

Abstract
BAR superfamily domains shape cellular membranes through poorly understood mechanisms. To gain mechanistic understanding, we solved structures of F-BAR modules bound to flat and curved bilayers using electron (cryo)microscopy. We show that membrane tubules form when F-BARs polymerize into helical coats that are held together by lateral and tip-to-tip interactions. Notably, on gel-state membranes or after mutation of residues along the lateral interaction surface, F-BARs adsorb onto bilayers via surfaces other than their concave face. We conclude that membrane-binding is separable from membrane-bending, and that imposition of the module’s concave surface forces fluid-phase bilayers to bend locally. Furthermore, exposure of the domain’s lateral interaction surface through a change in orientation serves as the crucial trigger for assembly of the helical coat and propagation of bilayer-bending. The geometric constraints and sequential assembly of the helical lattice explain how F-BAR and classical BAR domains segregate into distinct microdomains, and provide insight into the spatial regulation of membrane invagination.
INTRODUCTION
Vast and largely unexplored, the interfaces between biological membranes and the compartments they delimit are the loci of diverse and essential processes, including cellular motility, intra- and inter-cellular communication, cell division, and the biogenesis of organelles. Great strides have been made in characterizing membrane dynamics, but a mechanistic understanding of these processes remains in its infancy. Missing from the analysis to date are structural descriptions of membrane-associated macromolecules and their interactions with the bilayer. To advance our understanding of these fundamental mechanisms, we have exploited the versatility of (cryo)electron microscopy to directly image a membrane-bound protein module whose interactions with the bilayer are critical players in membrane remodeling processes.
The BAR (Bin, Amphiphysin, Rvs) domain superfamily of proteins—including “classical” BAR domains, F-BAR (FCH-BAR or EFC Extended-FCH) and I-BAR (Inverse-BAR) domains—have emerged as important actors in membrane-remodeling processes throughout eukarya. Members of the superfamily are recruited from the cytoplasm to trigger the formation of plasma membrane extensions, invaginations, tubular organelles and transport intermediates, including endocytic vesicles (Itoh et al., 2005; Kamioka et al., 2004; Lee et al., 2002; Mattila et al., 2007; Peter et al., 2004; Tsujita et al., 2006). Much of what is known about the structure-function relationships of the BAR superfamily has emerged from crystallographic studies, showing that members of the family are elongated dimers formed by the anti-parallel association of α-helical coiled coils (Casal et al., 2006; Gallop et al., 2006; Henne et al., 2007; Lee et al., 2007; Li et al., 2007; Masuda et al., 2006; Mattila et al., 2007; Millard et al., 2005; Peter et al., 2004; Shimada et al., 2007; Tarricone et al., 2001; Weissenhorn, 2005; Zhu et al., 2007). Individual members of the BAR superfamily differ in their overall degree of curvature, where F-BARs have an elongated and gentle crescent-shape and I-BARs have a nearly flat zeppelin-shape, in comparison with the banana-shape of “classical” BAR modules (Henne et al., 2007; Lee et al., 2007; Millard et al., 2005; Shimada et al., 2007; Tarricone et al., 2001). Despite these differences, all BAR superfamily dimers have a surface on which clusters of positive charges are positioned to interact with negatively-charged phosopholipid headgroups of the membrane.
Based on biophysical and theoretical work and the visual cues provided by crystallographic studies, two distinct—but not exclusive—mechanisms have been proposed for the curvature-inducing activity of BAR superfamily domains. First, the “scaffolding” hypothesis intuitively posits that the modules bend membranes by simply imposing their charged, curved shapes via electrostatic attraction (Mattila et al., 2007; Peter et al., 2004; Shimada et al., 2007). Until now, there had been no direct evidence for this shape-based “scaffolding” hypothesis, other than that purified domains generate tubules in vitro whose curvature correlates with the concavity of their quaternary structure: classical BARs generate narrower tubules than elongated and gently-curved F-BARs (Farsad et al., 2001; Henne et al., 2007; Itoh et al., 2005; Peter et al., 2004; Shimada et al., 2007; Takei et al., 1999). Conversely, I-BAR modules appear to generate filopodia in vitro and in living cells (tubules of the opposite curvature) by binding to the plasma membrane via a convex surface (Mattila et al., 2007). The second mechanism proposes that protein-induced curvature is induced as a “buckling” response to the insertion of amphipathic sequences into the cytosolic leaflet of the bilayer (Zimmerberg and Kozlov, 2006). Reminiscent of the mechanism of curvature induction by epsin-family proteins (Ford et al., 2002), biochemical (Farsad et al., 2001; Henne et al., 2007) and spectroscopic (Gallop et al., 2006) data indicate that the eponymous N-terminal helix of endophilin and amphiphysin act like “wedges” that penetrate into one bilayer leaflet.
Both the “scaffolding” and the amphipathic “wedge” mechanism place emphasis on the interaction between protein modules and their target membranes. Regardless of the molecular mechanism, however, the energetic requirement for extensive membrane deformation—like endocytosis of coated pits or extension of membrane tubules—exceeds the interaction energy between an individual protein and the bilayer by at least one order of magnitude and therefore require the collective activity of many proteins (Ayton et al., 2007; Zimmerberg and McLaughlin, 2004). In support of this idea, a continuous filament of F-BAR domains (Itoh et al., 2005) has been proposed to encircle tubular membranes (Shimada et al., 2007), while direct protomer interactions enable polymerization of spherical or cylindrical coats for proteins like clathrin and its adaptors (Brett and Traub, 2006) or the GTPase dynamin (Hinshaw and Schmid, 1995; Takei et al., 1995; Zhang and Hinshaw, 2001). Moreover, theoretical studies show that even in the absence of protein-protein interactions, protein-induced changes in bilayer properties may create attractive forces that cause microscopically bent bilayer regions to coalesce into macroscopic curvature domains (Ayton et al., 2007; Bruinsma and Pincus, 1996; Reynwar et al., 2007). Scaffolding, amphipathic wedges, and ensemble activity may each contribute to curvature generation or stabilization by a specific domain, but testing these hypotheses directly requires molecular-scale visualization of the proteins in their membrane-bound contexts.
Using a structural approach that allows for the presence of a membrane holds the potential to advance a mechanistic description of membrane remodeling by answering four immediate questions: first, it would visualize directly how members of the BAR superfamily interact with the bilayer. Second, it would provide insight into the ensemble component of protein-induced membrane curvature. Third, it would explain whether and how the same type of domain accommodates a spectrum of different membrane curvatures. Fourth, structures may suggest how spatial regulation of membrane deformation is achieved. By showing directly how F-BARs employ a combination of scaffolding and ensemble action to induce curvature, our study provides answers to these questions.
Results and Discussion
F-BAR PROTEINS SPONTANEOUSLY SEGREGATE FROM CLASSICAL BAR PROTEINS DURING MEMBRANE TUBULE FORMATION
High-level expression of fluorescently-labeled F-BAR proteins revealed that they generate membrane tubules inside living cells (Itoh et al., 2005; Tsujita et al., 2006). Less appreciated but presumably of functional significance, F-BAR and other BAR superfamily proteins physically segregated from each other on membrane surfaces during membrane remodeling as seen in Figure 1A (Itoh and De Camilli, 2006). While segregation is likely to be determined in part by the affinity of a given BAR superfamily domain for a specific degree of curvature, the dynamic alternation of F-BAR and N-BAR microdomains (Figure 1A) suggested that stereotyped protein-protein interactions enabled members of the BAR superfamily to distinguish and recruit self-similar domains during membrane remodeling. In support of this hypothesis, when two different F-BAR proteins from the Toca family (transducer of Cdc42-dependent actin assembly (Ho et al., 2004)), namely CIP4/Toca-3 and FBP17/Toca-2, were co-expressed they co-localized on the same tubules (Figure 1B). While interactions between highly homologous TOCA proteins may involve more than a single domain, it is most likely that co-localization of these proteins was driven by their highly conserved and structural homologous membrane-binding F-BAR domains (Shimada et al., 2007).
Figure 1. F-BAR versus N-BAR Tubulation in Living Cells: Spontaneous Segregation, Differences in Diameter and Rigidity.
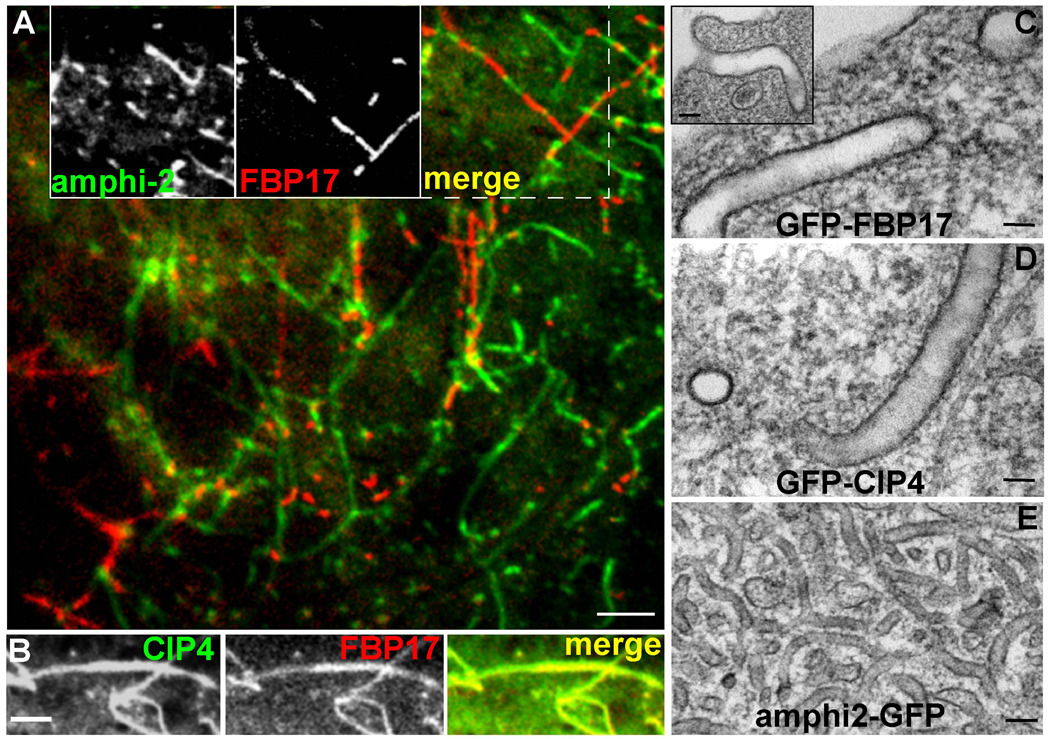
A) COS7 cell simultaneously transfected with amphiphysin2-GFP (green) and RFP-FBP17 (red) produces tubular networks in which the two proteins segregate from each other. Insets show the GFP, RFP, and merged channels. B) High magnification image of a cell transfected with GFP-CIP4 (left) and mRFP-FBP17 (middle) demonstrating the absence of segregation between the two proteins (merge; right). C) large invaginations of the plasma membrane observed by electron microscopy of thin-sections from COS7 cells transfected with full-length human GFP-FBP17 and D) GFP-CIP4; in comparison with the smaller tubules formed by amphiphysin2-GFP (E). Bars (A) 1 µm, (B) 0.5 µm, (C–E) 70 nm.
F-BAR TUBULES ARE LARGER THAN N-BAR TUBULES IN LIVING CELLS
When analyzed in living cells, F-BAR tubules were >3-fold wider in diameter than tubules formed by N-BARs, as shown by thin-section electron microscopy of COS7 cells expressing GFP-FBP17 (Figure 1C and inset), GFP-CIP4 (Figure 1D) and GFP-amphiphysin-2 (Figure 1E). This observation mirrors data that were previously obtained in vitro (Henne et al., 2007; Itoh et al., 2005; Shimada et al., 2007; Tsujita et al., 2006), emphasizing that tubulation in vitro generates biologically relevant structures. In addition, the striking differences in tubule diameters supported the scaffolding hypothesis, as the size difference between F-BAR and N-BAR tubules in living cells correlated directly with the difference in the radii of curvature for the respective domains (Casal et al., 2006; Shimada et al., 2007).
IN VITRO RECONSTITUTION OF MEMBRANE TUBULATION BY F-BAR DOMAINS FOR STRUCTURAL ANALYSIS BY ELECTRON CRYO-MICROSCOPY
To structurally analyze F-BAR induced membrane deformation, we generated mixed populations of tubules in vitro whose range of diameters were consistent with the range observed in living cells (57–85 nm in vitro compared with 64–113 nm in living cells, Figure 2A–B). Notably, micrographs of unstained liposomes caught in the process of tubule formation illustrated that tubulation involved at least two intermediate steps that correlated with the re-organization F-BAR domains into a defined coat following their adsorption onto the membrane. In Figure 2A, a bare bilayer is clearly resolved to the right of the yellow arrow and more clearly in the 2X enlarged inset. Immediately to the left of the yellow arrow, the outer surface of the bilayer is decorated by bound F-BAR domains but the curvature of the membrane has changed little if at all, in comparison with the naked membrane to the right. Between the yellow and cyan arrows, the F-BAR domains have clearly self-organized into a structured coat, and it is the organization of the coat that appears to transform the spherical liposome into a cylindrical tubule. Under the same solution conditions, when an N-BAR domain protein was mixed with an F-BAR domain protein prior to incubation with liposomes, homogeneous microdomains with a constant diameter—corresponding with the curvature of the F-BAR domain—were contiguous with equally homogenous but distinct tubules whose smaller diameter corresponded with the curvature of the N-BAR domain (Figure 2E–F). These in vitro observations were in accord with segregation observed in living cells.
Figure 2. Reconstitution of CIP4 F-BAR Induced Tubulation and Segregation from Endophilin N-BAR Domains in vitro.
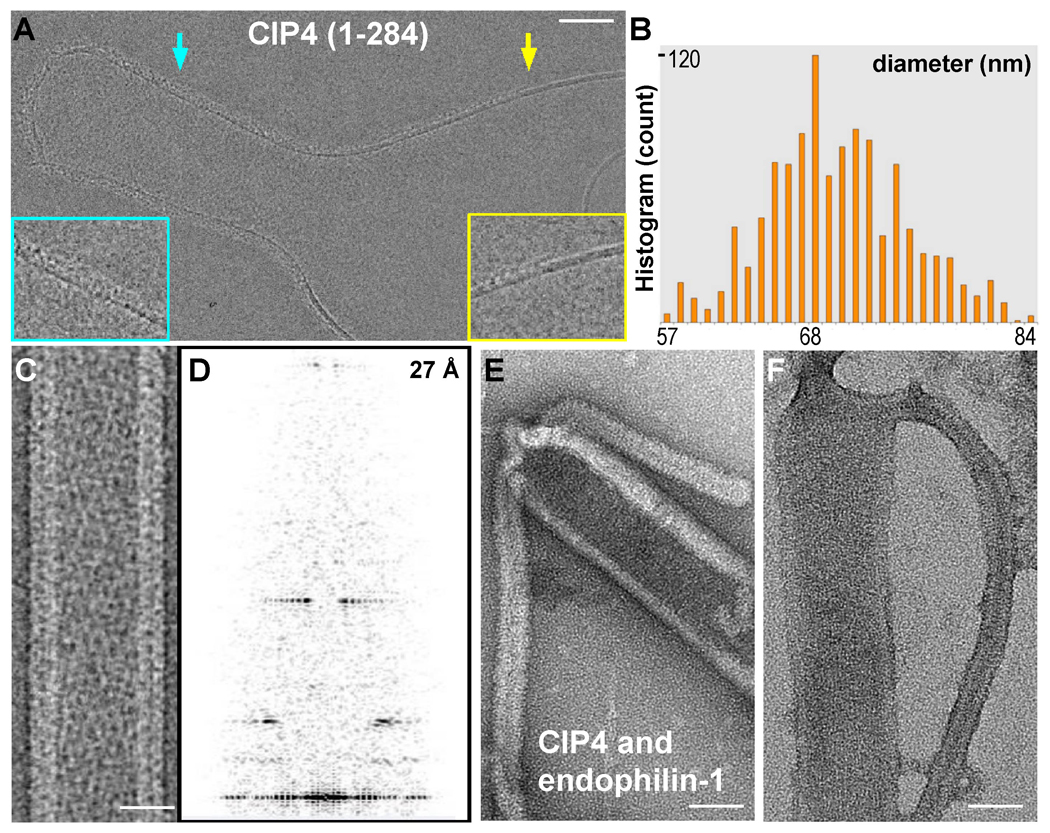
A) electron (cryo)micrograph of a nascent tubule generated in vitro by F-BAR domains (human CIP4, residues 1–284). The yellow arrow points to the demarcation between the membrane surface with and without F-BAR domains, revealing a smooth bilayer to the right and adsorbed protein to the left, as seen in the 2x enlarged inset surrounded by the yellow box. To the left of the yellow arrow, the curvature of the membrane has changed little, if at all, despite the presence of bound proteins. Induction of tubule formation accompanies self-organization of F-BAR domains into a helical coat (cyan arrow and enlarged inset). B) Histogram of tubule widths generated in vitro measured from electron (cryo)micrographs. C) electron (cryo)micrograph of a tubule following temperature annealing and its corresponding Fourier Transform (D), which displays high signal-to-noise ratio layerlines beyond ~27 Å. E,F) Liposomes co-incubated with F-BAR (CIP4) and N-BAR (endophilin-1) proteins in vitro observed after negative staining with uranyl formate (E) or uranyl acetate (F), displaying contiguous membrane tubules whose change in diameter corresponds with the change in the radius of curvature for F-BAR versus N-BAR domains. Bars (A) 300 Å; (C) 25 nm; (E,F) 40 nm.
The heterogeneity within populations of tubules, subtle changes in diameter along individual tubules, and the loss of lattice coherence over long distances presented significant obstacles to 3D reconstruction. Fortuitously, the long range order of the CIP4 F-BAR coat could be improved by subjecting F-BAR tubules to a period of slow temperature annealing before vitrification (see methods). In electron (cryo)micrographs of annealed tubules the helical nature of the F-BAR coat was obvious (Figure 2C) and Fourier transforms of these images revealed strong layer-lines (Figure 2D). Temperature annealing was only used to generate images suitable for structure determination. For all other in vitro experiments reported here, tubulation reactions were performed at room temperature for up to 30 minutes.
ITERATIVE HELICAL REAL SPACE RECONSTRUCTION OF F-BAR TUBULES
We employed the Iterative Helical Real Space Reconstruction algorithm (Egelman, 2000) to reconstruct volumes from individual tubules composed of up to ~3000 F-BAR domains, after preliminary efforts with Fourier-Bessel reconstruction failed to achieve the desired resolution (see methods and Figs S1,S2,S3). This approach enabled us to resolve individual F-BAR dimers and the contacts defining the helical coat (Figure 3A–C). To our knowledge, this is the first reconstruction of a membrane-binding protein with sufficient resolution to unambiguously identify individual protein subunits adsorbed onto an underlying membrane. The membrane itself appeared relatively smooth, with a hydrophobic core that was ~26Å thick and phosphocholine headgroup regions that were ~12Å thick (Figure 3C). The correspondence between the dimensions of the bilayer in our reconstructions and measurements of similar synthetic lipid mixtures strongly supported the validity of these results (Rodriguez et al., 2007; Wang et al., 2006). Notably, in reconstructions calculated from images of tubules with broken-open ends (Figure S2B), there was additional unstructured density along the surface of the inner leaflet. Since 3D reconstruction depends on averaging, we cannot rule out entirely that randomly distributed lipid protrusions were responsible for this layer of unstructured density. However, since this additional layer was observed only in tubes that were broken open, and given that the F-BAR domain was the only protein added to the reaction mixture, it seemed more likely that the additional densities represented a disordered layer of protein. This observation was significant because it, unexpectedly, conveyed that F-BARs could apparently bind to membranes with convex curvature. Mechanistically, this reinforced the idea that membrane-binding and membrane-bending are separable events (Figure 2A).
Figure 3. Single Particle Helical Reconstruction of a CIP4 F-BAR Domain-Induced Membrane Tubule.
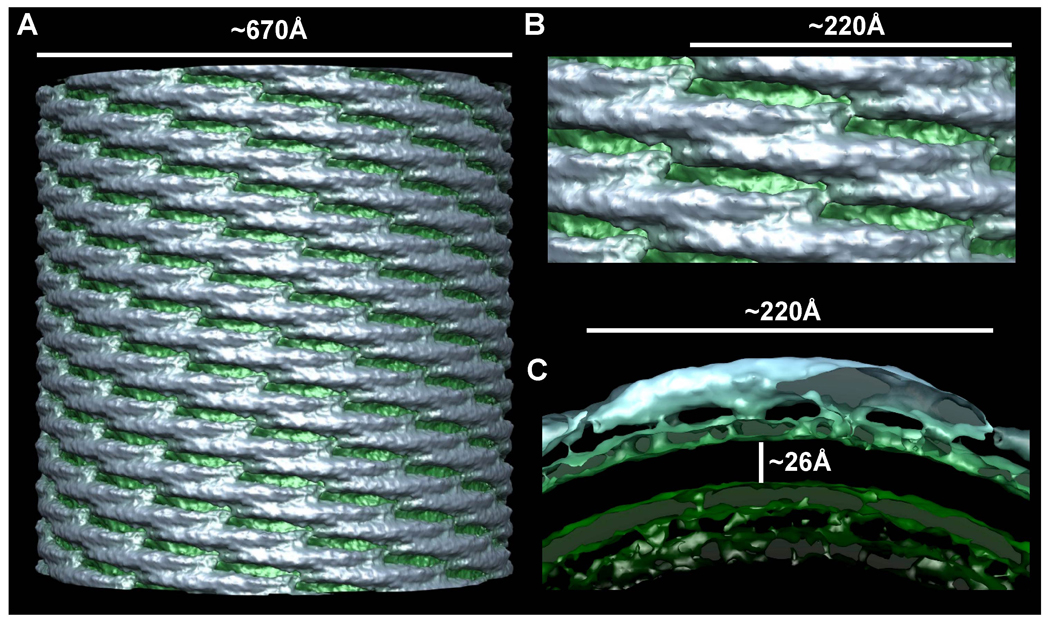
A) Surface of a ~67 nm diameter membrane tubule at ~17 Å resolution. The protein coat is colored blue-gray and the underlying membrane is green. B) Zoom in on the lattice seen orthogonal to the cylindrical axis, highlighting the tip-to-tip interactions and the broad contacts between laterally-adjacent dimers. C) Cross-sectional slab through one dimer parallel with the plane of the tip-to-tip interaction. There are four clearly resolved points of membrane binding. The hydrophobic core of the phospholipid bilayer is ~26 Å thick and the headgroup regions are ~12Å thick.
DIRECT VISUALIZATION OF SCAFFOLDING BY F-BAR DOMAINS
Like other BAR superfamily domains, F-BAR tubulation requires the presence of anionic headgroups to be present in the membrane at >10 mol% (Itoh et al., 2005; Tsujita et al., 2006). Moreover, tubulation was inhibited by increasing solution ionic strength, such that tubule formation was blocked at >300mM [NaCl] (data not shown), demonstrating that membrane deformation by F-BARs depended on electrostatic interactions. Consistent with these observations, the scaffolding hypothesis predicts that defined points of contact between the protein’s clusters of cationic residues and the phospholipid headgroups constrain the membrane to match the curvature of the domain. Proving this model, the 3D reconstruction visualized how through four points of close apposition the F-BAR dimer imposed its own shape on the underlying bilayer (Figure 3C, 4B). Moreover, there was no significant difference between the curvature of the F-BAR dimer bound to tubules and the structure obtained from x-ray crystallography in the absence of lipids (Figure 4B). This observation established the additional feature of the scaffolding hypothesis positing that protein scaffolds must be more rigid than the membrane.
Figure 4. Fitting F-BAR Crystal Structures into the CryoEM Map Reveals Membrane-Binding Residues and Possible Lattice Contacts.
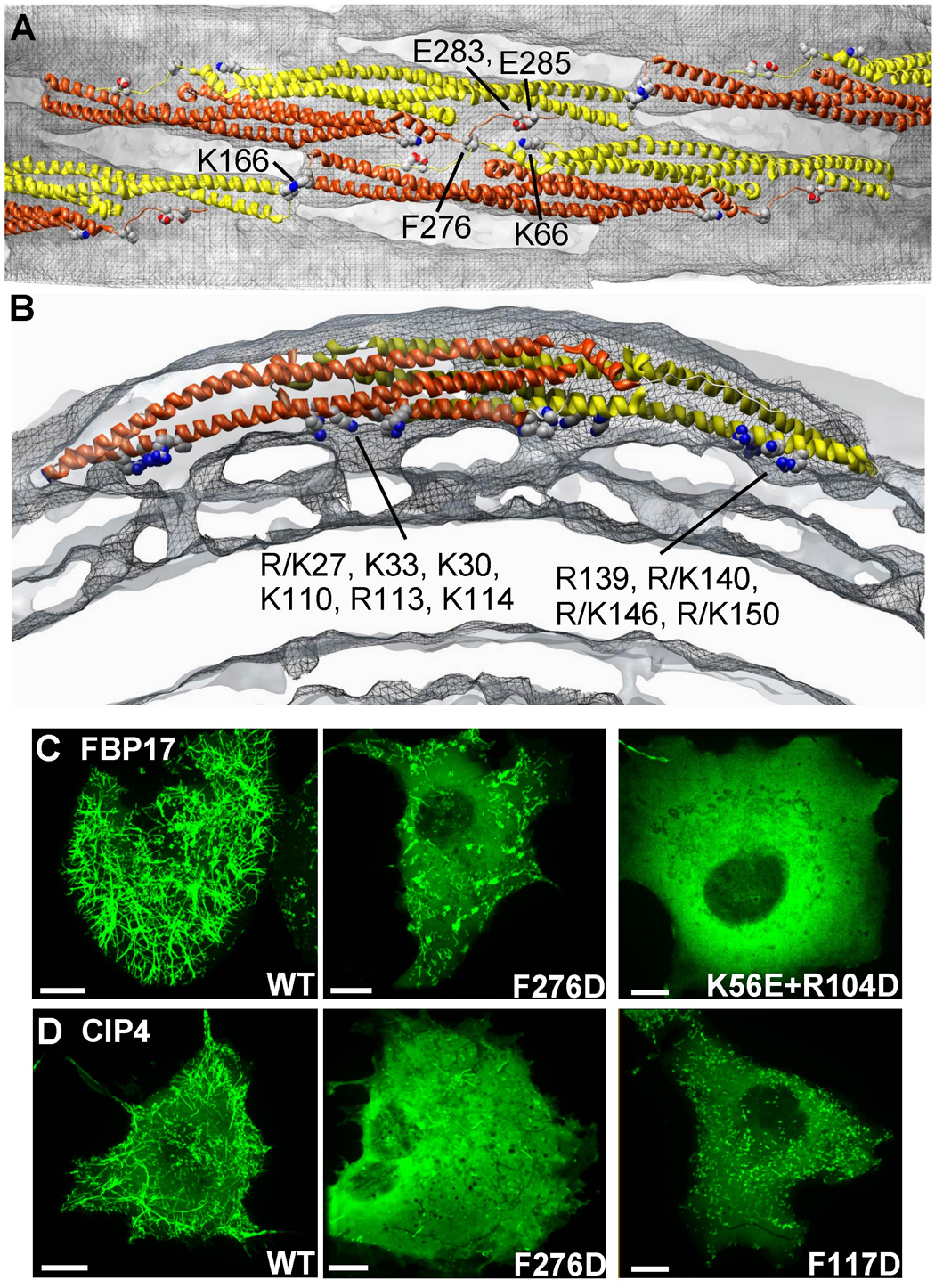
A) Surface representation of a membrane tubule perpendicular to the cylindrical axis, focused on the interactions between four neighboring F-BAR molecules. The underlying membrane is colored in grey and the protein coat in grey mesh. One monomer of each F-BAR module is in yellow, the other in orange-red. Conserved residues hypothesized to contribute to the tip-to-tip and lateral interactions are annotated and shown with space-filling atoms. B) Cross-sectional slab through one dimer parallel with the plane of the tip-to-tip interactions. The four resolved points of membrane binding correspond with clusters of conserved, cationic residues found along the concave faces of both dimers, where R/K indicates the amino acid found in CIP4 or FBP17, respectively (Shimada et al., 2007). C) Representative images of COS7 cells with high levels of expression of wild type or mutated constructs of GFP-FBP17 or D) GFP-CIP4. Some mutations completely abolish membrane localization, while others only compromise tubule formation. Bars 10 µm.
To identify which residues participated in membrane binding at these four sites, the atomic coordinates of the F-BAR domains of human FBP17 and CIP4 were fit into the map manually and then refined using algorithms implemented in UCSF Chimera (Pettersen et al., 2004) or the program SITUS (Pettersen et al., 2004; Wriggers et al., 1999) with equivalent results. The agreement for the fit of both structures was not surprising given that their crystal structures superimposed to within 2.15Å rmsd between corresponding Cα-atoms (Shimada et al., 2007) (see Figure S4). As illustrated in Figure 4, two regions of membrane binding near the center of the module appeared to correspond with the cationic clusters composed of R/K27, K30, K33, K110, R113, K114, and R/K150 (where R/K indicates the amino acid found in CIP4 or FBP17, respectively). Two additional areas of contact nearer the dimer’s tips appeared to correspond with a cluster of cationic residues composed of R139, R/K140, R/K146 and R/K150. Consistent with this interpretation of our map, mutating residues that line the concave face, including K33E, K33Q, R113Q and K114Q, compromise membrane binding and tubule formation (Shimada et al., 2007; Tsujita et al., 2006).
Importantly, there was no evidence at this resolution that extended amphipathic sequences were partially intercalated into the bilayer—distinguishing F-BAR mediated tubulation from the combination of scaffolding plus amphipathic “wedges” employed by N-BAR domains (Farsad et al., 2001; Gallop et al., 2006). However, it is possible, given the moderate resolution of this analysis, that isolated residues shallowly inserted into the outer leaflet of the bilayer. To explore this possibility, we noted from the fit of the atomic coordinates into our reconstruction that F117 faced the membrane from the concave surface of the domain and that it was surrounded by the hydrophobic alkane moieties of cationic residues that mediated binding to lipid headgroups (Figure S5C). To test whether possible insertion of F117 into the bilayer contributed to membrane-binding or tubulation, we mutated F117 to Ala and Asp, respectively. The F117A mutant had no observable defects in membrane tubule formation. Given the smaller volume occupied by the hydrophobic side chain of Ala, this suggested that tubulation did not require insertion of a bulky Phe amongst the acyl chains of the membrane. Further supporting this idea, when F117 was mutated to Trp in a Trp-less variant of the domain, no blue-shift of the fluorescence emission spectrum was detectable (data not shown, (Ladokhin et al., 2000)). In contrast, the F117D mutation in FBP17 F-BAR domains potently inhibited tubulation in every reaction condition tested in vitro (Figure 6A), while the corresponding mutation in full-length CIP4 also inhibited tubulation in living cells (Figure 4D). This observation suggested that the F117D mutant was defective in forming high-affinity interactions with the membrane surface via its concave surface. We speculate that the functional defect arose because the Asp strongly interacted with its neighboring cationic residues, partially neutralizing the surface potential and perhaps preventing conformational extensions of Lys and Arg residues toward the membrane surface. In support of the latter, we noted that the molecular envelope of the dimer was continuous with the bilayer exclusively in the four positions that, based on the fit of the crystal structure, were occupied by cationic clusters.
Figure 6. Mutant Phenotypes & Tubule Persistence Lengths.
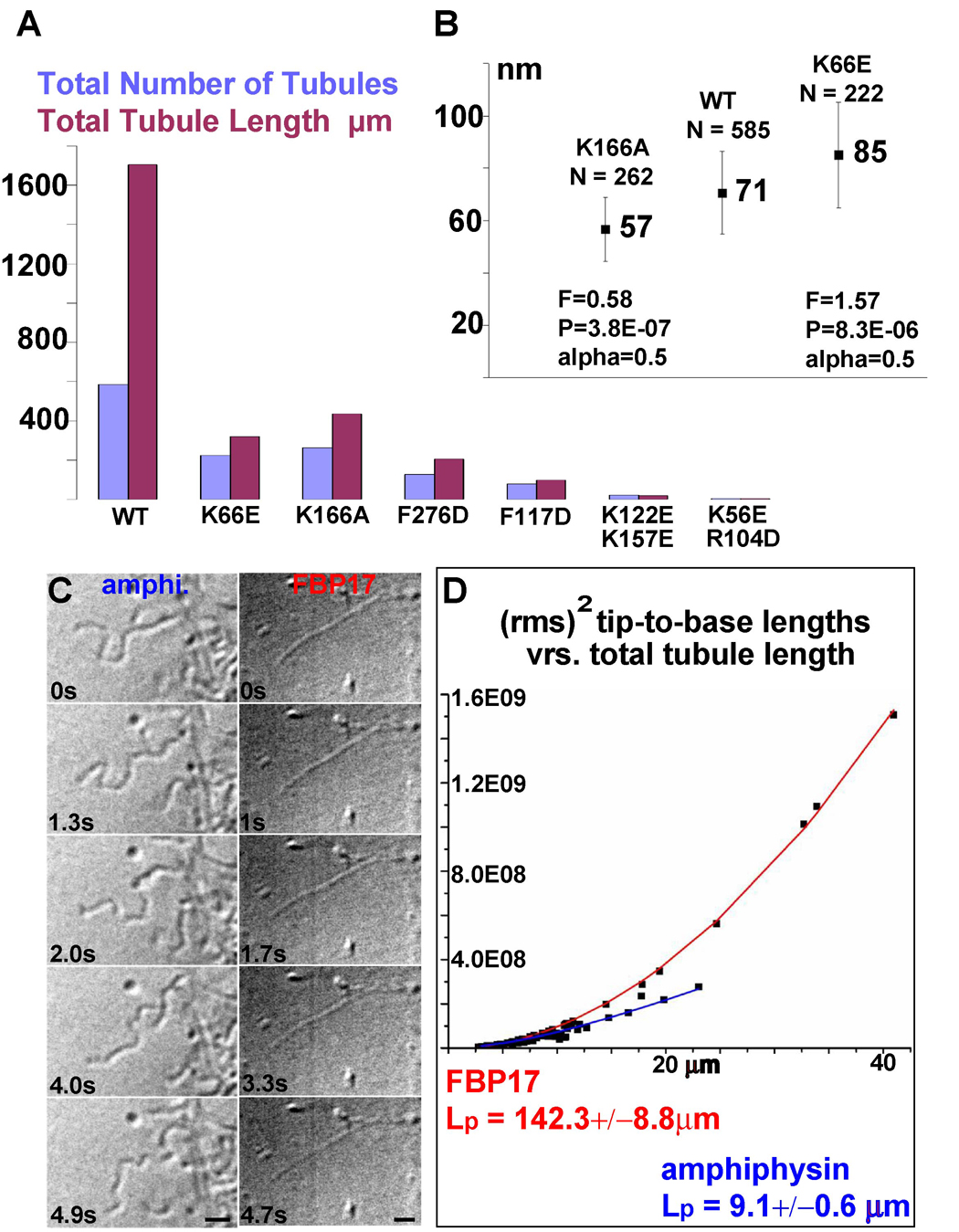
A) Quantification of total tubule number, total tubule length (sum of all tubule lengths measured) as determined from 50 low magnification images evenly sampling one EM grid. B) Quantification of mean tubule diameter from low-magnification images like those in Figure S5A–B. Error bars are the standard deviation for the population of tubules. C) Still images of video-DIC recordings of amphiphysin and FBP17. D) Each data point is the rms2 (root mean square to the square) of 100 tip-to-base measurements of a tubule as a function of total tubule length. The persistence length (Lp) was determined by fitting the data with a theoretical curve according to the equation: δR2(t) = 2(Lp)2[x/Lp − 1 + e(−x Lp)]; where δR2(t) is the rms2, x the total length and Lp the persistence length (Derenyi et al., 2002; Le Goff et al., 2002a; Le Goff et al., 2002b). Bars (A,B) 10 µm (C) 5 µm.
F-BAR DOMAINS SELF-ORGANIZE INTO HELICAL LATTICES TO INDUCE TUBULE FORMATION
An important finding of our study was that scaffolding by individual F-BARs was necessary but not sufficient for tubule formation. Specifically, the reconstruction demonstrated that tubule formation involved the collective assembly of F-BARs into a helical coat that propagated curvature around and along the growing tubule. The helical lattice was held together by tip-to-tip and extensive lateral interactions (Figure 3A–B, Figure 4A). Of the two, only the tip-to-tip interaction, mediated in part by K166 in the loop between the α3 and α4 helices, was predicted by the crystal structures (Shimada et al., 2007). Importantly, the 6.3Å translation and 40.3° rotation separating each dimer along the helical path defined by the tip-to-tip interaction did not allow preservation of the reported hydrogen bonding pattern, when the dimer was fit as a rigid body. In fact, attempts to maintain the hydrogen bonding pattern as seen in the crystal structure resulted in a ~50% decrease in the correlation coefficient between our map and the structure.
The possibility that the tip-to-tip interaction was flexible or underwent rearrangement during polymerization of the helical coat was consistent with the existence of an additional, lateral contact between neighboring dimers (Figure 3A–B, Figure 4A). This broad overlapping interaction involved 50% of the dimer’s lateral surface, including the loop between α2 and α3, segments of the lateral surface of α3 and α5, and the C-terminal extended peptide (Shimada et al., 2007). Notably, the near-atomic model generated by fitting the crystal structures into our reconstruction suggested specific contacts that may have been important for the formation of the lateral interactions. This included ionic interactions between K66 or K273 in one dimer and E285 or D286 in the other, as well as between D161 or N163 in one dimer and R47 or K51 in the other (Figure 4A and Figure S4). There also appeared to be hydrophobic interactions, including one between directly opposing F276 in both dimers (Figure 4A and Figure S4). As shown in the bottom panel of Figure S4, these surface exposed residues are among the most highly conserved throughout the evolution of the Toca proteins, and are oriented on the surface of the model such that if they do participate in lateral interactions, neighboring dimers would overlap by ~50% of their length.
Because of the large number of potential pairwise interactions, we did not probe the importance of all these residues for the purpose of this study. Nevertheless, analysis of a total of 14 mutants along the lateral interface (7 each for CIP4 and FBP17 respectively) revealed that most of them affected tubulation behavior to some extent. Shown here are examples illustrating the spectrum of effects that were observed in living cells with full-length CIP4 or FBP17 (Figure 4C–D) and in vitro with purified FBP17 FBAR domains (Figure 6A–B). Considering the large surface area involved in the lateral interaction, we were surprised that some point mutations did compromise tubule formation both in vitro and in living cells, at least as potently as the previously reported mutation of the tip-to-tip residue K166 to Ala (Figure 6A and (Shimada et al., 2007). Specifically, replacing F276 with the charged residue Asp in full-length FBP17 and CIP4 potently inhibited tubule formation in living cells (Figure 4C–D). Similarly, the same mutation strongly compromised tubulation in vitro using isolated F-BAR domains (Figure 6A). Similarly, reversing the charge of K66 inhibited tubule formation when compared with wild type FBP17 F-BAR domains when assayed under equivalent in vitro conditions (Figure 6A, see methods).
The importance of the lateral interactions was further underscored by electron microscopic examination of the filaments formed in the absence of liposomes by the F-BAR domains of FBP17 or CIP4 (Itoh et al., 2005). Being 12–13 nm thick and with a 4–5 nm repeat distance, they must be composed of both lateral and tip-to-tip interactions (Figure S7), which contrasts with a previous proposal that the length of the F-BAR dimer corresponds to one periodic repeat of these filaments (Shimada et al., 2007).
VARIABILITY IN COAT ARCHITECTURE ALLOWS A RANGE OF TUBULE DIAMETERS TO FORM
F-BAR domains generate different diameter tubules in vitro and in living cells (Figure 1C–D, Figure 2B). To determine the structural basis for this variability, we calculated independent reconstructions of tubules with different diameters. These volumes revealed that F-BARs rotated relative to the tubule's cylindrical axis while maintaining their intrinsic curvature (Figure 5). Specifically, tubules with a diameter near the population mean of 67 nm had ~9.5 tip-to-tip dimers around the circumference. In these cases, the long axis of the dimer was only slightly tilted relative to the cylindrical axis, such that a thread of tip-to-tip dimers wrapped around the tubule with a shallow, right-handed twist. Subtle variability in the tilt angle of the dimer still produced resolvable differences in the helical symmetry, and precluded averaging data from different tubules of the same apparent diameter (Figure 5B versus 5C). In contrast, the smallest tubule observed was ~57 nm in diameter and accommodated only ~8 dimers around its circumference (Figure 5A). In this case, fitting the dimers into the map suggested that each F-BAR had a left-handed tilt, relative to the cylindrical axis, and the tip-to-tip contacts apparently did not form (Figure 5A, white asterisks).
Figure 5. Independent Reconstructions of Tubules with Different Diameters and Symmetries.
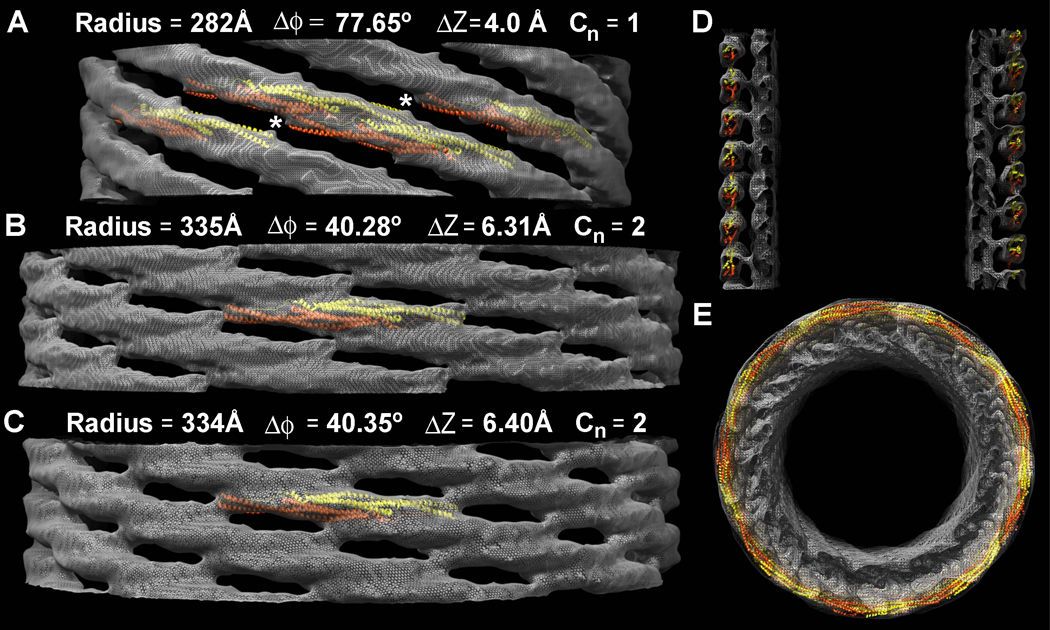
A) The narrowest tubule reconstructed is ~56 nm in diameter, with ~8 tip-to-tip dimers around its circumference. Tilting the long axis of the dimer relative to the cylindrical axis produces a narrower tubule. In this case, the dimers are so steeply tilted that the tip-to-tip contacts appear to be broken (white asterisks). The tubule has no rotational symmetry; the fundamental (J+1) helical symmetry does not describe an inter-molecular contact. Only the near side of the lattice is shown and the underlying membrane has been masked out to emphasize differences in the protein coat. Atomic models of F-BAR domains were fit into the map as rigid bodies. B&C) Two tubules with the same apparent diameter and ~9.5 tip-to-tip dimers around their circumference have resolvable differences in their helical symmetry. D) Central section along the longitudinal axis of the thinnest tubule shown in ‘A’, demonstrating that the density of the protein coat accommodates rigid atomic models of the F-BAR module that are tilted relative to the cylindrical axis, but whose radius of curvature is unchanged. E) View along the cylindrical axis of the thinner reconstruction shown in ‘A’ and ‘D’.
To test the hypothesis that F-BARs rigidly maintain their intrinsic degree of curvature—even when bound to tubules with smaller diameters—we used the tubule radii and the pitch of the left-handed helical path defined by the lateral interactions to calculate the helical arc length between the center of one dimer and its nearest lateral neighbors. The helical arc lengths for all three tubules—as measured from the reconstructed volumes—were calculated to be 114 +/− 1.5 Å (see methods). This indicates that F-BARs bound to the smaller tubules were not appreciably deformed and that they overlapped their neighbors by the same length, despite being tilted relative to the cylindrical axis (Figure 5A). Moreover, using a complete model of the protein coat, built from 128 copies of the CIP4 F-BAR domain structure, we observed that the density corresponding with the protein coat of the thinnest tubule could be entirely accounted for with rigid but tilted F-BAR modules, interacting via the same lateral contacts (Figure 5D–E). Finally, abolishing the tip-to-tip interaction with the K166A mutation, which produces some tubules in vitro but not in living cells (Shimada et al., 2007), appears to bias the population distribution of tubule diameters toward smaller diameters (Figure 6A–B). This was consistent with the hypothesis that in the absence of the tip-to-tip constraint, F-BAR modules are more likely to tilt relative to the tubule axis and thus produce narrower tubules. In contrast, compromising the formation of the lateral contacts with the K66E mutation biased the population distribution of tubule diameters toward larger diameters (Figure 6B). Taken together, these variations in coat structure, particularly in the angle between the dimer's long axis and the cylindrical axis, emphasized how plasticity in the lattice allows rigid dimers to accommodate a range of curvatures. To our knowledge, this is the first experimental demonstration of this previously predicted corollary of the scaffolding mechanism (Blood and Voth, 2006; Henne et al., 2007).
THE F-BAR COAT IS MORE RIGID THAN N-BAR OR DYNAMIN COATS
The extensive interactions between dimers observed in the reconstruction explained why F-BAR coated tubules seemed to be more rigid than tubules coated by classical BAR domains. To quantify the difference in the rigidities of N-BAR and F-BAR tubules in vitro, we calculated their persistence length, Lp, which is a measure of macromolecular rigidity expressed as the length over which correlations in the direction of the tangent are lost. For FBP17-coated tubules, we determined an Lp of 142.3±8.8 µm. In comparison, amphiphysin-coated tubules had an Lp of 9.1±0.6 µm while dynamin-coated tubules, which were used as a non-BAR superfamily control sample, had an Lp of 37.3±4.6 µm (Figure 6C–D, dynamin data not shown). The 16-fold smaller Lp for amphiphysin tubules compared with FBP17 tubules may have been due partly to their thinner diameter, though it would be of interest to determine whether N-BAR dimers form less extensive inter-molecular contacts than those of the F-BAR coat. Similarly, the 4-fold smaller Lp observed for dynamin, which also forms tubular coats, suggested that the contacts making up the dynamin coat were either less constraining than those of the F-BAR coat or, less-likely, that F-BAR dimers were more rigid than dynamin dimers (Chen et al., 2004; Zhang and Hinshaw, 2001).
F-BAR DOMAINS BIND TO FLAT MEMBRANES VIA A SURFACE OTHER THAN THEIR CONCAVE FACE
Raw micrographs of liposomes in the midst of being transformed into tubules by the F-BAR domain displayed regions of the membrane that were clearly decorated by bound protein molecules, but whose curvature had not yet appreciably changed (Figure 2A, enlarged insets). Fortuitously, we were able to observe this intermediate state directly by cooling liposomes below the Tm of the most abundant lipid species used in our experiments (palmitoyl-oleyl-phosphatidylserine), which presumably increased membrane rigidity (Figure S4A–B). Both FBP17 and CIP4 F-BARs bound avidly to these rigid membranes, and formed 2D arrays in which laterally-adjacent dimers aligned in almost perfect register while maintaining tip-to-tip interactions (Figure 7A–C, Figure S6A). Unexpectedly and regardless of whether crystals were negatively stained or vitrified, a dozen projection density maps calculated from similar but not identical crystals lacked any evidence of the two-fold symmetry that would be observed if the dimers were bound symmetrically to the bilayer via their concave surface (Table S1, Figure S8). The simplest explanation for this absence of a two-fold rotational symmetry was that the domains were bound to the membrane obliquely, such that their maximum curvature surface could not be imposed.
Figure 7. F-BAR Domains Bind to Flat Membranes via a Surface Other Than Their Concave Face.
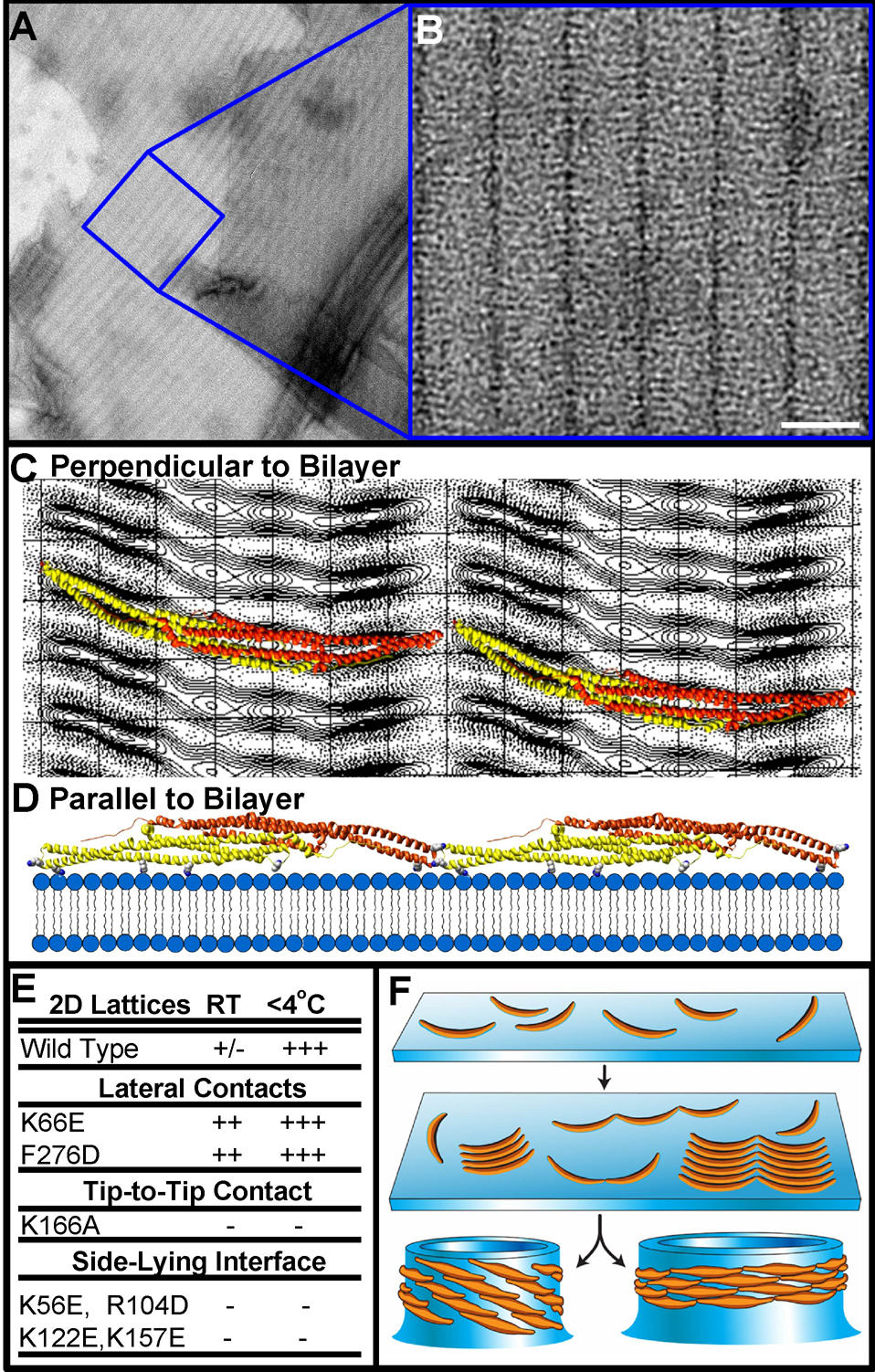
A) Electron micrograph of negatively-stained membranes that were pre-cooled before incubation with human FBP17 F-BAR domains (see Figure S6). B) Higher magnification of the 2D F-BAR lattice; unit cell a = 33Å b = 214Å γ = 91.0° C) Projection view of the F-BAR domain, calculated from a 3D data set composed of images from a single-axis tilt series over ±40° (representative lattice lines in Figure S7, crystal statistics in Table S1). Ribbon diagrams of the domain are superimposed over the projection image, as seen from the orientation with the highest correlation perpendicular to the membrane surface. D) Two dimers interacting tip-to-tip viewed parallel with the membrane surface, or rotated by 90° with respect to the view in ‘C’. Residues likely to mediate membrane-binding in this side-lying state are shown as space-filing atoms from left-to-right: K122, R104, K56, and K157 (see also Figures S4–S5). E) Table of mutant propensity for forming flat lattices at different temperatures. F) Proposed model in which tubule formation proceeds through observable intermediate steps. F-BARs can bind to flat or curved bilayers, clustering in arrays by forming intermolecular interactions. Following the transition to high-affinity binding of the dimer’s concave surface, formation of the lateral contacts triggers the vectorial assembly of the helical coat and drives membrane invagination.
To obtain an estimate for the orientation of the module bound to these rigid membranes, we used a reference library of evenly-spaced projection views of the FBP17 F-BAR domain atomic model (Shimada et al., 2007) to search for the highest correlation with the calculated 2D projection image (Figure 7C). A broad correlation peak was found for views in which the dimer’s two-fold axis was rotated by ~60° with respect to the membrane normal. In Figure 7C, ribbon diagrams of the domain in this orientation were superimposed over the projection image, as seen perpendicular to the membrane surface. In Figure 7D, two dimers interacting tip-to-tip are displayed as viewed parallel with the membrane surface (or rotated by 90° with respect to the view in ‘C’). In this “side-lying” state, the lateral interactions seen in the helical lattice were unable to form, while the relatively flat membrane-binding surface was composed almost entirely of one monomer (another indication that the intrinsic rigidity of F-BAR dimers exceeds the rigidity of the membrane). A similar side-lying state was recently predicted for N-BAR domains in molecular dynamics simulations, in which a range of curvatures were semi-stable depending on the degree to which the maximum curvature surface was directly imposed on the membrane (Blood and Voth, 2006).
Based on this orientation, our model predicted that the conserved residues K56, K/R104, K122, and K157 played an important role in mediating the electrostatic interaction with the membrane in this ‘side-lying state’ (Figure 7D, Figures S4, S5B,D–E). Point mutations of any of these residues to amino acids of the opposite charge abolished 2D lattice formation (Figure 7E), and were observed to have decreased tubulation efficiencies in vitro (Figure S5B). Moreover, mutating pairs of them (K122E + K157E or K56E + R104D) potently blocked tubulation in vitro with purified F-BAR domains (Figure 6A) and in living cells with full-length GFP-FBP17 (Figure 4C). The K56E + R104D mutation in full-length GFP-FBP17 in living cells was particularly striking, in that it apparently abolished membrane binding entirely and resulted in a diffuse distribution of the associated GFP-signal.
These observations suggested that F-BAR proteins may form small clusters on membrane surfaces, ready to induce tubule formation (Figure 7F). Importantly, in this side-lying state the lateral interaction surfaces of the F-BAR domain were obscured, which prevented formation of the helical lattice. In support of this idea, the 2D-lattices converted to tubular structures upon warming. The easiest explanation for this behavior would be that the reduced rigidity and membrane-bending energy above the Tm enabled individual dimers to force the bilayer to adopt their intrinsic curvature locally. Consequently, it was this transition to full imposition of the concave face that exposed the module's lateral interaction surfaces to neighboring dimers and allowed the helical coat to polymerize (Figure 7F). This proposed mechanism for tubulation predicted that inhibiting lateral interactions would shift the equilibrium away from tubule generation and towards the formation of 2D arrays. Consistent with this prediction and our model for the lateral contacts (Figure 4A, Figure S4), the mutants K66E and F276D shifted the equilibrium toward the formation of flat lattices even at temperatures >Tm of the principal lipid component (Figure S6D).
CONCLUSION
Together, the results reported here indicate that tubule formation by the F-BAR domain results through a shape-based scaffolding mechanism that is amplified by the self-assembly of a helical coat. There is no apparent contribution—at the resolution of this analysis—from the insertion of amphipathic sequences (Farsad et al., 2001; Gallop et al., 2006). As shown here, F-BAR modules readily bind flat membranes and generate curvature de novo and hence, they are more than just curvature sensors or stabilizers. There is also no obvious need to invoke membrane curvature-mediated attractive forces (Bruinsma and Pincus, 1996; Reynwar et al., 2007) since the dimers interact directly and extensively with each other. At the same time, the structural determinants of tubule formation serve to spatially segregate F-BAR activity from other membrane-binding domains. The work presented here is an important first step towards the structural exploration of membrane remodeling.
Experimental Procedures
Detailed Methods Accompany this Manuscript Online
F-BAR Domain Purification & Mutagenesis
cDNA fragments encoding human FBP17 (1–303) and CIP4 (1–284) were subcloned into pGEX6P-1 (Amersham Biosciences, Piscataway, NJ) with codons for six additional histidine residues inserted at the C-terminus via PCR. Fusion proteins were bacterially expressed and purified first on a nickel affinity resin and then on a GST-glutathione affinity column. The GST tag was cleaved, followed by gel filtration chromatography in buffer (350 mM NaCl/250mM Imidazole/20mM HEPES/1 mM DTT, pH 7.4). Aliquots of 1–3 mg/ml protein were stored at −80° C. Site-directed mutation of was performed via the quick-change protocol (Stratagene, La Jolla, CA).
Electron Microscopy
2D and helical arrays were screened using 1% uranyl acetate-stained samples and a Philips Tecnai 12 microscope operating at 120 kV. Cryo images were taken at a sample temperature of −172°C under low-dose conditions on a Philips Tecnai F20 microscope equipped with a field emission gun and operating at an accelerating voltage of 160–200 kV, nominal magnifications of 29–50kx, and defocus values of −1,500 to −22,000 Å.
2D Crystal Image Processing
Images of 2D crystals were corrected for lattice distortions, effects of the contrast transfer function, and astigmatism using the MRC image-processing software package (Crowther et al., 1996; Smith, 1999). A single-axis tilt series including 13 images from ±40° of a single negatively stained crystal was acquired. Applying the proper tilt geometry, all 13 images were brought to their common phase origin, merged and subjected to an additional round of origin and geometry refinement. After fitting a final set of lattice lines (Figure S8), the projection structure was calculated from the 3D-data set using a B-factor of −5000Å−2.
Helical Image Processing
Fourier–Bessel reconstruction proved to be limited in recovering high resolution features from these tubules because of flexibility and the multiple image interpolations required for axis alignment and straightening. Moreover, helices with the same apparent diameter proved to have distinct helical symmetries, precluding reciprocal space averaging. We therefore reconstructed individual tubes with an extended version of the Iterative Helical Real Space Reconstruction (IHRSR) single particle algorithm as implemented in SPIDER (Egelman, 2000; Frank et al., 1996).
Supplemental Data
Supplemental information, including a full methods section, eight additional figures, and one table accompany this manuscript online.
Supplementary Material
Acknowledgments
We thank Derek Toomre for his direction with the live cell imaging experiments. We thank Bridget Carragher and the staff at the National Resource for Automated Molecular Microscopy—which is supported by the National Institutes of Health (NIH) through the National Center for Research Resources (NCRR) P41 program (RR17573)—where some of the data was collected. We also thank Robert Bjornson and Nicholas Carriero at the Yale Center for High Performance Computation in Biology and Biomedicine, which is supported by NIH grant NCRR 19895-02. Finally, we thank Fred Sigworth and David Chester for their advice and helpful discussions. This work was supported by a Pre-Doctoral Research Training Fellowship from the Epilepsy Foundation (A.F.); the European Molecular Biology Organization Long-Term Postdoctoral Fellowship program and the Cross-Disciplinary Fellowship program of the Human Frontier Science Program (A.R.); and a G. Harold and Leila Y. Mathers Charitable Foundation grant (P.D.C). This work was also supported in part by the following NIH grants: MSTP TG 5T32GM07205 (A.F.), GM071590 (V.M.U.), EB001567 (E.H.E.), CA46128 and DK45735 (P.D.C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ayton GS, Blood PD, Voth GA. Membrane remodeling from N-BAR domain interactions: insights from multi-scale simulation. Biophys J. 2007;92:3595–3602. doi: 10.1529/biophysj.106.101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood PD, Voth GA. Direct observation of Bin/amphiphysin/Rvs (BAR) domain-induced membrane curvature by means of molecular dynamics simulations. Proc Natl Acad Sci U S A. 2006;103:15068–15072. doi: 10.1073/pnas.0603917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett TJ, Traub LM. Molecular structures of coat and coat-associated proteins: function follows form. Curr Opin Cell Biol. 2006;18:395–406. doi: 10.1016/j.ceb.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Bruinsma R, Pincus P. Protein aggregation in membranes. Current Opinion in Solid State & Materials Science. 1996;1:401–406. [Google Scholar]
- Casal E, Federici L, Zhang W, Fernandez-Recio J, Priego EM, Miguel RN, DuHadaway JB, Prendergast GC, Luisi BF, Laue ED. The crystal structure of the BAR domain from human Bin1/amphiphysin II and its implications for molecular recognition. Biochemistry. 2006;45:12917–12928. doi: 10.1021/bi060717k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Zhang P, Egelman EH, Hinshaw JE. The stalk region of dynamin drives the constriction of dynamin tubes. Nat Struct Mol Biol. 2004;11:574–575. doi: 10.1038/nsmb762. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Henderson R, Smith JM. MRC image processing programs. J Struct Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Derenyi I, Julicher F, Prost J. Formation and interaction of membrane tubes. Phys Rev Lett. 2002;88:238101. doi: 10.1103/PhysRevLett.88.238101. [DOI] [PubMed] [Google Scholar]
- Egelman E. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, McMahon HT. Mechanism of endophilin N-BAR domain-mediated membrane curvature. Embo J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Kent HM, Ford MGJ, Hegde BG, Daumke O, Butler PJG, Mittal R, Langen R, Evans PR, McMahon HT. Structure and Analysis of FCHo2 F-BAR Domain: A Dimerizing and Membrane Recruitment Module that Effects Membrane Curvature. Structure. 2007;15:1–4. doi: 10.1016/j.str.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Hinshaw JE, Schmid SL. Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature. 1995;374:190–192. doi: 10.1038/374190a0. [DOI] [PubMed] [Google Scholar]
- Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Itoh T, De Camilli P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim Biophys Acta. 2006;1761:897–912. doi: 10.1016/j.bbalip.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Itoh T, Erdmann K, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Kamioka Y, Fukuhara S, Sawa H, Nagashima K, Masuda M, Matsuda M, Mochizuki N. A novel dynamin-associating molecule, formin-binding protein 17, induces tubular membrane invaginations and participates in endocytosis. J Biol Chem. 2004;279:40091–40099. doi: 10.1074/jbc.M404899200. [DOI] [PubMed] [Google Scholar]
- Ladokhin A, Jayasinghe S, White S. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother. Anal Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- Le Goff L, Amblard F, Furst EM. Motor-driven dynamics in actin-myosin networks. Phys Rev Lett. 2002a;88:018101. doi: 10.1103/PhysRevLett.88.018101. [DOI] [PubMed] [Google Scholar]
- Le Goff L, Hallatschek O, Frey E, Amblard F. Tracer studies on f-actin fluctuations. Phys Rev Lett. 2002b;89:258101. doi: 10.1103/PhysRevLett.89.258101. [DOI] [PubMed] [Google Scholar]
- Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- Lee S, Kerff F, Chereau D, Ferron F, Klug A, Dominguez R. Structural Basis for the Actin-Binding Function of Missing-in-Metastasis. Structure. 2007;15:145–155. doi: 10.1016/j.str.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Mao X, Dong LQ, Liu F, Tong L. Crystal Structures of the BAR-PH and PTB Domains of Human APPL1. Structure. 2007;15:525–533. doi: 10.1016/j.str.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, Mochizuki N. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. Embo J. 2006;25:2889–2897. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, Pykalainen A, Saarikangas J, Paavilainen VO, Vihinen H, Jokitalo E, Lappalainen P. Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism 10.1083/jcb.200609176. J Cell Biol. 2007;176:953–964. doi: 10.1083/jcb.200609176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard TH, Bompard G, Heung MY, Dafforn TR, Scott DJ, Machesky LM, Futterer K. Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. Embo J. 2005;24:240–250. doi: 10.1038/sj.emboj.7600535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, Ferrin T. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Reynwar BJ, Illya G, Harmandaris VA, Muller MM, Kremer K, Deserno M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 2007;447:461–464. doi: 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- Rodriguez Y, Mezei M, Osman R. Association free energy of dipalmitoylphosphatidylserines in a mixed dipalmitoylphosphatidylcholine membrane. Biophys J. 2007;92:3071–3080. doi: 10.1529/biophysj.106.089078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Niwa H, Tsujita K, Suetsugu S, Nitta K, Hanawa-Suetsugu K, Akasaka R, Nishino Y, Toyama M, Chen L, et al. Curved EFC/F-BAR-Domain Dimers Are Joined End to End into a Filament for Membrane Invagination in Endocytosis. Cell. 2007;129:761–772. doi: 10.1016/j.cell.2007.03.040. [DOI] [PubMed] [Google Scholar]
- Smith JM. Ximdisp--A visualization tool to aid structure determination from electron microscope images. J Struct Biol. 1999;125:223–228. doi: 10.1006/jsbi.1998.4073. [DOI] [PubMed] [Google Scholar]
- Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- Takei K, Slepnev VI, Haucke V, De Camilli P. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat Cell Biol. 1999;1:33–39. doi: 10.1038/9004. [DOI] [PubMed] [Google Scholar]
- Tarricone C, Xiao B, Justin N, Walker PA, Rittinger K, Gamblin SJ, Smerdon SJ. The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. 2001;411:215–219. doi: 10.1038/35075620. [DOI] [PubMed] [Google Scholar]
- Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T. Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tubulation domain of PCH proteins is involved in endocytosis 10.1083/jcb.200508091. J Cell Biol. 2006;172:269–279. doi: 10.1083/jcb.200508091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Bose PS, Sigworth FJ. Using cryo-EM to measure the dipole potential of a lipid membrane. Proc Natl Acad Sci U S A. 2006;103:18528–18533. doi: 10.1073/pnas.0608714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W. Crystal structure of the endophilin-A1 BAR domain. J Mol Biol. 2005;351:653–661. doi: 10.1016/j.jmb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Zhang P, Hinshaw JE. Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol. 2001;3:922–926. doi: 10.1038/ncb1001-922. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chen J, Liu J, Brunzelle JS, Huang B, Wakeham N, Terzyan S, Li X, Rao Z, Li G, Zhang XC. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. Embo J. doi: 10.1038/sj.emboj.7601771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, McLaughlin S. Membrane Curvature: How BAR Domains Bend Bilayers. Current Biology. 2004;14:R250–R252. doi: 10.1016/j.cub.2004.02.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.