

Abstract
In Helicobacter pylori gastritis gastric epithelium plays a central role in the innate immunity to H. pylori. However, epithelial receptors interacting with H. pylori have been poorly characterized so far. Recently a new triggering receptor expressed on myeloid cells-1 (TREM-1) has been identified on human neutrophils and monocytes. On these cells TREM-1 triggers innate immunity by stimulating the secretion of interleukin (IL)-8 and tumour necrosis factor (TNF)-α and thus amplifies bacterial-induced inflammation. In this study expression and function of TREM-1 in gastric epithelium exposed to H. pylori has been investigated. TREM-1 mRNA and protein were expressed on gastric epithelial cell lines as demonstrated by reverse transcription–polymerase chain reaction (RT–PCR) and fluorescence activated cell sorter analysis. Gastric epithelial TREM-1 expression was up-regulated directly by H. pylori and was independent of epithelial IL-8 induced by H. pylori. Immunohistochemistry and tissue RT–PCR demonstrated significantly stronger TREM-1 expression in H. pylori gastritis compared with the non-inflamed gastric mucosa supporting in vivo that epithelial TREM-1 is up-regulated during H. pylori infection. Stimulation of gastric epithelial TREM-1 receptor resulted in IL-8 up-regulation on mRNA and protein level, as shown by real-time PCR and immunoassay. This is the first study localizing TREM-1 on gastric epithelium. Functional data suggest that TREM-1 expressed on gastric epithelium amplifies inflammation of the underlying gastric mucosa by up-regulation of IL-8.
Keywords: gastric epithelium, H. pylori gastritis, H. pylori, innate immunity, TREM-1
Introduction
Infection of the gastric mucosa by Helicobacter pylori is one of the most common human bacterial infections worldwide. During infection H. pylori colonizes the mucus layer overlying the gastric surface epithelium and causes inflammation of the underlying mucosa, called chronic active gastritis. In this scenario, gastric epithelium plays the central role in first-line defence against H. pylori. H. pylori infection leads to epithelial secretion of proinflammatory cytokines, which trigger innate immunity of the gastric mucosa [1–4]. Recently, a novel receptor involved in innate immunity, the triggering receptor expressed on myeloid cells-1 (TREM-1) has been described. TREM-1 has been found to be expressed on neutrophils and subsets of monocytes and tissue macrophages and signals upon association with DAP12, an immunoreceptor tyrosine-based activation motif (ITAM)-containing transmembrane adaptor protein. The natural ligand of TREM-1 has not yet been identified. However, stimulation of the TREM-1 receptor on neutrophils and monocytes with agonistic antibodies leads to the rapid and enhanced secretion of proinflammatory mediators such as interleukin (IL)-8 and tumour necrosis factor (TNF)-α, which enables TREM-1 to contribute to innate immunity [5,6]. Therefore, in this study we analysed gastric epithelium for TREM-1 expression and characterized the role of gastric epithelial TREM-1 expression in H. pylori infection. For the first time, we were able to detect TREM-1 receptor expression on gastric epithelium in vitro and in vivo. Further experiments suggest that TREM-1 expressed by gastric epithelium plays a functional role in mucosal innate immunity during H. pylori infection.
Materials and methods
Gastric epithelial cell lines and gastric tissue specimens
For in vitro experiments gastric epithelial cell lines HM02, 23 132, 2957, 4433, 3051, KatoIII and 2474 were used. Gastric epithelial cell lines 23 132 [commercially available from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), no. ACC 201], 2957, 4433, 3051 and 2474 were established as described [7].
The cell line KatoIII was a gift from Professor H. P. Vollmers (Institut für Pathologie, Universität Würzburg, Germany) and the cell line HM02 was from Professor S. Suerbaum (Institut für Hygiene und Mikrobiologie, Medizinische Hochschule Hannover).
Surgical tissue specimens from gastric antrum and corpus mucosa of five patients (four with tumour of the pancreas, one with oesophageal carcinoma) without H. pylori gastritis and of 12 patients with chronic active H. pylori gastritis were investigated by immunohistochemistry and reverse transcription–polymerase chain reaction (RT–PCR) for TREM-1 expression.
In the patients without H. pylori gastritis no inflammatory cells were detectable in the antrum and the corpus mucosa, and H. pylori colonization was excluded in both compartments by a modified Giemsa stain. Tissue specimens from patients with chronic active H. pylori gastritis showed moderate to severe chronic inflammatory infiltrate of T and B lymphocytes, plasma cells and monocytes and mild to severe activity with neutrophils in the lamina propria, within the gastric epithelium and the foveolar lumen. H. pylori colonization was mild to severe as determined by a modified Giemsa stain.
This study was approved by a local ethics committee.
Measurement of TREM-1 by semiquantitative RT–PCR
Total RNA from gastric epithelial cell lines and gastric mucosa tissue specimens was extracted with TRIzol (Sigma, Taufkirchen, Germany) and treated with RNAse free DNAse I (MBI fermentas, St Leon-Rot, Germany) according to the manufacturer's protocol.
DNAse-treated total RNA from aliquots of 1 μl was taken for reverse transcription using oligo-dT 15–18 primers (Invitrogen, Karlsruhe, Germany). Subsequent PCR amplification of the RT reaction using the following primer pair was performed as follows: TREM-1 upstream primer: 5′-AGG GGC CAC ACC AAC CTT CTG-3′, TREM-1 downstream primer: 5′-AGT GCC TGC CTC AAT GTC TCC A-3′, annealing temperature: 65°C; product length: 364 base pairs (bp).
All PCR reactions were performed in the GeneAmp PCR System 2400 (Perkin Elmer Cetus, Emeryville, CA, USA) in a final volume of 25 μl containing 2 μl cDNA, 2·5 μl 10× buffer without MgCl2, 1·25 μl of 50 mM MgCl2 (final concentration 2·5 mM), 5 μl of deoxynucleotide triphosphate (dNTP)-mix, 1 mM of each nucleotide (final concentration 0·2 mM each) (Invitrogen, Karlsruhe, Germany), 10 pmol of each primer (final concentration 0·4 μM each), 0·1 μl of Taq DNA polymerase (5 U/μl) (Invitrogen, Karlsruhe, Germany).
PCR conditions were as follows: initially at 94°C for 2 min, 94°C for 30 s, annealing at the primer-specific temperature for 30 s, 30 s at 72°C for 40 cycles and finally at 72°C for 5 min.
Ten μl of each PCR reaction was size-fractionated by gel electrophoresis in a 2% agarose gel and analysed. Amounts of TREM-1 product were estimated and compared with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a rough control of the estimated amount. In gastric biopsies, after equalizing GAPDH m-RNA levels, TREM-1 mRNA expression was grouped semiquantitatively in none, weak and strong expression.
Measurement of TREM-1 expression on gastric epithelial cell lines by fluorescence activated call sorter (FACS) analysis
Gastric epithelial cell lines were washed with phosphate-buffered saline (PBS) containing 2% fetal calf serum (FCS) and reacted for 30 min with monoclonal mouse anti-TREM-1 antibody (R&D Systems, Wiesbaden, Germany) in an appropriate dilution. As control, a related isotype antibody was used with the same protein concentration as the primary antibody. Human peripheral neutrophils, which were isolated according to an already established protocol [8], were used as a positive control for TREM-1 receptor expression. After washing, fluorescence staining amplification was performed (Staining Amplification Kit; Caltag, Hamburg, Germany) according to the manufacturer's instructions. Stained cells were analysed on a FACScan flow cytometer (Becton Dickinson Biosciences, Mountain View, CA, USA) with appropriate gatings using CellQuestPro software.
Co-incubation of gastric epithelial cell lines with H. pylori, H. pylori lipolysaccharide (LPS) and IL-8
For the co-incubation experiments H. pylori strain G27, a cagA-positive clinical isolate [9], the corresponding isogenic cagA deletion mutant [10] and isolated H. pylori LPS were used. Isolation of H. pylori LPS was performed as described previously [11].
When recovered from frozen stocks, H. pylori strains were grown under microaerophilic conditions (Oxoid, Wesel, Germany) on Columbia agar plates containing 5% horse blood, 0·2% cyclodextrin and Dent's or Skirrow's antibiotic supplement at 37°C for 3 days. After passaging on fresh plates, bacteria were cultured in a 5% CO2/95% air atmosphere for another 24 h at 37°C.
For TREM-1 measurement, gastric epithelial cell lines were co-incubated with viable bacteria at a multiplicity of infection (MOI) of 50 bacteria per cell at 37°C with 10% CO2 at three different time-points (3 h, 24 h and 48 h) in RPMI-1640/10% FCS. Gastric epithelial cell line HM02 was co-incubated with H. pylori LPS from a cagA-positive H. pylori strain at a concentration of 100 ng/ml for 24 h. Additionally, gastric epithelial cell lines were stimulated with IL-8 (Biomol, Hamburg, Germany) at different concentrations (1, 5 and 10 ng/ml) for 10 h, 24 h and 48 h.
As negative control, in every experiment epithelial cell lines were incubated with RPMI-1640/10% FCS medium without H. pylori or IL-8 under the same conditions.
Measurement of IL-8 mRNA and TREM-1 mRNA by real-time PCR
Total RNA was obtained from harvested epithelial cells using the TRIzol (Sigma, Taufkirchen, Germany) method. cDNA synthesis was performed by RNA priming with oligo-dT primers using REVERSE-IT™ RTase blend (ABgene, Epsom, UK) following the manufacturer's directions. Quantification of IL-8 was performed by real-time PCR standardization with SYBR Green (ABgene) versus a housekeeping gene (β-microglobulin) and to standard dilution curves. Tests were performed in triplicate in two independent experiments. The primers used for these analyses were IL-8 S: 5-TGTAAACATGACTTCCAAGCTG-3; IL-8 AS: 5-ACTCCTTGGCAAAACTGCAC-3; β-microglobulin S: 5-TTCCTGAATTGCTATGTGTCTG-3; AS: 5-ACAAGTCTGAATGCTCCACTTTT-3. Reaction conditions were 10 min at 95°C, 40 cycles for 30 s at 95°C and 1 min at 60°C. For TREM-1 quantification primers were used at 300 nm, probes at 25 μM, on serial three- to threefold dilutions in triplicate of each RNA (range = 200 ng−1 ng). Quantification was performed versus RPL11 as housekeeping gene. The primers used for these analyses were purchased from Sigma (Taufkirchen, Germany); TREM-1 S: 5-TGGTCTTCTCTGTCCTGTTTG-3; TREM-1 AS: 5-ACTCCCTGCCTTTTACCTC-3; TREM-1 probe: 5-FAM-ACTTCCAGCCACATCCATCTGGCAGT-TAMRA-3; RPL11S: 5-AACTTCGCATCCGCAAAC-3; RPL11 AS: 5-AACTTCGCATCCGCAAAC-3; RPL11 probe: 5-FAM–CAGGGCAGACCCCTGTGTTTTCCAAA-TAMRA-3. Reaction conditions for TREM-1 were 10 min at 95°C, 40 cycles for 30 s at 95°C and 1 min at 60°C.
TREM-1 receptor stimulation on gastric epithelial cell lines
TREM-1 receptor stimulation was performed by an anti-TREM-1 agonistic antibody with the cross-linking technique, as described previously [5]. Briefly, gastric epithelial cell lines 23 132 and 4433 were incubated with an agonistic monoclonal mouse anti-TREM-1 antibody (R&D Systems) at a concentration of 10 μg/ml. Cross-linking was induced by adding a goat anti-mouse-specific antibody (R&D Systems) at a concentration of 50 μg/ml. The TREM-1 receptor stimulation was performed in two independent experiments on different days.
Cells were collected after 3 h (IL-8) and tested for IL-8 mRNA by real-time PCR (triplets). Supernatant was collected after 12 h, 24 h and 48 h and tested for IL-8 protein by a bead immunoassay (doublets). TNF-α was measured in the supernatant after 24 h by the enzyme-linked immunosorbent assay (ELISA) technique.
Measurement of IL-8 and TNF-α protein
Supernatants of the gastric epithelial cell culture were cleared of cellular debris by centrifugation for 1 min at 10 000 g and stored at −20°C prior to protein measurement.
IL-8 protein measurement was performed by a bead immunoassay kit (BioSource, Camarillo, CA, USA) in doublets according to the instructions provided by the manufacturer (sensitivity: 3 pg/ml). TNF-α release into the cell supernatants was quantified by a highly sensitive (mean minimal detectable dose of TNF-α 1·6 pg/ml) commercially available ELISA kit (R&D Systems) as described recently [8].
Immunohistochemistry
For immunohistological analyses tissue specimens were fixed in 10% formalin buffered at pH 7·0 for 24 h and paraffin-embedded. Monoclonal TREM-1 antibody and Toll-like receptor (TLR)-4 antibody (both R&D Systems) were used at a dilution of 1:10 and 1:500, respectively. Immunohistochemistry has been performed as described recently [12,13]. To exclude non-specific staining, in each case the primary antibody was replaced by an isotype control antibody. Using this procedure no staining of the gastric epithelium was detectable. Slides were counterstained by haematoxylin and eosin (H&E).
Results
TREM-1 mRNA and protein are expressed on gastric epithelial cell lines
TREM-1 expression has been investigated on seven gastric epithelial cell lines by RT–PCR. TREM-1 receptor mRNA was detectable in five of seven gastric epithelial cell lines (HM02, 23 132, 2957, 4433 and 2474 (data not shown).
Using FACS analysis three of four cell lines tested showed TREM-1 protein expression (HM02, 23 132 and 4433); 30–40% of the total cell population expressed the TREM-1 receptor (Fig. 1). These results demonstrate that not only myeloid cells, but also gastric epithelial cells are able to express TREM-1 receptor.
Fig. 1.

Triggering receptor expressed on myeloid cells-1 (TREM-1) protein is expressed on gastric epithelial cell lines determined by fluorescence activated cell sorter analysis. TREM-1 protein is expressed on gastric epithelial cell lines, as shown exemplarily for cell line 23 132. In this experiment, 38·2% of the cells of the total population expressed the TREM-1 receptor. The x-axis indicates fluorescence intensity measured on the log10 scale, and the y-axis indicates event counts per channel on a linear scale. The filled figure shows the isotype matched control.
TREM-1 on gastric epithelial cell lines is up-regulated by H. pylori but not by H. pylori LPS
As on monocytes, stimulation with bacterial ligands results in TREM-1 up-regulation [6,14], the influence of H. pylori on the gastric epithelial TREM-1 receptor expression has been investigated.
The TREM-1 expressing gastric epithelial cell lines (HM02, 23 132, 2957, 4433 and 2474) were co-incubated with a cagA-positive H. pylori strain and the isogenic cagA-negative deletion mutant for three different time-points (3 h, 24 h and 48 h). In all cell lines tested co-incubation with H. pylori resulted in an up-regulation of TREM-1 mRNA, as determined by real-time PCR (shown exemplarily for cell lines 23 132 and 4433 in Fig. 2a). TREM-1 expression increased after H. pylori co-incubation on average approximately two- to fourfold with a maximum increase in cell line 23 132 after 48 h. Compared to the cagA-positive H. pylori strain, the cag-negative deletion mutant showed a slightly lower TREM-1 up-regulation, although it was still clearly higher than in the control (Fig. 2a).
Fig. 2.
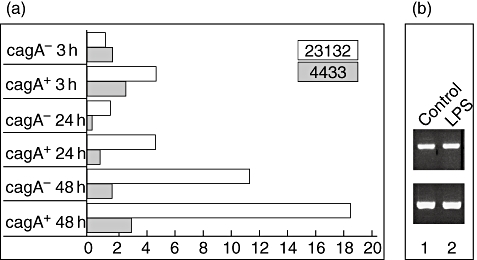
(a) The triggering receptor expressed on myeloid cells-1 (TREM-1) receptor on gastric epithelial cell lines is up-regulated by Helicobacter pylori determined by real-time polymerase chain reaction. The TREM-1-expressing gastric epithelial cell lines were incubated for 3 h, 24 h and 48 h with a cagA+H. pylori strain and the corresponding cagA- isogenic deletion mutant. TREM-1 expression increased after H. pylori co-incubation on average approximately two- to fourfold with a maximum increase in cell line 23 132 after 48 h. The cagA+H. pylori strain showed a slightly stronger TREM-1 up-regulation than the corresponding cagA- deletion mutant, but these results need further confirmation. The x-axis demonstrates fold change of TREM-1 mRNA over unstimulated control, the y-axis the time-points for measurement and the H. pylori strain. (b) H. pylori lipopolysaccharide (LPS) does not regulate TREM-1 expression. In contrast, co-incubation with isolated H. pylori LPS (lane 2) has no influence on TREM-1 regulation after 24 h compared with the medium control (lane 1). Data are shown exemplarily for the gastric epithelial cell line HM02.
Co-incubation with isolated H. pylori LPS had no influence on TREM-1 regulation after 24 h, as tested exemplarily on the gastric epithelial cell line HM02 (Fig. 2b).
TREM-1 expression on gastric epithelial cell lines is not influenced by IL-8
Because in the literature IL-8, which is known to be induced in gastric epithelial cells directly by H. pylori[15], is also described to up-regulate TREM-1 [16], the influence of IL-8 on the up-regulation of TREM-1 has also been investigated. In the two cell lines tested (23 132, 4433) IL-8 did not regulate TREM-1 expression at three different time-points (10 h, 24 h and 48 h) at different concentrations, as determined by RT–PCR (data not shown).
These data suggest that TREM-1 up-regulation on gastric epithelial cells is dependent directly on H. pylori and does not occur via an IL-8-driven pathway.
Stimulation of the TREM-1 receptor on gastric epithelial cell lines results in increased IL-8 expression
To demonstrate a functional role of TREM-1 on gastric epithelium the proinflammatory cytokines IL-8 and TNF-α were measured after stimulation of the TREM-1 receptor. The rationale for this experiment is that cross-linking of TREM-1 expressed on neutrophils and monocytes with agonistic antibodies enhances secretion of IL-8 and TNF-α[5,6].
Stimulation of gastric epithelial TREM-1 resulted in up-regulation of IL-8 transcription in both gastric epithelial cell lines tested (23 132, 4433), as tested by real-time PCR. The amount of IL-8 mRNA increased by 69·5% (4433) and 26·5% (23 132) (Fig. 3a) compared with the unstimulated control cell line.
Fig. 3.

Stimulation of the triggering receptor expressed on myeloid cells-1 (TREM-1) receptor expressed on gastric epithelial cell lines results in up-regulation of interleukin (IL)-8 as well on mRNA as on protein level. For receptor stimulation the epithelial TREM-1 receptor was cross-linked with an agonistic antibody on two gastric epithelial cell lines (23 132, 4433). IL-8 expression after TREM-1 stimulation was compared with IL-8 expression of an unstimulated control cell line. Standard deviations (bars) are given for two independent experiments. (a) The amount of IL-8 mRNA increased by 69·5% (4433) and 26·5% (23 132) compared with the unstimulated control cell line. IL-8 mRNA was measured after 3 h by real-time polymerase chain reaction. The x-axis demonstrates percentage increase of IL-8 m-RNA expression over unstimulated control; the y-axis shows the tested cell line. (b) Increase of IL-8 mRNA was confirmed by up-regulation of IL-8 protein tested by bead immunoassay. TREM-1 stimulation resulted in both cell lines (4433, 23 132) at all three tested time-points (12, 24, 48 h) in an IL-8 protein up-regulation. The maximal increase of IL-8 protein for cell line 4433 was at 24 h (41·1%) and for cell line 23 132 at 48 h (31·8%) after TREM-1 receptor stimulation. The x-axis shows the time-points for measurement; the y-axis demonstrates IL-8 protein expression (pg/ml).
Increase of IL-8 mRNA was confirmed by up-regulation of IL-8 protein tested by bead immunoassay. TREM-1 stimulation resulted in both cell lines at all three tested time-points in an IL-8 protein up-regulation. The IL-8 protein level was maximal at 24 h (41·1% for cell line 4433) or 48 h (31·8% for cell line 23 132), respectively (Fig. 3b). In contrast, TREM stimulation on gastric epithelial cell lines did not result in an increased TNF-α production tested by ELISA (data not shown).
TREM-1 is expressed in vivo by gastric epithelium in H. pylori gastritis and expression is significantly stronger in H. pylori gastritis than in the non-inflamed gastric mucosa.
To demonstrate TREM-1 expression by gastric epithelium in vivo gastric mucosa of patients with chronic active H. pylori gastritis and of patients without H. pylori infection was investigated by immunohistochemistry and RT–PCR.
TREM-1 expression was localized by immunohistochemistry on gastric epithelium in nine of 12 patients with H. pylori gastritis (Fig. 4a), whereas epithelial TREM-1 expression was accentuated in the glandular neck region but usually extended to the luminal surface. In contrast, no patients with non-inflamed gastric mucosa (n = 5, Fig. 4b) showed TREM-1 expression of the gastric epithelium.
Fig. 4.
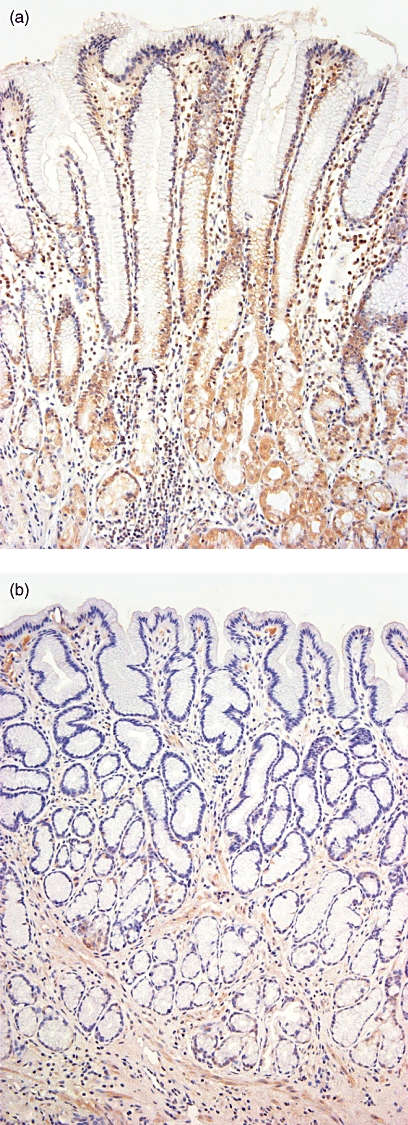
Triggering receptor expressed on myeloid cells-1 (TREM-1) is expressed on gastric epithelium in Helicobacter pylori gastritis in vivo as shown by immunohistochemistry. Slides were counterstained by haematoxylin and eosin. TREM-1 is expressed clearly on gastric epithelium in chronic active H. pylori gastritis, whereby epithelial TREM-1 expression was accentuated in the glandular neck region but usually extended to the luminal surf ace (Fig. 4a). In contrast, in the non-inflamed gastric mucosa no TREM-1 expression is detectable (Fig. 4b). Magnification: (a) ×400; (b) ×200.
In concordance with the immunohistochemical data, TREM-1 mRNA was expressed significantly more strongly in H. pylori gastritis than in uninflamed gastric mucosa. In H. pylori gastritis TREM-1 mRNA expression was strong in nine of 12 patients, whereas a strong TREM-1 mRNA expression could not be detected in any of the patients with uninflamed gastric mucosa. Table 1 shows TREM-1 mRNA signal-strength in H. pylori gastritis and in uninflamed gastric mucosa in detail. It should be noted that some amount of the detected TREM-1 mRNA may be due to mucosa-infiltrating monocytes and neutrophils. However, by immunohistochemistry TREM-1 expression could be localized mainly to gastric epithelium and only to a much lesser extent to monocytes and neutrophils.
Table 1.
Triggering receptor expressed on myeloid cells-1 (TREM-1) mRNA is expressed significantly more strongly in gastric mucosa specimens of patients with Helicobacter pylori gastritis than in patients without inflammation tested by reverse transcription–polymerase chain reaction.
| TREM-1 mRNA expression | |||
|---|---|---|---|
| None | Weak | Strong | |
| Uninflamed gastric mucosa (n = 5) | 3 | 2 | 0 |
| H. pylori gastritis (n = 12) | 1 | 2 | 9 |
TREM-1 mRNA intensity was grouped in no, weak and strong expression. In H. pylori gastritis TREM-1 expression was strong in nine of 12 patients, whereas in none of the patients with uninflamed gastric mucosa could a strong TREM-1 expression be detected.
These data demonstrate TREM-1 expression by gastric mucosal epithelium in H. pylori gastritis in vivo and confirm in vivo that gastric epithelial TREM-1 is up-regulated in H. pylori infection.
Discussion
Infection of the gastric mucosa by H. pylori, which colonizes the mucus overlaying the gastric surface epithelium, causes chronic active gastritis. A central role in first-line defence against H. pylori contributes to the gastric mucosal epithelium, which triggers inflammation of the underlying mucosa.
Most recently, a new group of activating receptors, called TREM, has been identified on human neutrophils and monocytes. TREM-1 is known to trigger innate immunity by phagocyte secretion of proinflammatory chemokines and cytokines such as IL-8 and TNF-α[5,6,17]. In this manner TREM-1 amplifies the inflammation induced by bacteria [5,6].
Because other receptors of the innate immunity, such as TLRs, expressed by gastric epithelium trigger mucosal inflammation, the question arises as to whether TREM-1 is involved in the epithelial innate immune response to H. pylori.
For the first time, in this study we were able to localize TREM-1 expression in vitro on gastric epithelial cell lines as well as in vivo on gastric epithelium of patients with H. pylori gastritis. This demonstrates that TREM expression is not restricted to myeloid cells. However, in this study gastric epithelial TREM-1 expression was significantly lower in patients without H. pylori infection, tempting us to speculate that H. pylori might be responsible for TREM-1 up-regulation. Indeed, we could also demonstrate in vitro a clear TREM-1 up-regulation on gastric epithelium by H. pylori. These results are in line with recent data that bacteria are able to up-regulate TREM-1 on phagocytes [14].
Because in the literature IL-8, which is known to be induced by H. pylori, is also described to up-regulate TREM-1 [16], the question arises as to whether IL-8 may contribute to TREM-1 up-regulation. In this study we could show that H. pylori itself, and not IL-8, is responsible for TREM-1 up-regulation on gastric epithelium.
To eludicate further the mechanism of epithelial TREM-1 receptor up-regulation by H. pylori, we investigated the influence of the H. pylori virulence factor cagA and of H. pylori LPS. Interestingly, epithelial TREM-1 up-regulation tended to be stronger by H. pylori strains bearing the virulence factor cagA, but further studies are necessary to confirm these results. LPS did not influence TREM-1 regulation. This is in contrast to TREM-1 expression on monocytes, which is described to increase after stimulation with LPS in vitro[14].
By studying the role of TREM-1 in H. pylori gastritis we were able to show that stimulation of gastric epithelial TREM-1 resulted in an increased gastric epithelial IL-8 production. These findings demonstrate that TREM-1 amplifies the proinflammatory immune response not only via myeloid cells, but also via mucosal epithelium, and underlines the functional role of gastric epithelial TREM-1 expression.
In this study, TREM-1 expession on gastric epithelium in patients with H. pylori gastritis has been demonstrated for the first time. Functional data suggest that TREM-1 enables gastric epithelium to ‘cross-talk’ with H. pylori and to trigger innate mucosal immunity during H. pylori infection. The pivotal role of mucosal epithelium in the innate immune response becomes increasingly evident. Recently, TLRs, other receptors involved in the innate immunity, which were described originally on professional immune cells, were also demonstrated to play a functional role during infection in the mucosal epithelium [18]. Most interestingly, on phagocytes TLRs were found to synergize with TREM receptors in amplifying the innate immune response [6].
Further studies are needed to show whether a synergistic effect of TREM and TLRs in the stomach is also necessary for sufficient innate mucosal immunity.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft grant EC 203/1-3. We thank E. Bachmann, E. Schmitt, J. Helfrich and B. Meyer for their excellent technical assistance.
References
- 1.Crabtree JE. Role of cytokines in pathogenesis of Helicobacter pylori-induced mucosal damage. Dig Dis Sci. 1998;43(Suppl. 9):46–55. [PubMed] [Google Scholar]
- 2.Dixon MF. Helicobacter pylori and peptic ulceration: histopathological aspects. J Gastroenterol Hepatol. 1991;6:125–30. doi: 10.1111/j.1440-1746.1991.tb01451.x. [DOI] [PubMed] [Google Scholar]
- 3.Nedrud JG, Blanchard SS, Czinn SJ. Helicobacter pylori inflammation and immunity. Helicobacter. 2002;7(Suppl. 1):24–9. doi: 10.1046/j.1523-5378.7.s1.4.x. [DOI] [PubMed] [Google Scholar]
- 4.Tummala S, Keates S, Kelly CP. Update on the immunologic basis of Helicobacter pylori gastritis. Curr Opin Gastroenterol. 2004;20:592–7. doi: 10.1097/00001574-200411000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–5. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- 6.Colonna M, Facchetti F. TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J Infect Dis. 2003;187(Suppl. 2):397–401. doi: 10.1086/374754. [DOI] [PubMed] [Google Scholar]
- 7.Vollmers HP, Stulle K, Dämmrich J, et al. Characterization of four new gastric cancer cell lines. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;63:335–43. doi: 10.1007/BF02899281. [DOI] [PubMed] [Google Scholar]
- 8.Schmausser B, Josenhans C, Endrich S, et al. Downregulation of CXCR1 and CXCR2 expression on human neutrophils by Helicobacter pylori: a new pathomechanism in H. pylori infection? Infect Immun. 2004;72:6773–9. doi: 10.1128/IAI.72.12.6773-6779.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang Z, Censini S, Bayeli PF, et al. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun. 1995;63:94–8. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beier D, Spohn G, Rappuoli R. Identification and characterization of an operon of Helicobacter pylori that is involved in motility and stress adaptation. J Bacteriol. 1997;179:4676–83. doi: 10.1128/jb.179.15.4676-4683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hynes S, Ferris J, Szponar B, et al. Comparative chemical and biological characterization of the lipopolysaccharides of gastric and enterohepatic helicobacters. Helicobacter. 2004;9:313. doi: 10.1111/j.1083-4389.2004.00237.x. [DOI] [PubMed] [Google Scholar]
- 12.Schmausser B, Andrulis M, Endrich S, et al. Expression and subcellular distribution of Toll-like receptors TLR4, TLR5 and TLR9 on the gastric epithelium in Helicobacter pylori infection. Clin Exp Immunol. 2004;136:521–6. doi: 10.1111/j.1365-2249.2004.02464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmausser B, Endrich S, Brändlein S, et al. The chemokine receptor CCR7 is expressed on epithelium of non-inflamed gastric mucosa, Helicobacter pylori gastritis, gastric carcinoma and its precursor lesions and up-regulated by H. pylori. Clin Exp Immunol. 2005;139:323–7. doi: 10.1111/j.1365-2249.2005.02703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knapp S, Gibot S, Vos A, Versteeg HH, Colonna M, Poll T. Cutting edge: expression patterns of surface and soluble triggering receptor expressed on myeloid cells-1 in human endotoxemia. J Immunol. 2004;173:7131–4. doi: 10.4049/jimmunol.173.12.7131. [DOI] [PubMed] [Google Scholar]
- 15.Crabtree JE, Lindley IJ. Mucosal interleukin-8 and Helicobacter pylori-associated gastroduodenal disease. Eur J Gastroenterol Hepatol. 1994;6(Suppl. 1):33–8. [PubMed] [Google Scholar]
- 16.Radsak MP, Salih HR, Rammensee HG, Schild H. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J Immunol. 2004;172:4956–63. doi: 10.4049/jimmunol.172.8.4956. [DOI] [PubMed] [Google Scholar]
- 17.Bleharski JR, Kiessler V, Buonsanti C, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol. 2003;170:3812–18. doi: 10.4049/jimmunol.170.7.3812. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen G, Andresen L, Matthiessen MW, Rask-Madsen J, Brynskov J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin Exp Immunol. 2005;141:298–306. doi: 10.1111/j.1365-2249.2005.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]