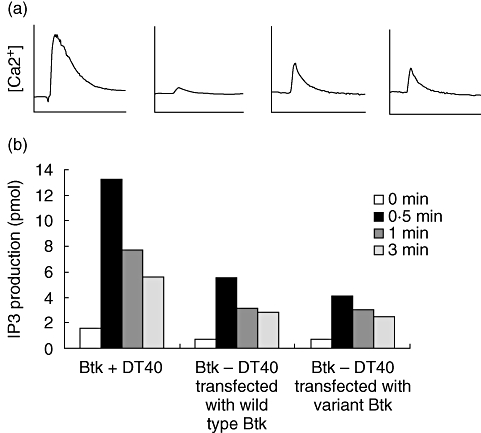
The functional consequences of the mutation in Btk were evaluated in a chicken B cell line, DT40, stimulated with anti-IgM. (a) Calcium flux in wild-type (Btk+) DT40, Btk– DT40, Btk– DT40 transfected with wild-type Btk and Btk– DT40 transfected with the variant Btk (left to right). (b) Inositol triphosphate (IP3) generation at the time intervals shown.
Discussion
In the family described in this paper, the proband presented with clinical signs and symptoms that are typical of XLA. The fact that his affected older brother had not yet come to medical attention because infection is not unusual, as both affected children were less than 5 years old at the time of diagnosis. The premature stop codon in exon 18 of Btk in these boys clearly confirmed the diagnosis of XLA in both children. The finding of an additional alteration, a tyrosine to histidine substitution in the ATP binding site, was unexpected. The fact that DNA from the asymptomatic maternal grandfather was positive for this mutation was more surprising.
The amino acid substitution in the ATP binding site of Btk may result in decreased binding of ATP or decreased phosphate transfer. The functional studies in the Btk– DT40 chicken B cell line suggest that the mutation at this site impairs Btk function, but the decrease in function was not great. Studies performed in Btk-deficient mice indicate that the requirements for the amount and function of Btk are very stringent. Btk-deficient mice that expressed 25–50% of the normal amount of Btk from either a gene therapy vector or a transgene had improved B cell numbers but did not make antibody to T cell-independent antigens [29,30]. Mice with a constitutively active Btk had markedly reduced numbers of peripheral B cells [31]. These findings suggest that modest but measurable decreases or increases in Btk function have physiological consequences.
The most sensitive indicator of impaired Btk function in the human appears to be reduced numbers of peripheral blood B cells. Normal or near-normal concentrations of serum immunoglobulins have been reported in 5–10% of patients with XLA [3,32]. A small number of patients with mutations in Btk have been described who were able to make some antibody to protein antigens but not polysaccharide antigens [8,10,13]. All these patients had markedly reduced numbers of peripheral blood B cells. One of these patients [10], like the grandfather of our patient, had easily detectable CD27+ memory B cells, indicating that Btk plays a less critical role in late stages of B cell differentiation. It is important to note that the majority of the mutations found in these patients with milder disease have also been reported in patients with more typical XLA. However, the maternal grandfather of our patient is the first individual to be reported who has the most consistent feature of XLA, markedly reduced numbers of B cells, but normal capacity to make antibodies to polysaccharide antigens and no clinical disease.
The tyrosine to histidine substitution in codon 418 has been reported in a patient with severe manifestations of XLA (http://bioinf.uta.fi/BTKbase/) and the Cardiff mutation data base (http://www.hgmd.cf.ac.uk/ac/index.php reporting the same patient). The difference between that patient and the maternal grandfather reported in this paper could be explained by modifying genetic factors that amplify the severity of the defect in the patient with typical disease or factors that ameliorate the defect in the grandfather. The question of which of these possibilities is more likely has implications for genetic counselling both families. Functional studies showing that the amino acid substitution has modest effects in the DT40 chicken B cell line suggest that it is more likely that environmental or genetic factors have contributed to the severity of the patient with typical disease. A better understanding of modifying genetic factors that influence B cell development and function in patients with XLA may reveal polymorphisms that are important in autoimmunity and antibody response to vaccine antigens.
Acknowledgments
We thank the family for their willingness to participate in this study and Julie Carter for help in preparation of the figures and the manuscript. These studies were supported in part by grants from the National Institute of Health AI25129, National Cancer Institute Grant P30 CA21765, American Lebanese Syrian Associated Charities, and by funds from the Federal Express Chair of Excellence.
References
- 1.Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr. 2002;141:566–71. doi: 10.1067/mpd.2002.127711. [DOI] [PubMed] [Google Scholar]
- 2.Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of 96 patients. Medicine. 1985;64:145–56. [PubMed] [Google Scholar]
- 3.Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol. 2002;104:221–30. doi: 10.1006/clim.2002.5241. [DOI] [PubMed] [Google Scholar]
- 4.Smith CIE, Satterthwaite A, Witte ON. X-linked agammaglobulinemia: a disease of Btk tyrosine kinase. In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases: a molecular and genetic approach. Oxford: Oxford University Press; 2007. pp. 279–303. [Google Scholar]
- 5.Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-linked agammaglobulinemia is a member of the src family of protein–tyrosine kinases. Nature. 1993;361:226–33. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 6.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–90. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 7.Saffran DC, Parolini O, Fitch-Hilgenberg ME, et al. A point mutation in the SH2 domain of Bruton's tyrosine kinase in atypical X-linked agammagobulinemia. N Engl J Med. 1994;330:1488–91. doi: 10.1056/NEJM199405263302104. [DOI] [PubMed] [Google Scholar]
- 8.Bykowsky MJ, Haire RN, Ohta Y, et al. Discordant phenotype in siblings with X-linked agammaglobulinemia. Am J Hum Genet. 1996;58:477–83. [PMC free article] [PubMed] [Google Scholar]
- 9.Kornfeld SJ, Haire RN, Strong SJ, et al. A novel mutation (Cys145-stop) in Bruton's tyrosine kinase is associated with newly diagnosed X-linked agammglobulinemia in a 51-year-old male. Mol Med. 1996;2:619–23. [PMC free article] [PubMed] [Google Scholar]
- 10.Wood PM, Mayne A, Joyce H, Smith CI, Granoff DM, Kumararatne DS. A mutation in Bruton's tyrosine kinase as a cause of selective anti-polysaccharide antibody deficiency. J Pediatr. 2001;139:148–51. doi: 10.1067/mpd.2001.115970. [DOI] [PubMed] [Google Scholar]
- 11.Stewart DM, Tian L, Nelson DL. A case of X-linked agammaglobulinemia diagnosed in adulthood. Clin Immunol. 2001;99:94–9. doi: 10.1006/clim.2001.5024. [DOI] [PubMed] [Google Scholar]
- 12.Broides A, Yang W, Conley M. Genotype/phenotype correlations in X-linked agammaglobulinemia. Clin Immunol. 2006;118:195–200. doi: 10.1016/j.clim.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Granados E, Perez dD, Ferreira CA, Fontan CG, Garcia Rodriguez MC. A genotype–phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol. 2005;116:690–7. doi: 10.1016/j.jaci.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 14.Smith CIE, Baskin B, Humire-Greiff P, et al. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152:557–65. [PubMed] [Google Scholar]
- 15.Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton's tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114:141–9. doi: 10.1046/j.1365-2141.2001.02905.x. [DOI] [PubMed] [Google Scholar]
- 16.Perez de Diego R, Bravo J, Allende LM, et al. Identification of novel non-pathogenic mutation in SH3 domain of Btk in an XLA patient. Mol Immunol. 2008;45:301–3. doi: 10.1016/j.molimm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Valiaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat. 2006;27:1209–17. doi: 10.1002/humu.20410. [DOI] [PubMed] [Google Scholar]
- 18.Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patients with defects in early B cell development. Immunol Rev. 2005;203:216–34. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 19.Rohrer J, Minegishi Y, Richter D, Eguiguren J, Conley ME. Unusual mutations in Btk: an insertion, a duplication, an inversion and four large deletions. Clin Immunol. 1999;90:28–37. doi: 10.1006/clim.1998.4629. [DOI] [PubMed] [Google Scholar]
- 20.Conley ME, Partain JD, Norland SM, Shurtleff SA, Kazazian HH., Jr Two independent retrotransposon insertions at the same site within the coding region of BTK. Hum Mutat. 2005;25:324–5. doi: 10.1002/humu.9321. [DOI] [PubMed] [Google Scholar]
- 21.Futatani T, Miyawaki T, Tsukada S, et al. Deficient expression of Bruton's tyrosine kinase in monocytes from X-linked agammaglobulinemia as evaluated by a flow cytometric analysis and its clinical application to carrier detection. Blood. 1998;91:595–602. [PubMed] [Google Scholar]
- 22.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Clin Immunol. 1999;93:190–7. doi: 10.1006/clim.1999.4799. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) [DOI] [PubMed] [Google Scholar]
- 23.Conley ME, Mathias D, Treadaway J, Minegishi Y, Rohrer J. Mutations in Btk in patients with presumed X-linked agammaglobulinemia. Am J Hum Genet. 1998;62:1034–43. doi: 10.1086/301828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobbs AK, Yang T, Farmer D, Kager L, Parolini O, Conley ME. A hypomorphic mutation in Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect in B cell development. J Immunol. 2007;179:2055–9. doi: 10.4049/jimmunol.179.4.2055. [DOI] [PubMed] [Google Scholar]
- 25.Ishiai M, Kurosaki M, Pappu R, et al. BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity. 1999;10:117–25. doi: 10.1016/s1074-7613(00)80012-6. [DOI] [PubMed] [Google Scholar]
- 26.Saha BK, Curtis SK, Vogler LB, Vihinen M. Molecular and structural characterization of five novel mutations in the Bruton's tyrosine kinase gene from patients with X-linked agammaglobulinemia. Mol Med. 1997;3:477–85. [PMC free article] [PubMed] [Google Scholar]
- 27.Takata M, Kurosaki T. A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184:31–40. doi: 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamadori T, Baba Y, Matsushita M, et al. Bruton's tyrosine kinase activity is negatively regulated by Sab, the Btk-SH3 domain-binding protein. Proc Natl Acad Sci USA. 1999;96:6341–6. doi: 10.1073/pnas.96.11.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satterthwaite AB, Cheroutre H, Khan WN, Sideras P, Witte ON. Btk dosage determines sensitivity to B cell antigen receptor cross- linking. Proc Natl Acad Sci USA. 1997;94:13152–7. doi: 10.1073/pnas.94.24.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conley ME, Rohrer J, Rapalus L, Boylin EC, Minegishi Y. Defects in early B-cell development: comparing the consequences of abnormalities in pre-BCR signaling in the human and the mouse. Immunol Rev. 2000;178:75–90. doi: 10.1034/j.1600-065x.2000.17809.x. [DOI] [PubMed] [Google Scholar]
- 31.Maas A, Dingjan GM, Grosveld F, Hendriks RW. Early arrest in B cell development in transgenic mice that express the E41K Bruton's tyrosine kinase mutant under the control of the CD19 promoter region. J Immunol. 1999;162:6526–33. [PubMed] [Google Scholar]
- 32.Minegishi Y, Rohrer J, Conley ME. Recent progress in the diagnosis and treatment of patients with defects in early B-cell development. Curr Opin Pediatr. 1999;11:528–32. doi: 10.1097/00008480-199912000-00010. [DOI] [PubMed] [Google Scholar]