

Abstract
During 25 years of research since HIV-1 was first identified in Paris, there have been great advances in our understanding of the virus and of the immune system. Practical advances include the early development of diagnostic tests of infection that made blood donation safe, and since 1996, combination anti-retroviral therapy that has great reduced incidence of AIDS in HIV-infected people who have access to the drugs. HIV prevention through behavioural change has been successful, and we do not yet have any safe and efficacious microbicides or vaccines.
Keywords: AIDS, HIV, immunopathogenesis, therapy, vaccine
Introduction
It is fitting, on the 25th anniversary of the discovery of HIV-1 [1], to look back on past achievements and to look forward to the daunting challenges we still face in order to overcome the AIDS pandemic. In this perspective, I shall not be so foolhardy as to attempt to provide a comprehensive review of HIV and AIDS; for that I recommend Jay Levy's book [2]. Rather, I provide a personal view of salient discoveries and remaining gaps in our knowledge. There have, of course, been magnificent advances in diagnosis and therapy, and in gaining insight into the immunopathogenesis of AIDS. However, despite sustained efforts by many talented investigators, we still appear to be near the starting block for controlling HIV through prevention.
The appearance of a novel type of immune deficiency was presaged in the summer of 1981 when a handful of young homosexual men in New York, San Francisco and Los Angeles were diagnosed with Pneumocystis carinii pneumonia and Kaposi's sarcoma (KS) [3]. Epidemiologists at the Center for Disease Control and Prevention in Atlanta detected this unusual clustering of patients and set out to investigate its provenance. It was soon recognized that the underlying immune deficiency of ‘gay compromise syndrome’ was associated with a selective depletion of CD4+ T helper cells [4,5]. In 1982 it became apparent that there must be an infectious agent inducing the disease when it was also found in injecting drug users and in recipients of blood transfusions, and thus the disease acquired the name ‘acquired immune deficiency syndrome’[6]. With the manifestation of AIDS in patients with haemophilia [7], the speculations were on some kind of virus as the cause because other microbes were unlikely to taint pooled clotting factors. In fact, the epidemiologists had elucidated the risk groups and modes of transmission accurately before the discovery of HIV itself.
In the early days, AIDS was a disease about which the patients often knew more than their doctors [8]. Because AIDS affected gay men disproportionately, there was an articulate, well-educated and assertive body of people with strong networks who could ask awkward questions and challenge any whiff of patronizing attitudes among physicians. They established advocacy groups to lobby on social and medical issues affecting HIV and AIDS. It was gay men at risk of AIDS who pioneered the electronic exchange of medical knowledge, which is now a commonplace source of information for any disease. It is a curious coincidence that the burgeoning of the HIV pandemic has been paralleled by the exponential expansion of the internet. The downside is that the worldwide web is also a superb medium through which to perpetuate myths of HIV denial (or blame and conspiracy theories) concerning AIDS. It is a tragedy that these siren voices of the so-called AIDS dissidents won the sympathy of the leader of a nation with more HIV-infected people than any other, despite our efforts to correct the situation [9–11].
Discovery of HIV-1
On 23 May 1983 Françoise Barré-Sinoussi and colleagues in Paris, led by Luc Montagnier at the Institut Pasteur, published a description of a previously unknown virus isolated from a patient with lymphadenopathy call ‘Bru’[1]. Because virus replication was associated with reverse transcriptase (RT) activity, it was assumed to be a retrovirus (Fig. 1). This virus was cytopathic and it could be propagated serially – with tender loving care and to low titre – only by adding medium harvested from dying cultues to fresh cultures of activated peripheral blood mononuclear cells (PBMC). We would now call Bru a primary ‘R5’ isolate because it has a tropism for primary T cells and macrophages that express CCR5, and it will not grow in T cell lines that express CXCR4.
Fig. 1.
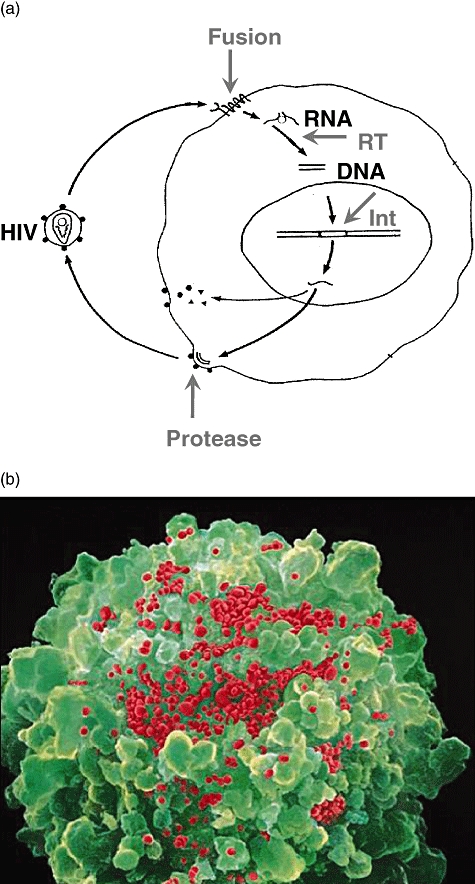
(a) Simplified replication cycle of HIV. (b) Scanning electronic micrograph of a lymphocyte releasing progeny HIV.
In retrospect, insufficient note was initially taken of Barré-Sinoussi's paper because it was but one of many candidate infectious agents. Indeed, an accompanying paper in the same issue of Science from Bob Gallo's and Max Essex's groups suggested another type of retrovirus, human T cell leukaemia virus (HTLV-1), as a possible cause of AIDS [12,13]. The detection of HTLV-1 was genuine enough, but in hindsight it turned out to be a ‘passenger’ virus in about 10% of patients with immunodeficiency. By April 1984, Montagnier's group had published on further HIV isolates [14], including one from an AIDS patient ‘Lai’ that grew to much higher titre and which we now call an ‘X4’ isolate because it can grow in T cell leukaemic cell lines expressing CXCR4. In addition, Montagnier's electron microscopic study [15] indicated that HIV resembled animal lentiviruses rather than deltaviruses such as HTLV-1. This interpretation was vindicated firmly in 1985, when Simon Wain-Hobson and colleagues sequenced and interpreted correctly the open reading frames of the genomes of both HIV and the prototype lentivirus, Maedi-Visna virus (MVV), of sheep [16,17].
Shortly after Montagnier's second publication, on HIV Lai [14], Gallo and Popovic at the National Institutes of Health (NIH) [18], and then Levy in San Francisco [19] published on their isolates of HIV. Each group gave their virus a different name. Eventually DNA cloning and genome sequencing (which were still labourious processes) confirmed that all these viruses belonged to a single species, but revealed that genuinely different isolates were surprisingly variable in sequence. This genome diversity later allowed Wain-Hobson to perform a neat forensic DNA analysis on how the Lai isolate had become an opportunistic contaminating agent in several laboratories, including mine [20]. The AIDS virus was variously called LAV [1], IDAV [14], HTLV-III [18] and ARV [19]. The term HIV was not coined until 1986, when an international committee chaired by Harold Varmus sought to rationalize the confusing terminology [21].
Oddly enough, it was a consortium of investigators in London that first demonstrated that Gallo and Montagnier were studying the same type of virus. Rachanee Cheingsong-Popov brought Lai from Paris to my laboratory in February 1984 and I obtained IIIB from NIH 3 months later. We found that both viruses grew to high titre in the CEM T cell line, and with Richard Tedder we established a competition radioimmune assay [22] employing HIV antigens from CEM cells to detect serum antibodies in British patients with AIDS and subjects in AIDS risk groups collected by Brian Gazzard, Tony Pinching, Jonathan Weber and Ian Weller. The same samples reacted positively to both viruses, just as Levy [19] had found for LAV and ARV, so we performed a simple, old-fashioned experiment using immunofluorescence: when antibodies were absorbed onto excess cells infected with HTLV-III, all reactivity to Lai was also removed from the serum sample [22].
Our serological test helped to establish that AIDS was not just a western disease, but was spreading rapidly in Africa. In 1984, patients with symptoms similar to HIV had been noted in both sexes in Congo [23] and Rwanda [24], and Montagnier's group had isolated HIV from one of them [25]. In Uganda, people realized that a novel affliction had first appeared among them in 1982 in the wake of Milton Obote's army, which liberated the country from Idi Amin's grip and opened up the truck routes for traders. They called it ‘Slim’ disease because the wasting syndrome and diarrhoea were its most notable symptoms. Although KS had always been a relatively frequent tumour in Africa, a new aggressive form in young adults had sprung up. Together with Anne Bayley in Lusaka and David Serwadda and Nelson Sewankambo in Kampala, we showed by serology that both aggressive KS [26] and Slim disease [27] were associated with HIV infection. At first we thought that our antibody test was not sufficiently specific because 10% of the control sera taken from healthy hospital staff yielded positive results [27]. It was an awesome moment when the penny dropped and we realized that they really were infected.
The disastrous spread of HIV in southern Africa, with even higher prevalence rates, occurred later, in the 1990s. The most recent UNAIDS estimate [28] is that some 36 million people worldwide are living with HIV infection, not counting the 25 million who have already died as AIDS was recognized. Although the overall estimate of global HIV prevalence has fallen in recent years owing to more refined monitoring methods, HIV mortality has overtaken that of malaria and it is superseded only by tobacco.
Compared with the identification of the SARS coronavirus by three independent groups in 2003, the path to discovery of HIV 20 years earlier appears tentative and erratic. However, molecular techniques such as reverse transcription–polymerase chain reaction (RT–PCR) were not yet available, virus isolation needed fastidious culture in CD4-positive lymphocytes, and it was less easy to apply the classical evidence of Koch's postulates to an illness with a long incubation period. Nevertheless, by 1984, some 3 years after the recognition of AIDS as a new disease in western countries, and 1 year after the pioneering first description of HIV-1 by Barré-Sinoussi et al. [1], there was sufficient incriminating evidence to satisfy a jury of epidemiologists, virologists and immunologists (but not everyone else) that this virus was guilty beyond reasonable doubt of causing AIDS.
HIV-2, simian immunodeficiency viruses and HIV diversity
The use of animal models for human disease is well recognized for the insight that may be gained into pathogenesis, and for the investigation of candidate drugs and vaccines. However, it was found that HIV-1, like hepatitis B virus, was able to infect only chimpanzees, and even these close relatives to humans did not succumb to AIDS. As a Convention on International Trade in Endangered Species of Wild Flora and Fauna endangered species, experimental infection of the great apes was soon embargoed. Therefore, simian immunodeficiency viruses (SIV) of macaques became one of the few experimental models of HIV, and more recently recombinant hybrid HIV/SIV viruses known as SHIV have been developed for challenge tests of vaccines. It is worth recalling that SIVmac was not discovered until 1985 [29], that is, 2 years after HIV-1, and feline immunodeficiency virus at around the same time. One can state without too much irony that HIV has been a great model for veterinary lentivirus infections.
Natural infection of chimpanzees by SIVcpz was not discovered until the 1990s. The genome organization and sequence similarity to HIV-1 revealed SIVcpz to be the natural precursor of HIV-1. Recent investigations indicate which chimpanzee subspecies in which location (Gabon) is likely to have been the origin of HIV-1 Group M, the pandemic strain [30], whereas Group O-related SIV is present both in chimpanzees and gorillas [31]. Like SIVcpz becoming HIV-1 [32], SIVsm has only recently crossed species from the sooty mangabey to rhesus and cynomolgus macaques (SIVmac) and to humans (HIV-2). The emergence of SIVmac occurred in captivity, presumably when African and Asian species were housed together in primate centres in the United States. Again, like HIV-1, SIVmac is pathogenic in its new host, whereas SIVsm causes little harm to mangabeys.
The elucidation of the provenance of SIVmac played a role in the discovery of HIV-2. Max Essex and Suleyman M'Boup found that serum samples from Senegalese patients with AIDS-like symptoms reacted more strongly with SIV than with HIV-1 [33]. Then in 1986 Montagnier's group isolated HIV-2 successfully from patients in the Cap Verde islands and Senegal [34]. It appears that SIVsm has transferred from mangabeys to humans in West Africa on at least six occasions. HIV-2 is said to be less pathogenic than HIV-1 in humans, with a much slower progression to AIDS. This is, in a sense, true and HIV-2 tends to have a lower viral load, is less transmissible and mother-to-child transmission has not been demonstrated. However, the development of AIDS is bimodal: the majority of HIV-2 infected people in West Africa do not become ill (they are genuine long-term non-progressors), whereas a minority progress to AIDS at much the same rate as untreated HIV-1-infected individuals [35]. Whether the virus or the host are determinants of the different courses of infection is not yet understood, and in my opinion HIV-2 research merits further attention.
The genetic diversity of HIV has become legendary [36]. HIV-1 group M has radiated into subtypes A–K, and numerous circulating recombinant forms (Fig. 2), and the quasispecies of HIV-1 genomes in a single individual 6 years after infection is as large as the global variation in H3N2 influenza virus.
Fig. 2.
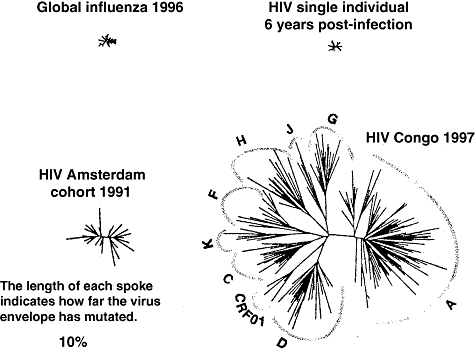
Genetic diversity of human immunodeficiency virus envelope glycoprotein gp120 compared with that of H3N2 influenza virus haemagglutinin [36].
Translating discovery into intervention
The most rapid practical health outcome following the discovery of HIV was the development of blood tests to determine who is infected. By late 1985, several diagnostic kits were marketed to detect specific antibodies to HIV. With no treatment for infection, the benefit of any of these to the individual of knowing their HIV status was debatable. For screening blood donors, however, there was an immediate public health benefit. Antibody detection still left a period between infection and seroconversion during which an individual became infectious to others. This window was first filled by testing for p24 antigen in plasma, and subsequently with the development of RT–PCR detection for viral genomes. Quantitative RT–PCR and related techniques led to the measurement of plasma viral load, which proved to be a useful prognostic marker and guide to clinical management after effective anti-retroviral drugs combinations were introduced. Later, genotypic markers of drug resistance also proved their worth. Thus molecular techniques for detection, quantification and characterization of viral genomes have played an important role in screening and in treatment.
Pharmacological treatment has also been a resounding success for those with access to it. The first anti-retroviral drug to go into clinical trial was azidothymidine (Zidovudine), which is a chain terminator for nascent DNA during reverse transcription. A phase I/II trial on HIV-infected patients in the United States in 1986 apeared so promising that the placebo arm was stopped prematurely. However, the Anglo–French Concorde trial showed that the drop in viral load and benefit to the patient was short-lived owing to the rapid emergence of Zidovudine-resistant HIV mutants in vivo[37]. Further RT inhibitors and protease inhibitors were introduced, and recently cell-entry inhibitors and integrase inhibitors have been licensed. Thus the understanding of molecular events in the virus replication cycle led to the rational design of anti-retroviral drugs (Fig. 1).
The development of new anti-retroviral drugs which did not show cross-resistance with Zidovudine took several years, but a number of RT inhibitors and protease inhibitors were developed. When these were combined as three or more drugs taken together, the era of highly active anti-retroviral therapy (HAART) was born in 1996 and the effect was dramatic. As shown in Fig. 3a, mortality fell by almost 70%, the infectious disease wards in hospitals emptied and HIV infection became a treatable condition in sexually transmitted infections (STI) departments and physicians' offices. There was even some over-optimistic speculation that HAART might eradicate HIV from the infected person's body whereas, in fact, a resurgence of viral replication occurs as soon as patients take a ‘drug holiday’.
Fig. 3.
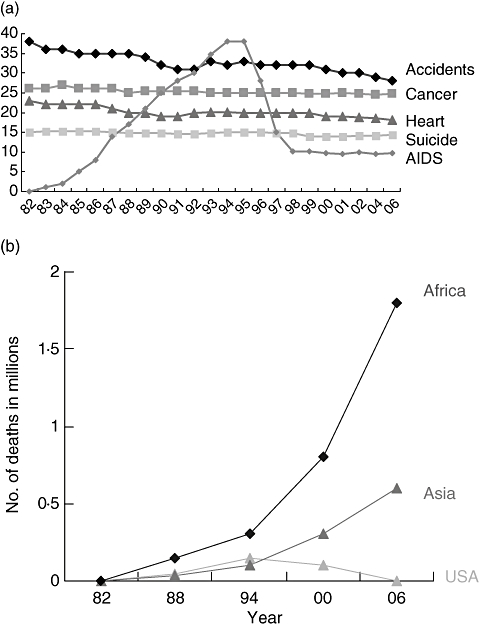
(a) Changes in five leading causes of mortality in the United States, both sexes aged 25–44 years. (b) Acquired immune deficiency virus mortality in Africa, Asia and the United States 1982–2006.
Two big challenges remain. First, will HIV in patients on HAART eventually acquire multiple resistance to the available drugs? So far, there is little evidence of multiple resistance occurring, yet increasingly drug-resistant HIV strains are being transmitted in susceptible populations. Therefore, novel drugs and drug targets are likely to be required. There continues to be debate as to when to start anti-retroviral therapy. Treatment during primary infection might improve the initial clearance of infection and immune responses to HIV [38]. Postponing initiation of treatment until the late stages of infection may delay the emergence of resistance.
The second, larger challenge is whether HAART should be rolled out to those in greatest need of treatment, often in the poorest of countries and settings. Figure 3b shows that HAART has not yet dented the estimates of AIDS mortality in sub-Saharan Africa and Asia. The logistics of delivering HAART in developing countries is more complex than providing packets of pills. A diagnosis has to be made, and the viral load and CD4 T cell measurements that help to inform treatment regimens in countries with well-developed health systems are expensive in terms of resources and trained personnel.
Behavioural and epidemiological interventions
In the absence of a safe and efficacious prophylactic vaccine against HIV, a number of proxy methods to reduce or prevent in the spread of HIV have been promoted, with mixed results. The ABC nostrum of the US Aid agency (abstinence, be faithful, and if you can't, use condoms) is a most worthy aspiration, but adherence can be difficult, especially if the partner refuses to comply. My favourite safe-sex slogan is from the Harcon AIDS Campaign in Mumbai. Its Kamasutra prescription for HIV prevention states:
Many postures with one
Better than one with many.
Safe sex advice to gay men also helped during the period when to acquire HIV infection was to be placed on death row for an indefinite period but with no hope of release. One of the downsides to HAART has been to diminish the perceived threat of HIV because it is a treatable condition.
Clean needle and syringe supply in exchange for old ones have helped reduce the risk of parenteral transmission among injecting drug users. This pragmatic approach also offended moralists, who viewed needle exchange as condoning or even encouraging illicit drug habits, so it was introduced in western Europe years before the United States.
One of the more imaginative interventions to be trialled was to target co-factors that exacerbate risk of HIV transmission. STI such Neisseria gonorrhea or Haemophilus ducreyi cause local inflammation or ulceration, and therefore STI are associated with increased HIV transmission. Different trials on the prophylatic use of inexpensive antibiotics and of acyclovir to control genital herpes have yielded mixed results for reducing HIV incidence [39]. A potential problem with prophylaxis against non-HIV STI is that the selection of resistant strains of HSV-2 and bacterial STI may eventually emerge.
Male circumcision is associated with a lower rate of HIV transmission to men [40]. The mucosal surface of the foreskin is relatively rich in HIV target cells such as CD4+ T lymphocytes and Langerhans cells [41]. In addition, lack of circumcision may be associated with more frequent or longer-lasting inflammation because of STI and adventitious infections, again heightening the risk of HIV acquisition. It has been remarkable to see in recent years how these observational epidemiological findings have been translated into intervention trials, and that young men have been willing to be assigned into randomized (if not blinded) groups for circumcision or no intervention. The results show a significant protective effective of circumcision [42].
There is much interest in vaginal microbicides which could be applied discreetly by women and prevent transmission in either direction [43]. Unfortunately, the first to be tested, the spermicide nonoxynol 9, actually increased the risk of infection in women because it had a slight inflammatory effect. Other microbicides such as polyaniomic macromolecules have not yet shown efficacy, but the notion of chemically blocking HIV at the transmission point is a good one that should not be abandoned [43].
Immunopathogenesis of AIDS
Cellular tropism and receptors
When AIDS was first recognized in 1981 immunologists had recently distinguished T helper cells from T killer or effector cells and used the T4 (CD4) and T8 (CD8) surface antigens to discriminate between them. Thus it was soon found that AIDS was associated with a disappearance of CD4 cells in the peripheral blood [4,5]. Following the discovery of HIV, David Klatzmann in Paris showed that, in vitro, HIV replicated selectively and caused a cytopathic effect in CD4 cells but not in CD8 cells [44]. This allowed his group [45] and ours [46] to demonstrate that CD4 antigen itself is the binding receptor for HIV. This sequence of findings seems logical in retrospect, but actually we had no reason to think that HIV would use exactly the same marker as that chosen by clinical immunologists for its receptor. Our study [46] benefited from a fruitful collaboration between such as Peter Beverley and Mel Greaves, and immunologists virologists such as Dorothy Crawford and myself. We used all the lymphocyte cell surface markers available at the time − some 160 monoclonal antibodies (mAbs) to CD and other antigens [including 14 anti-CD4 mAbs] − to pinpoint CD4.
After the cDNA for CD4 had been cloned, we were able to confirm our immunological findings with transfection studies. This revealed that while CD4 was needed for HIV infection and was sufficient for HIV binding to the cell surface, some other component was required for virus penetration [47]. It took a further 10 years to identify the HIV co-receptors or entry factors as chemokine receptors. The first clue came from Gallo's laboratory which found that CC chemokines regulated uopn activation normal T cell expressed and secreted (CCL5) and macrophage inflammatory protein (MIP)-αP (CCL3) inhibited HIV infection [48]. Then Ed Berger at NIH discovered, through expression cloning, that CXCR4 is the co-receptor on T cell lines [49], followed quickly by the identification of CCR5 as the co-receptor on PBMC and macrophages [50]. Identifying CCR5 as the co-receptor for the majority of transmissible strains of HIV led to the development of entry inhibitors and to discerning CCR5 polymorphisms as resistance factors, discussed later.
Macrophages were first shown to be infected by HIV by Susan Gartner in Gallo's laboratory [51]. The dogma at the time was that retroviruses could replicate only in proliferating cells, because the pre-integration complex could not cross the nuclear membrane, and required mitosis to access chromosomes. However, that proved to be true for oncogenic retroviruses but not for lentiviruses, where Vpu and other core proteins have nuclear location signals [52].
In fact, it was known that the prototype lentivirus, MVV, of sheep infects macrophages but not T helper lymphocytes [53]. It is my opinion that this observation in comparative virology provides some insight into HIV pathogenesis. MVV causes severe wasting diseases, neurodegeneration and pulmonary dysfunction, but not T cell immunodeficiency. I would postulate, therefore, that the wasting syndrome as well as AIDS dementia is essentially a disease of macrophages in human AIDS. However, sheep susceptible to MVV suffer a remorselessly progressive disease leading to death. If this represents the underlying pathogenesis common to most, if not all, lentiviral infections, then protecting CD4 T cell numbers and function without protecting macrophages will not ultimately save the patient.
The targeting of dendritic cells (DC) by HIV was more debatable than infection of macrophages. In England, Stella Knight had claimed since the 1980s that HIV infects DC [54] but was disbelieved by Ralph Steinman, although he is now a convert [55]. This controversy has been resolved largely by discerning a differential sensitivity of two types of DC, plasmacytoid (pDC) and meyloid (mDC), just as the distinction between CD4 and CD8 T lymphocytes two decades earlier helped to pinpoint which was susceptible to HIV infection [44]. In London, Steve Patterson showed that mDC express CD4 and CCR5 and hence support HIV entry and replication, whereas pDC allow binding of HIV to DC-SIGN without viral replication [56], except possibly by X4 strains during maturation. Nevertheless, the attachment HIV to DC-SIGN allows pDC to deliver HIV to susceptible CD4 T cells upon migration to the lymph nodes. The immunological synapse between pDC and CD4+ cells not only activates the T helper lymphocyte (making it more permissive to HIV replication) but also delivers the HIV particles across the synapse [57].
The course of HIV infection
A ‘typical’ course of HIV is shown in Fig. 4. Primary infection via the mucosal or parenteral route results in high viraemia, accompanied sometimes by symptoms such as fever, diarrhoea and lymphadenopathy [58]. This state of active replication and high virus load then resolves to a lower set point, and the level of this point is predictive of the rate of progression to AIDS in untreated individuals: the higher level the worse the prognosis [59].
Fig. 4.
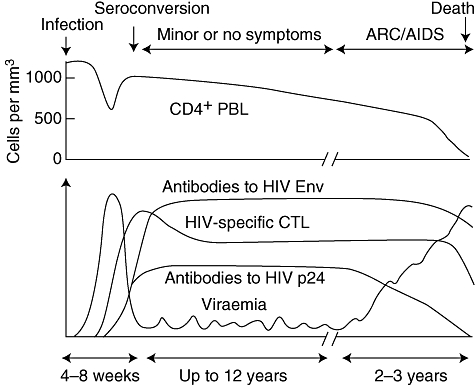
Typical cause of human immunodeficiency virus infection [58].
The picture in the blood is, however, but a faint image indicative of much higher activity in lymphoid tissue [60], as shown first by Tenner-Racz in 1988 [61]. Experimental vaginal or cervical infection of macaques by SIVmac shows that there is local replication of HIV in dermal macrophages [62]. From there, DCs migrating to lymph nodes will deliver virus to T helper cells, as already discussed. Recent findings show that the most active site of HIV replication early in infection is the largest ‘lymphoid organ’ of all, the mucosal associated lymphoid tissue (MALT) of the gut [63].
The partial clearance of virus following seroconversion is ascribed most often to cell-mediated immunity because specific cytotoxic T cells first appear at this time, as well as specific CD4-helper cells. Indeed, the patients with the lowest set points and longest survival, the so-called ‘elite controllers’ of infection, show the strongest specific CD4 cell help against HIV [64]. There are, however, other features that may contribute to fall in viral load. The role of humoral immunity tends to be ignored because neutralizing antibodies appear only some months after seroconversion; but we forget that neutralization assays represent an artificial, in vitro measure of ‘protective’ antibodies. In vivo, antibodies circulate in a pool of complement (C′). My colleagues Marlen Aasa-Chapman et al. [65] have shown that C′-mediated HIV inactivation (lysis of the viral envelope) by specific anti-gp41 and anti-gp120 antibodies occurs concomitantly with the appearance of cytotoxic lymphocytes and the fall in viral load. These findings have been corroborated by Alexandra Trkola's group [66]. Similar non-neutralizing antibodies may also destroy HIV-infected cells through anti-dependent cellular cytotoxicity. Thus a humoral component in the clearance of primary infection merits serious consideration.
Another contributor to the fall in viral load may not be a result of specific immunity at all. The infection of CD4 cells in the MALT is so severe that depletion of cells susceptible to HIV infection might account for the apparent clearance of infection [67]. Conversely, one could argue that the abundance of susceptible cells permits the high virus load at peak viraemia, which never reappears. It will therefore be interesting to investigate what proportion of CD4 cells in MALT are CCR5-positive and in an activated state to support HIV replication.
The long clinically asymptomatic period that follows seroconversion is deceptive, because HIV infection is not latent at all. The introduction of HAART in 1996 provided an opportunity to analyse the dynamics of virus replication and CD4+ T cell turnover [68,69]. It became apparent that the ‘steady state’ in CD4 cell counts actually represented a balance between rapid cell destruction and powerful restoration within the immune system [70] (somewhat like watching a duck glide across a pond without seeing the activity of its webbed feet). Eventually, the capacity for immune regeneration becomes exhausted, and the level of CD4+ cells drops below a threshold of about 200 cells/μl when opportunistic infections can overwhelm the patient.
There are many aspects of HIV pathogenesis that remain to be investigated. For example, why do X4 variants arise late in infection, and why are they seen more frequently in western patients infected with Clade B strains of HIV? It is thought that X4 viruses are more pathogenic than R5 strains and are therefore harbingers of AIDS; but this is a chicken-and-egg dilemma. I would argue that X4 strains are ‘opportunistic’ infections that emerge because immune control diminishes. Such viruses appear to be relatively unfit for person-to-person transmission and they are more sensitive to immune control, particularly to humoral immunity. Once they do emerge, however, they may well exacerbate immune deficiency, analogous to other persistent virus infections. As my colleague Paul Griffiths has shown, cytomegalovirus is both an opportunist and a driver of AIDS [71].
Another puzzle is why simians naturally infected with SIV, e.g. chimpanzees and mangabeys, can sustain viral replication without becoming ill. One difference is that HIV in humans and SIVmac in macaques induce a chronic immune activation, and these ‘danger signals’ lead eventually to immune exhaustion [2]. An intriguing model for the difference is possible mimicry of the HIV envelope gp120 C5 region with human major histocompatibility complex molecules [72].
Host susceptibility to HIV and AIDS
There are a number of different host proteins that affect susceptibility to infections by HIV or to progression to AIDS. Some of these, such as the class I and class II major histocompatibility antigens, are polymorphic in human populations and some alleles predispose to disease while others reduce the risk of infection, or progression [73]. On the other hand, the restriction factor Trim5α discovered in macaques [74], while polymorphic in humans, acts more to restrict zoonoses; that is, the risk of SIV transferring to humans [75]. Despite a report on human single nucleotide polymorphisms (SNPs) for Trim5α[76] they do not appear to have a marked affect on HIV or AIDS [77]. Similarly, human variation in gene for the restriction factor APOBECG3 has not revealed major changes in susceptibility, as the Vif protein of all HIV-1 strains seems able to abrogate its restrictive effects [78]. Recently, whole genome scanning has revealed additional polymorphisms associated with HIV susceptibility [79], although care will need to be taken to distinguish them from linkage to known risk genes.
In contrast to these uncertainties, the genetic polymorphisms of human suppressive chemokines and their receptors do have major effects on susceptibility to HIV infection, and on rates of progression to AIDS [80]. The most dramatic illustration of receptor polymorphism came rapidly after the discovery that CCR5 was the major co-receptor for HIV. It was found that several long-term exposed, uninfected people in unprotected sexual relationships with HIV+ partners were homozygous for a 32 base-pair deletion in the CCR5 gene. The CCR5Δ32 homozygotes lived in good health without a functional CCR5 protein, but were genetically resistant to infection by HIV [81]. The few homozygous individuals who became HIV-positive carried X4 variants of the virus. Individuals who are heterozygous CCR5Δ32 are susceptible to infection (although probably at a lower risk) but have a significantly slower rate of disease progression. The CCR5Δ32 mutation is found only in Caucasians of European descent. There has been speculation as to whether a previous pandemic pathogen such as smallpox or plague might have selected for the mutation's high frequency in Europeans, but there is no strong evidence to implicate a particular pathogen.
As mentioned earlier, CC chemokines can compete with HIV for interaction with the CCR5 receptor. Therefore the higher the plasma levels of chemokines, and the lower the density of CCR5 on the target cell surface, the greater the effect of the ligand-receptor module on HIV. In particular, CCL3L1 (MIP-1αS) varies in gene copy number across human populations, whereas an SNP in the CCR5 promoter affect levels of co-receptor expression. Sunil Ahuja's group in Texas has shown [82] a synergistic effect of high CCL3L1 and low CCR5 to delay disease progression (Fig. 5).
Fig. 5.

Time-line of milestones and setbacks in the history of human immunodeficiency virus/acquired immune deficiency virus.
Prospect for HIV vaccine
The biggest disappointment in the field of HIV research has been the failure to date to develop an efficaceous vaccine to prevent infection. Neither envelope-based vaccines designed to elicit neutralizing antibodies nor DNA and vector-based vaccines designed to prime and boost cell-mediated immunity have shown efficacy in field trials [83,84]. In fact, strong immune responses to an adenovirus 5 vector carrying HIV immunogenic genes may exacerbate the risk of HIV infection.
There have, however, been instances of protection in macaques. Passive transfusion of antibody can protect against challenge with the homologous SIV strain [85]. A xenogeneic or allogeneic cell-based vaccine can effect broader protection [86]. Live, attenuated SIV strains, e.g. with deletion in nef, can protect adult macaques against challenge with a virulent strain [87], although the mechanism of protection has yet to be elucidated satisfactorily. The anecdotal evidence that a small proportion of sex workers in African cities acquired protective immunity attracted a lot of attention [88], but the protection appeared to be short-lived if they went off the game.
The sheer variability of the millions of HIV-1 variants in the 36 million currently infected people (Fig. 2) means that vaccine-induced immunity will need to be broadly reactive, at least within a clade [89]. There are some common features among these myriad strains, e.g. the CD4 binding site on gp120, but even here the epitope varies. For example, Denis Burton's b12 recognizes the CD4 binding site [90]; it is the most broadly cross-neutralizing human mAb described to date, but it neutralizes few clade C isolates, and none from clades A, D or circulating recombinant form A/G.
There is growing interest in ‘therapeutic’ vaccines, although I prefer the term ‘immunotherapy’. Given that progression to AIDS can be controlled to a significant degree by anti-retroviral drugs, the idea is to improve the immune response to act synergistically with the anti-retroviral therapy. One idea was that this might be achieved by encouraging natural immune mechanisms, such as allowing a drug ‘holiday’ with a temporary resurgence of viraemia to act as a boost, but it has not yielded promising results. Another is to immunize infected people with modified HIV antigens made to appear more foreign, an idea akin to immunotherapy in cancer. It is too early to say whether such an approach will be beneficial.
Conclusions
Scientifically, the study of HIV and AIDS over the past 25 years has been fascinating (Fig. 5). It has led to prevention through blood screening and to highly successful anti-retroviral therapy for the majority of infected people who have access to treatment. It has led us to a better understanding of the complexities of the human immune system; but it has not led to a cure for infection, and we do not yet have any really promising leads for microbicides or for vaccines.
During the 1990s there was debate, especially among AIDS ‘activists’, on whether sufficient research funds were being spent on therapeutics (in order to treat currently infected people) as opposed to prophylactic vaccines (in order to protect future generations). Happily, the cumulative funds from governments, charitable foundations and pharmaceutical companies available for AIDS research and development is not a stumbling block today. What we need is a little humility in the face of this insidious foe, HIV, further intensive and extensive investigations and a startling, perhaps serendipitous breakthrough.
References
- 1.Barré-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–71. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Levy JA. HIV and the pathogenesis of AIDS. 3. Washington, DC: American Society for Microbiology; 2007. [Google Scholar]
- 3.CDC. Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men − New York City and California. MMWR. 1981;30:305–8. [PubMed] [Google Scholar]
- 4.Gottlieb MS, Schroff R, Schanker HM, et al. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med. 1981;305:1425–31. doi: 10.1056/NEJM198112103052401. [DOI] [PubMed] [Google Scholar]
- 5.Masur H, Michelis MA, Greene JB, et al. An outbreak of community-acquired Pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. N Engl J Med. 1981;305:1431–8. doi: 10.1056/NEJM198112103052402. [DOI] [PubMed] [Google Scholar]
- 6.CDC. Possible transfusion-associated acquired immune deficiency syndrome (AIDS) − California. MMWR. 1982;31:652–4. [PubMed] [Google Scholar]
- 7.Ragni MV, Lewis JH, Spero JA, Bontempo FA. Acquired-immunodeficiency–like syndrome in two haemophiliacs. Lancet. 1983;1:213–14. doi: 10.1016/s0140-6736(83)92589-8. [DOI] [PubMed] [Google Scholar]
- 8.Shilts R. In: And the band played on: politics, people and the AIDS epidemic. Inn Stonewall., editor. London, Penquin: Saint Martin's Press Inc.; 1994. [Google Scholar]
- 9.Anonymous. The Durban declaration. Nature. 2000;406:15–16. doi: 10.1038/35017662. [DOI] [PubMed] [Google Scholar]
- 10.AIDSTruth. Available at: http://www.aidstruth.org/
- 11.Nattrass N. Mortal combat: AIDS denialism and the struggle for antiretrovirals in South Africa. Durban: University of KwaZulu-Natal Press; 2007. [Google Scholar]
- 12.Gelmann EP, Popovic M, Blayney D, et al. Proviral DNA of a retrovirus, human T-cell leukemia virus, in two patients with AIDS. Science. 1983;220:862–5. doi: 10.1126/science.6601822. [DOI] [PubMed] [Google Scholar]
- 13.Essex M, McLane MF, Lee TH, et al. Antibodies to cell membrane antigens associated with human T-cell leukemia virus in patients with AIDS. Science. 1983;220:859–62. doi: 10.1126/science.6342136. [DOI] [PubMed] [Google Scholar]
- 14.Vilmer E, Barré-Sinoussi F, Rouzioux C, et al. Isolation of new lymphotropic retrovirus from two siblings with haemophilia B, one with AIDS. Lancet. 1984;1:753–7. doi: 10.1016/s0140-6736(84)91275-3. [DOI] [PubMed] [Google Scholar]
- 15.Montagnier L, Dauguet C, Axler C, et al. A new type of retrovirus isolated from patients presenting with lymphadenopathy and acquired immune deficiency syndrome: structural and antigenic relatedness with equine infectious anaemia virus. Ann Virol. 1984;135E:119–34. [Google Scholar]
- 16.Wain Hobson S, Sonigo P, Danos O, Cole S, Alizon M. Nucleotide sequence of the AIDS virus, LAV. Cell. 1985;40:9–17. doi: 10.1016/0092-8674(85)90303-4. [DOI] [PubMed] [Google Scholar]
- 17.Sonigo P, Alizon M, Staskus K, et al. Nucleotide sequence of the visna lentivirus: relationship to the AIDS virus. Cell. 1985;42:369–82. doi: 10.1016/s0092-8674(85)80132-x. [DOI] [PubMed] [Google Scholar]
- 18.Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984;224:497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- 19.Levy JA, Hoffman AD, Kramer SM, Landis JA, Shimabukuro JM, Oshiro LS. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science. 1984;225:840–2. doi: 10.1126/science.6206563. [DOI] [PubMed] [Google Scholar]
- 20.Wain-Hobson S, Vartanian JP, Henry M, et al. LAV revisited: origins of the early HIV-1 isolates from Institut Pasteur. Science. 1991;252:961–5. doi: 10.1126/science.2035026. [DOI] [PubMed] [Google Scholar]
- 21.Coffin J, Haase A, Levy JA, et al. Human immunodeficiency viruses. Science. 1986;232:697. doi: 10.1126/science.3008335. [DOI] [PubMed] [Google Scholar]
- 22.Cheingsong-Popov R, Weiss RA, Dalgleish A, et al. Prevalence of antibody to human T-lymphotropic virus type III in AIDS and AIDS-risk patients in Britain. Lancet. 1984;2:477–80. doi: 10.1016/s0140-6736(84)92562-5. [DOI] [PubMed] [Google Scholar]
- 23.Piot P, Quinn TC, Taelman H, et al. Acquired immunodeficiency syndrome in a heterosexual population in Zaire. Lancet. 1984;2:65–9. doi: 10.1016/s0140-6736(84)90241-1. [DOI] [PubMed] [Google Scholar]
- 24.Van de Perre P, Rouvroy D, Lepage P, et al. Acquired immunodeficiency syndrome in Rwanda. Lancet. 1984;2:62–5. doi: 10.1016/s0140-6736(84)90240-x. [DOI] [PubMed] [Google Scholar]
- 25.Ellrodt A, Barre-Sinoussi F, Le Bras P, et al. Isolation of human T-lymphotropic retrovirus (LAV) from Zairian married couple, one with AIDS, one with prodromes. Lancet. 1984;1:1383–5. doi: 10.1016/s0140-6736(84)91877-4. [DOI] [PubMed] [Google Scholar]
- 26.Bayley AC, Downing RG, Cheingsong-Popov R, Tedder RS, Dalgleish AG, Weiss RA. HTLV-III serology distinguishes atypical and endemic Kaposi's sarcoma in Africa. Lancet. 1985;1:359–61. doi: 10.1016/s0140-6736(85)91383-2. [DOI] [PubMed] [Google Scholar]
- 27.Serwadda D, Mugerwa RD, Sewankambo NK, et al. Slim disease: a new disease in Uganda and its association with HTLV-III infection. Lancet. 1985;2:849–52. doi: 10.1016/S0140-6736(85)90122-9. [DOI] [PubMed] [Google Scholar]
- 28.UNAIDS. Available at: http://www.unaids.org/en/KnowledgeCentre/HIVData/EpiUpdate/EpiUpdArchive/2007/default.asp.
- 29.Daniel MD, Letvin NL, King NW, et al. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science. 1985;228:1201–4. doi: 10.1126/science.3159089. [DOI] [PubMed] [Google Scholar]
- 30.Keele BF, Van Heuverswyn F, Li Y, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–6. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Heuverswyn F, Li Y, Neel C, et al. Human immunodeficiency viruses: SIV infection in wild gorillas. Nature. 2006;444:164. doi: 10.1038/444164a. [DOI] [PubMed] [Google Scholar]
- 32.Heeney JL, Dalgleish AG, Weiss RA. Origins of HIV and the evolution of resistance to AIDS. Science. 2006;313:462–6. doi: 10.1126/science.1123016. [DOI] [PubMed] [Google Scholar]
- 33.Barin F, M'Boup S, Denis F, et al. Serological evidence for virus related to simian T-lymphotropic retrovirus III in residents of west Africa. Lancet. 1985;2:1387–9. doi: 10.1016/s0140-6736(85)92556-5. [DOI] [PubMed] [Google Scholar]
- 34.Clavel F, Guetard D, Brun Vezinet F, et al. Isolation of a new human retrovirus from West African patients with AIDS. Science. 1986;233:343–6. doi: 10.1126/science.2425430. [DOI] [PubMed] [Google Scholar]
- 35.Leligdowicz A, Yindom LM, Onyango C, et al. Robust Gag-specific T cell responses characterize viremia control in HIV-2 infection. J Clin Invest. 2007;117:3067–74. doi: 10.1172/JCI32380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korber B, Gaschen B, Yusim K, et al. Evolutionary and immunological implications of contemporary HIV-1 variation. In: Weiss RA, Adler M A, Rowland-Jones SL, editors. The changing face of HIV and AIDS. Oxford: Oxford University Press; 2001. pp. 19–42. [DOI] [PubMed] [Google Scholar]
- 37.Aboulker JP, Swart AM. Preliminary analysis of the Concorde trial. Lancet. 1993;341:889–990. doi: 10.1016/0140-6736(93)93096-j. Concorde Coordinating Committee. [DOI] [PubMed] [Google Scholar]
- 38.Hicks CB, Gay C, Ferrari G. Acute HIV infection: the impact of anti-retroviral treatment on cellular immune responses. Clin Exp Immunol. 2007;149:211–16. doi: 10.1111/j.1365-2249.2007.03437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van de Perre P, Segondy P, Foulongne V, et al. Herpes simplex virus and human immunodeficiency virus: deciphering a deadly synergy. Lancet Infect Dis. in press. [DOI] [PubMed]
- 40.Weiss HA, Quigley MA, Hayes RJ. Male circumcision and risk of HIV infection in sub-Saharan Africa: a systematic review and meta-analysis. AIDS. 2000;14:2361–70. doi: 10.1097/00002030-200010200-00018. [DOI] [PubMed] [Google Scholar]
- 41.Patterson BK, Landay A, Siegel JN, et al. Susceptibility to human immunodeficiency virus-1 infection of human foreskin and cervical tissue grown in explant culture. Am J Pathol. 2002;161:867–73. doi: 10.1016/S0002-9440(10)64247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss HA. Male circumcision as a preventive measure against HIV and other sexually transmitted diseases. Curr Opin Infect Dis. 2007;20:66–72. doi: 10.1097/QCO.0b013e328011ab73. [DOI] [PubMed] [Google Scholar]
- 43.Klasse PJ, Shattock R, Moore JP. Antiretroviral drug-based microbicides to prevent HIV-1 sexual transmission. Annu Rev Med. 2008;59:455–71. doi: 10.1146/annurev.med.59.061206.112737. [DOI] [PubMed] [Google Scholar]
- 44.Klatzmann D, Barre Sinoussi F, Nugeyre MT, et al. Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science. 1984;225:59–63. doi: 10.1126/science.6328660. [DOI] [PubMed] [Google Scholar]
- 45.Klatzmann D, Champagne E, Chamaret S, et al. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312:767–8. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 46.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–7. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 47.Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–48. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 48.Cocchi F, DeVico AL, Garzino Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–15. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 49.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 50.Alkhatib G, Combadiere C, Broder CC, et al. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–8. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 51.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233:215–19. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 52.Fassati A. HIV infection of non-dividing cells: a divisive problem. Retrovirology. 2006;3:74. doi: 10.1186/1742-4690-3-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryan S, Tiley L, McConnell I, Blacklaws B. Infection of dendritic cells by the Maedi-Visna lentivirus. J Virol. 2000;74:10096–103. doi: 10.1128/jvi.74.21.10096-10103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knight SC, Macatonia SE. Dendritic cells and viruses. Immunol Lett. 1988;19:177–81. doi: 10.1016/0165-2478(88)90140-x. [DOI] [PubMed] [Google Scholar]
- 55.Piguet V, Steinman RM. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 2007;28:503–10. doi: 10.1016/j.it.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patterson S, Rae A, Hockey N, Gilmour J, Gotch F. Plasmacytoid dendritic cells are highly susceptible to human immunodeficiency virus type 1 infection and release infectious virus. J Virol. 2001;75:6710–13. doi: 10.1128/JVI.75.14.6710-6713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med. 2004;199:283–93. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiss RA. How does HIV cause AIDS? Science. 1993;260:1273–9. doi: 10.1126/science.8493571. [DOI] [PubMed] [Google Scholar]
- 59.Mellors JW, Munoz A, Giorgi JV, et al. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann Intern Med. 1997;126:946–54. doi: 10.7326/0003-4819-126-12-199706150-00003. [DOI] [PubMed] [Google Scholar]
- 60.Pantaleo G, Graziosi C, Demarest JF, et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–8. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 61.Tenner-Racz K, Racz P, Dietrich M, Kern P. Altered follicular dendritic cells and virus-like particles in AIDS and AIDS-related lymphadenopathy. Lancet. 1985;1:105–6. doi: 10.1016/s0140-6736(85)91994-4. [DOI] [PubMed] [Google Scholar]
- 62.Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol. 2005;5:783–92. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- 63.Stebbing J, Gazzard B, Douek DC. Where does HIV live? N Engl J Med. 2004;350:1872–80. doi: 10.1056/NEJMra032395. [DOI] [PubMed] [Google Scholar]
- 64.Walker BD. Elite control of HIV infection: implications for vaccines and treatment. Top HIV Med. 2007;15:134–6. [PubMed] [Google Scholar]
- 65.Aasa-Chapman MM, Holuigue S, Aubin K, et al. Detection of antibody-dependent complement-mediated inactivation of both autologous and heterologous virus in primary human immunodeficiency virus type 1 infection. J Virol. 2005;79:2823–30. doi: 10.1128/JVI.79.5.2823-2830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huber M, Fischer M, Misselwitz B, et al. Complement lysis activity in autologous plasma is associated with lower viral loads during the acute phase of HIV-1 infection. PLoS Med. 2006;3:e441. doi: 10.1371/journal.pmed.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–9. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 68.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–6. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 69.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–22. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 70.Wu H, Zhu H, Miao H, Perelson AS. Parameter identifiability and estimation of HIV/AIDS dynamic models. Bull Math Biol. 2008 doi: 10.1007/s11538-007-9279-9. doi: 10.1007/s11538-007-9279-9. [DOI] [PubMed] [Google Scholar]
- 71.Deayton JR, Sabin CA, Johnson MA, Emery VC, Wilson P, Griffiths PD. Importance of cytomegalovirus viraemia in risk of disease progression and death in HIV-infected patients receiving highly active antiretroviral therapy. Lancet. 2004;363:2116–21. doi: 10.1016/S0140-6736(04)16500-8. [DOI] [PubMed] [Google Scholar]
- 72.Cadogan M, Austen B, Heeney JL, Dalgleish AG. HLA homology within the C5 domain promotes peptide binding by HIV-1 gp120. AIDS Res Hum Retroviruses. in press. [DOI] [PubMed]
- 73.Carrington M, O'Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–51. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 74.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–53. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 75.Towers GJ. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007;4:40. doi: 10.1186/1742-4690-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5alpha in humans. Curr Biol. 2006;16:95–100. doi: 10.1016/j.cub.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 77.Goldschmidt V, Bleiber G, May M, Martinez R, Ortiz M, Telenti A. Role of common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology. 2006;3:54. doi: 10.1186/1742-4690-3-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32:118–28. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 79.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Colobran R, Pujol-Borrell R, Armengol MP, Juan M. The chemokine network. II. On how polymorphisms and alternative splicing increase the number of molecular species and configure intricate patterns of disease susceptibility. Clin Exp Immunol. 2007;150:1–12. doi: 10.1111/j.1365-2249.2007.03489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–77. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 82.Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 83.Gilbert PB, Peterson ML, Follmann D, et al. Correlation between immunologic responses to a recombinant glycoprotein 120 vaccine and incidence of HIV-1 infection in a phase 3 HIV-1 preventive vaccine trial. J Infect Dis. 2005;191:666–77. doi: 10.1086/428405. [DOI] [PubMed] [Google Scholar]
- 84.Anonymous. HIV vaccine failure prompts Merck to halt trial. Nature. 2007;449:390.. doi: 10.1038/449390c. [DOI] [PubMed] [Google Scholar]
- 85.Mascola JR, Stiegler G, VanCott TC, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000;6:207–10. doi: 10.1038/72318. [DOI] [PubMed] [Google Scholar]
- 86.Stott J, Hu SL, Almond N. Candidate vaccines protect macaques against primate immunodeficiency viruses. AIDS Res Hum Retroviruses. 1998;14(Suppl. 3):S265–70. [PubMed] [Google Scholar]
- 87.Wyand MS, Manson K, Montefiori DC, Lifson JD, Johnson RP, Desrosiers RC. Protection by live, attenuated simian immunodeficiency virus against heterologous challenge. J Virol. 1999;73:8356–63. doi: 10.1128/jvi.73.10.8356-8363.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fowke KR, Dong T, Rowland-Jones SL, et al. HIV type 1 resistance in Kenyan sex workers is not associated with altered cellular susceptibility to HIV type 1 infection or enhanced beta-chemokine production. AIDS Res Hum Retroviruses. 1998;14:1521–30. doi: 10.1089/aid.1998.14.1521. [DOI] [PubMed] [Google Scholar]
- 89.Garber DA, Silvestri G, Feinberg MB. Prospects for an AIDS vaccine: three big questions, no easy answers. Lancet Infect Dis. 2004;4:397–413. doi: 10.1016/S1473-3099(04)01056-4. [DOI] [PubMed] [Google Scholar]
- 90.Zhou T, Xu L, Dey B, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–7. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]