

Abstract
X-linked agammaglobulinaemia (XLA) is characterized by absence of mature B cells because of mutations in the Bruton's tyrosine kinase (Btk) gene. Btk-deficient early B cell precursors experience a block in their differentiation potentially reversible by the addition of an intact Btk gene. Btk expression was measured in 69 XLA patients with 47 different mutations and normal expression was detected in seven. We characterized these Btk mutant forms functionally by transfection into a lymphoma cell line that lacks endogenous Btk expression (Btk−/− DT40 cells) and analysed the calcium flux in response to B cell receptor stimulation. To test whether co-expression of a mutated form could compromise the function of the intact Btk transfection, studies in wild-type (WT) DT40 cells were also performed. Study reveals that none of the seven Btk mutants analysed was able to revert the absence of calcium mobilization upon IgM engagement in Btk−/− DT40 cells, as does intact Btk. In addition, calcium mobilization by anti-IgM stimulation in DT40 Btk+/+ cells was unaffected by co-expression with Btk mutants. These results suggest that gene addition would be feasible not only for patients with XLA and mutations that prevent Btk expression, but for those with expression of a mutant Btk.
Keywords: Bruton's tyrosine kinase, calcium mobilization, DT40 cells, gene therapy, X-linked agammaglobulinaemia
Introduction
X-linked agammaglobulinaemia (XLA) is a primary immune deficiency characterized by lack of circulating mature B cells, hypogammaglobulinaemia and recurrent infections [1–7] because of mutations in the Bruton's tyrosine kinase (Btk) gene [8,9]. Btk is a member of the Tec family of kinases, which participates in several signalling pathways and is essential for early human B cell differentiation. It contains four interaction domains: pleckstrin homology, Tec homology, Src homology 3, Src homology 2, and the catalytic tyrosine kinase domain [10]. Mutations have been identified throughout the Btk gene and most XLA patients do not express detectable Btk protein [11]. Current treatment of XLA is palliative and consists of immunoglobulin substitution therapy and antibiotics.
Several lines of evidence suggest a strong selective advantage for B lineage cells expressing wild-type (WT) Btk when compared with mutant forms. Female carriers of XLA exhibit non-random X-inactivation of the mutant allele within the B cell compartment [12]. Similar observations were reported in an XLA mouse model using spleen B cells of X-linked immunodeficient (XID) females [13]. Transplantation of mixtures of CBA/J (WT) and CBA/N (XID) bone marrow cells into lethally irradiated XID mice also leads to the selective expansion and survival of WT B cells, although these results were not reproducible in other strains [14]. In addition, sublethal irradiation or high numbers of cells without myeloablation can rescue B lineage development in murine models [15–18]. Sustained correction of B cell development has been achieved with haematopoietic-targeted Btk gene addition in a XLA mouse model [19]. These studies provide the rationale for a gene therapy trial in XLA, although several preclinical studies are needed to improve the efficiency and safety. The success of this type of therapy in all XLA patients, which could provide them with normal life spans, would rely on the assumption that an intact Btk protein would also reconstitute the normal function in the presence of a mutated form. Both the capacity to interact with other molecules and the enzymatic function of Btk are essential for its role in signalling. The presence of a mutant Btk retaining some of these functions in some patients could potentially interfere with functional reconstitution by the intact Btk, a phenomenon called ‘dominant negative effect’. In this study we use Btk mutants expressed in patients with XLA and analyse their possible dominant negative effect in a cellular model. We report that seven Btk missense mutations located in different domains with protein expression do not exert a dominant negative effect in calcium mobilization, providing the first evidence that gene addition could be an efficient therapy for XLA patients with residual protein expression.
Materials and methods
Subjects
Seven unrelated patients diagnosed as having XLA, according to the criteria of the European Society for Immunodeficiencies/Pan-American Group for Immunodeficiency Scientific Group [20]. The ethics committee approved the protocol and written consent was obtained before blood was drawn. Samples were collected immediately before a new routine intravenous immunoglobulin dose and without any evidence of infection in the patients.
Btk mutation, Btk expression and Btk-specific phosphorylation studies
Bruton's tyrosine kinase sequence and expression studies from controls or XLA patients were performed by Western blot and flow cytometry as described [21,22]. Control non-B cells (C*) for Btk phosphorylation studies were obtained from the negative fraction of peripheral blood mononuclear cells (PBMC) sorted with CD19 Multisort microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), showing 0·06% CD19+ cells. For Btk phosphorylation antiphospho-Tyr223-Btk (Cell Signalling Technology, Beverly, MA, USA), peroxidase-conjugated goat anti-rabbit (Cell Signalling Technology, Beverly, MA, USA) and the enhanced chemiluminescence system (Amersham-Pharmacia-Biotech, Buckinghamshire, UK) were used. For chicken Btk detection, polyclonal anti-Btk, N-terminal (Sigma, St Louis, MO, USA) primary antibody was used. Btk levels and number of monocytes were equivalents both patients and controls (data not shown).
Site-directed mutagenesis
Site-directed mutagenesis was performed with QuikChange® II XL (Stratagene, La Jolla, CA, USA) in the plasmid pApuro-Btk (kindly provided by Tomohiro Kurosaki).
DT40 cell culture and transfections
Wild-type or Btk-deficient chicken DT40 cells (Riken Cell Bank, Ibaraki, Japan) were maintained per the manufaturer's recommendations. Cells were transfected by electroporation at 250 V and 960 μF in phosphate-buffered saline (107 cells/0·5 ml). Five micrograms of pApuro-Btk mutants, linearized with ScaI (New England Biolabs, Beverly, MA, USA), was transfected. Transfectants were selected in 0·5 mg/ml puromycin 24 h after electroporation. The presence of Btk was verified by Western blot analysis [23]. Btk phosphorylation in DT40 cells was detected by Western blot after stimulation with 10 μg/ml mouse anti-chicken IgM-BIOT Clone M4 (Southern Biotechnology, Birmingham, AL, USA) for 4 min.
Calcium flux
Fluo-4/Fura red (Molecular Probes, Invitrogen, Carlsbad, CA, USA) loaded Btk−/− DT40 (A) or WT DT40 (B) cells, transfected with the indicated construct, were stimulated with 5 μg/ml of mouse anti-chicken IgM or with 1 ng/μl ionomycin (Sigma) as positive control. Fluorescence measurements were performed with a fluorescence activated cell sorter (FACSCalibur) and data were analysed by FlowJo (Tree Star Inc., San Francisco, CA, USA). Data are representative of three different experiments.
Results
Btk expression and Btk kinase activity in XLA
In a previous study we reported the analysis of a group of 54 XLA patients [24]. At present, we have diagnosed up to 69 XLA patients from 52 unrelated families where we have found 47 different mutations in the Btk coding region (data not shown). Seven of these patients had a significant mutated protein expression (Fig. 1a and b) and presented different mutations in the Btk coding sequence (Table 1).
Fig. 1.
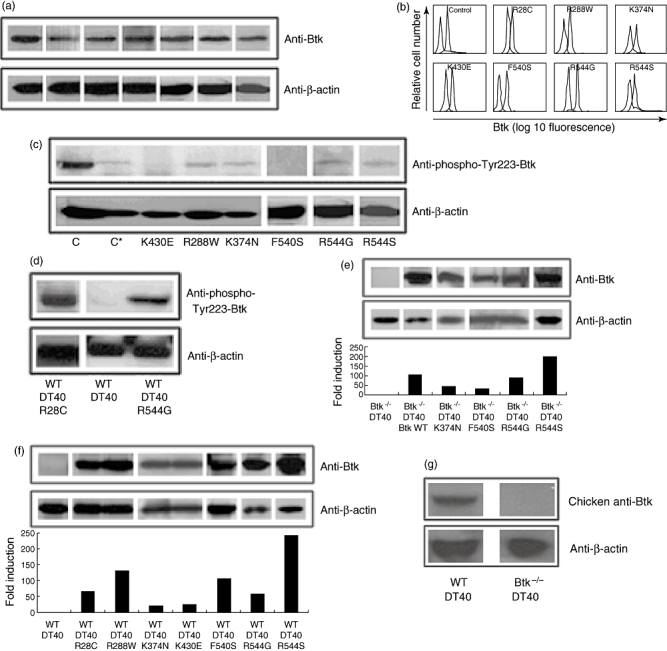
Bruton's tyrosine kinase (Btk) expression and kinase activity in X-linked agammaglobulinaemia (XLA) patient-derived peripheral blood mononuclear cells (PBMC). (a,b) Btk expression in PBMC by Western blot (a) and in monocytes by flow cytometry (b) in healthy controls and XLA patients. In (b), the thick line represents specific Btk staining, the thin line an irrelevant isotype control and the dashed line the secondary antibody alone. (c) Btk kinase activity tested by immunoblot with anti-Phospho-Tyr223-Btk in PBMC from XLA patients and a healthy control (C and C*). Sample in C contains full PBMC while C* contains B cell-depleted PBMCs. (d) anti-Phospho-Tyr223-Btk immunoblot of wild-type (WT) DT40 cells or WT DT40 cells transfected with R28C or R544G mutant Btk. (e, f) Western blot and densitometry analysis normalized to β-actin expression of WT or mutant Btk transfected in Btk−/− DT40 (e) or WT DT40 (f) cells. (g) anti-Btk N-terminal immunoblot of WT DT40 and Btk−/− DT40 cells.
Table 1.
Clinical and molecular data from patients.
| Patient | Age at diagnosis | IgG (mg/dl) | IgA (mg/dl) | IgM (mg/dl) | IgE (Ui/ml) | B cell (%) | Mutation in protein | Mutation in cDNA | Dom | Btk expression |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | 6 years | 50 | 53 | 16 | 255 | 0·1 | R28C | C214T | PH | Positive |
| Patient 2 | 8 months | 33 | <6 | 4 | <2 | 0·0 | R288W | C994T | SH2 | Positive |
| Patient 3 | 11 years | 200 | 11 | 14 | – | 1·0 | K374N | A1254C | SH2 | Positive |
| Patient 4 | 5 years | 18 | <6 | 15 | 9 | 0·0 | K430E | A1420G | TK | Positive |
| Patient 5 | 8 years | – | – | – | – | 0·0 | F540S | T1751C | TK | Positive |
| Patient 6 | 4 years | 26 | <6 | <4 | <2 | 0·0 | R544G | A1762G | TK | Positive |
| Patient 7 | 18 months | <33 | <6 | <4 | <2 | 0·0 | R544S | A1764T | TK | Positive |
IgG, IgA, IgM levels at diagnosis B, percentage of B cells in the total lymphocyte population. Btk, Bruton's tyrosine kinase; Dom, Btk domain affected by mutation; PH, pleckstrin homology; SH2, Src homology 2 and TK, tyrosine kinase.
Bruton's tyrosine kinase participates in signal transduction pathways of B lineage lymphoid cells, initiated by the binding of a variety of extracellular ligands to their cell surface receptors [25–28]. For example, engagement of the B cell antigen receptor triggers activation of a series of tyrosine kinases such as Lyn and Syk and induction of phospholipase C-γ2-mediated calcium mobilization [29]. As part of the activation process Lyn phosphorylates Btk in Tyr551, which is subsequently autophosphorylated in Tyr223 to achieve full activation [30–32]. Thus, the extent of Tyr223 phosphorylation represents a good measurement of Btk kinase activity. In monocytes, growth factor and Fc receptor engagement also induces Btk kinase activity [33]. We therefore determined Btk kinase capacity in our patients by measuring the amount of phospho-Tyr223 form of Btk in monocyte extracts by immunoblot (Fig. 1c). The most appropriate control for this experiment is a PBMC sample from a healthy individual depleted of B cells in which Btk is derived primarily from monocytes as in XLA patients. This sample is marked as C* in Fig. 1c. Two mutations (K430E and F540S) showed absence of Btk phosphorylation on Tyr223, while in four (R288W, K374N, R544G and R544S) at least residual kinase activity is present (Fig. 1c). We were unable to obtain blood from the patient with mutation R28C so we tested it by transfection of DT40 lymphoma cells, which have been shown previously to be amenable for complementation of endogenous Btk deletion by the expression of human Btk [34]. Anti-IgM stimulation of DT40 cells transfected with R28C or R544G induced autophosphorylation of these two Btk mutants on Tyr223 (Fig. 1d). These data are consistent with the fact that R28C mutation had been shown previously to have full kinase activity [35,36]. The band we detect in anti-phospho-Tyr223-Btk blots seems to measure primarily the transfected human Btk, as cross-reactivity with the chicken form of Btk, observed when loading extracts from untransfected DT40 cells, is very weak.
Btk mutants from XLA patients do not complement Btk−/− DT40 cells
When stimulated with anti-IgM antibodies or with the calcium ionophore ionomycin, WT DT40 cells trigger a calcium mobilization response detected by changes in fluorescence of the Fluo4 and Fura red dyes loaded into the cell. The anti-IgM response in DT40 cells is fully dependent upon Btk expression, while the response to ionomycin is independent of Btk function: Btk−/− DT40 cells show undetectable calcium flux in response to anti-IgM engagement [35]. We used this system to test the functionality of four of the Btk mutants that do not affect protein expression (K374N, F540S, R544G and R544S). Btk−/− DT40 cells were transfected with either WT human Btk control or the various Btk mutants by electroporation. Btk expression in each transfectant was assessed by Western blot, as shown in Fig. 1e. Calcium mobilization in the new Btk−/− DT40 transfectants was analysed, as shown in Fig. 2a. None of the mutations tested triggered any calcium flux in response to anti-IgM cross-linking, while all these transfectants maintained their ability to respond to ionomycin with the same efficiency. As a control, we observed that WT human Btk transfected into the same Btk−/− DT40 cells was able to complement the defect because anti-IgM cross-linking triggered calcium mobilization comparable to WT DT40 cells. Overall, these experiments show that calcium flux in response to B cell receptor (BCR) engagement is recovered in DT40 Btk-deficient cells when they are transfected with WT human Btk, but not when transfected with four different mutants, regardless of whether they show kinase activity (K374N, R544G and R544S) or not (F540S).
Fig. 2.
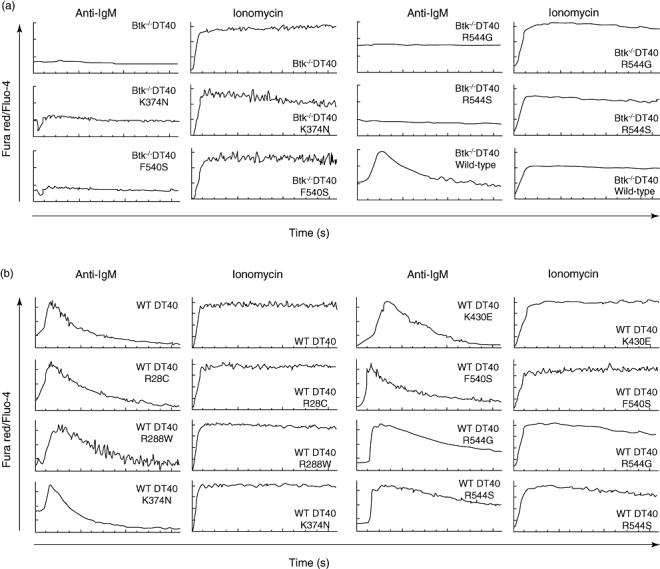
Calcium flux analysis in DT40 lymphoma cells transfected with Bruton's tyrosine kinase (Btk) mutants. (a, b) Calcium flux upon B cell receptor engagement in DT40 Btk−/− (a) and DT40 wild-type (b) transfected with the indicated construct in real time by flow cytometry. Arrows indicate the time (in seconds).
Mutant Btk expression has not dominant negative effect in WT DT40 transfectants
DT40 cells were transfected with seven different Btk constructs that contained the mutations described above. Btk expression in each of the transfectants was tested by Western blot, as shown in Fig. 1f. An antibody which recognizes chicken Btk is used to show endogenous Btk expression levels in WT DT40 cells. We do not use this antibody to test mutant forms because it also recognizes human Btk. As shown in Fig. 1g, endogenous Btk expression levels are less than Btk mutants.
We observed that all seven mutant Btk transfectants, even those with high expression levels, have the same response to calcium mobilization upon anti-IgM stimulation as the untransfected DT40 cells, and the control responses with ionomycin were also indistinguishable from that obtained with WT DT40 cells (Fig. 2b). We conclude therefore that none of the Btk mutants have a negative dominant effect over WT Btk function, albeit with the caveat that human Btk might not interact with endogenous chicken molecules with the same efficiency. We think this is unlikely, because human and chicken Btk are highly homologous proteins (85% amino acid identity), and the studies in Btk−/− DT40 transfected with human WT Btk show that WT human Btk can restore fully all the Btk functions tested in this system (Fig. 2a) [37].
Discussion
This study aims to contribute to the accumulation of the body of knowledge towards the design of a gene therapy trial for XLA. So far the general aim of preclinical gene therapy studies in XLA has been the reconstitution of cells with no measurable Btk protein with a WT Btk gene. Our studies, in a series of 69 XLA patients, showed that up to 15% of mutations allow normal protein expression [24], all of them being missense. We studied if a dominant negative effect towards the introduced Btk could be exerted by these Btk mutants, a phenomenon that could reduce the efficiency of a potential gene therapy. For this purpose we used a cellular model, the chicken DT40 lymphoma cell line, to test functional interactions between the transfected mutant forms of human Btk and the endogenous Btk using as readout the calcium mobilization in response to BCR stimulation.
Our results suggest that none of the XLA-associated Btk mutants with significant protein expression have a dominant negative effect over WT Btk function. This study focuses upon missense mutations with significant protein expression. Theoretically, Btk mutations affecting the mRNA splicing and leading to residual protein expression could be identified in XLA patients. If that were the case, a putative dominant negative effect should be also tested, as this phenomenon has been associated with Btk mutations in leukaemia [38]. Our experiments also show that the DT40 system is a reliable and fast way to test the effect of naturally occurring Btk mutations that lead to the XLA phenotype.
Acknowledgments
We wish to thank all the families involved in the study for their invaluable contribution to the project. This work was supported by the ‘Fondo de Investigación Sanitaria’ (FIS) Grant No. 020982, by the CICYT (BMC2002-00437) and by the Intramural Research Program of NIH/NIAID. R. P. dD. is the recipient of a fellowship from the FIS.
References
- 1.Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of 96 patients. Medicine (Baltimore) 1985;64:145–56. [PubMed] [Google Scholar]
- 2.McKinney RE, Katz SL, Wilfert CM. Chronic enteroviral meningoencephalitis in agammaglobulinemic patients. Rev Infect Dis. 1987;9:334–56. doi: 10.1093/clinids/9.2.334. [DOI] [PubMed] [Google Scholar]
- 3.Ochs HD, Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine (Baltimore) 1996;75:287–99. doi: 10.1097/00005792-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Rawlings DJ, Saffran DC, Tsukada S, et al. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–61. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 5.Thomas JD, Sideras P, Smith CI, Vorechovský I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–8. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 6.Sideras P, Smith CI. Molecular and cellular aspects of X-linked agammaglobulinemia. Adv Immunol. 1995;59:135–223. doi: 10.1016/s0065-2776(08)60631-8. [DOI] [PubMed] [Google Scholar]
- 7.Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–33. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 8.Tsukada S, Rawlings DJ, Witte ON. Role of Bruton's tyrosine kinase in immunodeficiency. Curr Opin Immunol. 1994;6:623–30. doi: 10.1016/0952-7915(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 9.Yamada N, Kawakami Y, Kimura H, et al. Structure and expression of novel protein-tyrosine kinases, Emb and Emt, in hematopoietic cells. Biochem Biophys Res Commun. 1993;192:231–40. doi: 10.1006/bbrc.1993.1404. [DOI] [PubMed] [Google Scholar]
- 10.Smith CIE, Islam TC, Mattson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammaliam Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23:436–46. doi: 10.1002/bies.1062. [DOI] [PubMed] [Google Scholar]
- 11.Gaspar HB, Conley ME. Early B cell defects. Clin Exp Immunol. 2000;119:383–9. doi: 10.1046/j.1365-2249.2000.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conley ME, Puck JM. Definition of the gene loci in X-linked immunodeficiencies. Immunol Invest. 1998;17:425–63. doi: 10.3109/08820138809049847. [DOI] [PubMed] [Google Scholar]
- 13.Nahm MH, Paslay JW, Davie JM. Unbalanced X chromosome mosaicism in B cells of mice with X-linked immunodeficiency. J Exp Med. 1983;158:920–31. doi: 10.1084/jem.158.3.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sprent J, Bruce J. Physiology of B cells in mice with X-linked immunodeficiency (xid) III: disappearance of xid B cells in double bone marrow chimeras. J Exp Med. 1984;160:711–23. doi: 10.1084/jem.160.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohrer J, Conley ME. Correction of X-linked immunodeficient mice by competitive reconstitution with limiting numbers of normal bone marrow cells. Blood. 1999;94:3358–65. [PubMed] [Google Scholar]
- 16.Porpiglia AS, Rohrer J, Conley ME. Reconstitution of B cell function in murine models of immunodeficiency. Clin Immunol. 2003;107:90–7. doi: 10.1016/s1521-6616(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 17.Quan ZS, Dick RF, Regueiro B, Quintans J. B cell heterogeneity, II: transplantation resistance in xid mice which affects the ontogeny of B cell sub-populations. Eur J Immunol. 1981;11:643–9. doi: 10.1002/eji.1830110810. [DOI] [PubMed] [Google Scholar]
- 18.Quintans J. The immune response of CBA/N mice and their F1 hybrids to 2,4,6-trinitrophenylated (TNP) antigens, I: analysis of the response to TNP-coupled lipopolysaccharide in vivo and at the clonal level. Eur J Immunol. 1979;9:67–71. doi: 10.1002/eji.1830090114. [DOI] [PubMed] [Google Scholar]
- 19.Yu PW, Tabuchi RS, Kato RM, et al. Sustained correction of B-cell development and function in a murine model of X-linked agammaglobulinemia (XLA) using retroviral-mediated gene transfer. Blood. 2004;104:1281–90. doi: 10.1182/blood-2003-09-3044. [DOI] [PubMed] [Google Scholar]
- 20.Conley ME. Diagnostic guidelines − an international consensus document. Clin Immunol. 1999;93:189. doi: 10.1006/clim.1999.4798. [DOI] [PubMed] [Google Scholar]
- 21.García Rodríguez MC, López-Granados E, Ferreira A, Fontán G. Molecular analysis of Bruton's tyrosine kinase gene in Spain. Mutation brief. Hum Mutat. 2001;18:84–8. doi: 10.1002/humu.1155. [DOI] [PubMed] [Google Scholar]
- 22.Pérez de Diego R, López-Granados E, Pozo M, et al. Bruton's tyrosine kinase is not essential for LPS-induced activation of human monocytes. J Allergy Clin Immunol. 2006;117:1462–9. doi: 10.1016/j.jaci.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 23.Futatani T, Miyawaki T, Tsukada S, et al. Deficient expression of Bruton's tyrosine kinase in monocytes from X-linked agammaglobulinemia as evaluated by a flow cytometric analysis and its clinical application to carrier detection. Blood. 1998;91:595–602. [PubMed] [Google Scholar]
- 24.López-Granados E, Pérez de Diego R, Ferreira Cerdán A, Fontán Casariego G, García Rodríguez MC. A genotype-phenotype correlation study in a group of 54, X-linked agammaglobulinemia (XLA) patients. J Allergy Clin Immunol. 2005;116:690–7. doi: 10.1016/j.jaci.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 25.Matsuda T, Takahashi-Tezuka M, Fukada T, et al. Association and activation of Btk and Tec tyrosine kinases by gp130, a signal transducer of the interleukin-6 family of cytokines. Blood. 1995;85:627–33. [PubMed] [Google Scholar]
- 26.Sato S, Katagiri T, Takaki S, et al. IL-5 receptor-mediated tyrosine phosphorylation of SH2/SH3-containing proteins and activation of Bruton's tyrosine and Janus 2 kinases. J Exp Med. 1994;180:2101–11. doi: 10.1084/jem.180.6.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt U, van den Akker E, Parren-van Amelsvoort M, et al. Btk is required for an efficient response to erythropoietin and for SCF-controlled protection against TRAIL in erythroid progenitors. J Exp Med. 2004;199:785–95. doi: 10.1084/jem.20031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Feng J, Wen R, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 29.Kurosaki T, Kurosaki M. Transphosphorylation of Bruton's tyrosine kinase on tyrosine 551 is critical for B cell antigen receptor function. J Biol Chem. 1997;272:15595–8. [Google Scholar]
- 30.Mao C, Zhou M, Uckun FM. Crystal structure of Bruton's tyrosine kinase domain suggest a novel pathway for activation and provides insights into the molecular basis of X-linked agammaglobulinemia. J Biol Chem. 2001;276:41435–43. doi: 10.1074/jbc.M104828200. [DOI] [PubMed] [Google Scholar]
- 31.Nisitani S, Kato RM, Rawlings D, Witte ON, Wahl MI. In situ detection of activated Bruton's tyrosine kinase in the Ig signalling complex by phosphopeptide-specific monoclonal antibodies. Proc Natl Acad Sci USA. 1999;96:2221–6. doi: 10.1073/pnas.96.5.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satterthwaite AB, Witte ON. The role of Bruton's tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–7. [PubMed] [Google Scholar]
- 33.Horwood NJ, Mahon T, McDaid JP, et al. Bruton's tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med. 2003;197:1603–11. doi: 10.1084/jem.20021845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurosaki T, Maeda A, Ishiai M, Hashimoto A, Inabe K, Takata M. Regulation of the phospholipase C-gamma2 pathway in B cells. Immunol Rev. 2000;176:19–29. doi: 10.1034/j.1600-065x.2000.00605.x. [DOI] [PubMed] [Google Scholar]
- 35.Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005;203:216–34. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 36.Fukuda M, Kojima T, Kabayama H, Mikoshiba K. Mutation of the pleckstrin homology domain of Bruton's tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5–tetrakisphosphate binding capacity. J Biol Chem. 1996;271:30303–6. doi: 10.1074/jbc.271.48.30303. [DOI] [PubMed] [Google Scholar]
- 37.Takata M, Kurosaki T. A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184:31–40. doi: 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feldhahn N, Rio P, Soh BN, et al. Deficiency of Bruton's tyrosine kinase in B cell precursor leukemia cells. Proc Natl Acad Sci USA. 2005;102:13266–71. doi: 10.1073/pnas.0505196102. [DOI] [PMC free article] [PubMed] [Google Scholar]