

Abstract
Hepatitis C virus (HCV) is a major cause of hepatic disease and of liver transplantation worldwide. Mannan-binding lectin (MBL), encoded by the MBL2 gene, can have an important role as an opsonin and complement activating molecule in HCV persistence and liver injury. We assessed the MBL2 polymorphism in 102 Euro–Brazilian patients with moderate and severe chronic hepatitis C, paired for gender and age with 102 HCV seronegative healthy individuals. Six common single nucleotide polymorphisms in the MBL2 gene, three in the promoter (H/L, X/Y and P/Q) and three in exon 1 (A, the wild-type, and B, C or D also known as O) were evaluated using real-time polymerase chain reaction with fluorescent hybridization probes. The concentration of MBL in plasma was measured by enzyme-linked immunosorbent assay. The frequency of the YA/YO genotype was significantly higher in the HCV patients compared with the controls (P = 0·022). On the other hand, the genotypes associated with low levels of MBL (XA/XA, XA/YO and YO/YO) were decreased significantly in the patients with severe fibrosis (stage F4), when compared with the patients with moderate fibrosis (stage F2) (P = 0·04) and to the control group (P = 0·011). Furthermore, MBL2 genotypes containing X or O mutations were found to be associated with non-responsiveness to pginterferon and ribavirin treatment (P = 0·023). MBL2 polymorphisms may therefore be associated not only with the development of chronic hepatitis C, but also with its clinical evolution and response to treatment.
Keywords: HCV, hepatitis C, liver fibrosis, mannan-binding lectin, MBL
Introduction
Since its discovery in 1989, hepatitis C virus (HCV) has been recognized as a major public health problem worldwide because of the high prevalence of chronic hepatitis C, the high risk of progression to cirrhosis, liver failure and hepatocarcinoma [1,2]. HCV is transmitted parenterally, and transmission results typically in a chronic infection. There are six HCV genotypes, with genotype 1 being widespread and the most difficult to treat [3].
Hepatitis C virus is the leading cause of liver transplantation in developed countries, and the most common chronic bloodborne infection in the United States [4]. Epidemiological studies have indicated that approximately 60–85% of HCV infected cases (approximately 200 million people) fail to eradicate the virus and progress subsequently to chronic hepatitis. This high rate of viral chronicity may be related to a failure in the host immune response or to the ability of HCV of overcoming the host's defence [5,6]. A previous report examining an HCV outbreak in a plasmapheresis centre identified a wide pattern of disease progression in a group of patients infected at the same time with the same virus strain [7]. This sustains the idea that host factors are involved critically in the development of fibrosis.
Mannan-binding lectin (MBL) is an important element of the innate response. MBL acts as a recognition molecule for pathogen-associated molecular patterns (PAMPs) having a role in the initiation, regulation and amplification of the immune response [8]. Its role in HCV pathogenesis has been discussed recently [9].
The MBL molecule displays recognition domains for sugars such as mannose, N-acetyl-D-glucosamine, N-acetyl manosamine, fucose and glucose that decorate the surface of different microorganisms, including viruses. After interacting with PAMPs on the pathogen's surface, MBL leads to the activation of the complement system by MBL-associated serine proteases (MASP-1, -2 and -3), with consequent destruction of the pathogen by the membrane attack complex or by complement-mediated phagocytosis mediated by the deposition of opsonic C3b fragments. MBL is also able to cause direct opsonization and phagocytosis of the infecting agent to modulate the release of proinflammatory cytokines, and to promote the removal of apoptotic cells [9–11].
Mannan-binding lectin deficiencies are caused partly by three single nucleotide polymorphisms (SNPs) in the first exon of the gene: MBL2*D (Arg52Cys), B (Gly54Asp) and C (Gly57Glu). The D, B and C SNPs have been labelled collectively O, whereas the major allele at these loci has been called A. The O amino acid changes disturb the polymerization of the polypeptide resulting in low levels of high-order oligomeric MBL in plasma, probably because of short half-life, but an increase of low-molecular-mass MBL, without any known activity, is also seen [12,13]. The concentration of the protein in serum is also modulated by two SNPs in the promoter region: MBL2*H,L[located 550 base pairs (bp) before the transcription start site] and X,Y (located 221 bp before the transcription start site). The SNP P,Q (not coding SNP located 4 bp after the transcription start site) has a marginal effect [14,15]. Linkage disequilibrium between the SNPs in the promoter region and exon 1 is responsible for seven common haplotypes, which are associated with decreasing MBL concentration in plasma: MBL2*HYPA > LYQA > LYPA > LXPA >> HYPD = LYPB = LYQC[15–17]. The most frequent MBL2 haplotypes in most populations encode proteins fully capable of complement activation and pathogen opsonization: HYPA in populations of European and Asian ancestry and in North American Native populations, and LYQA in African populations. The LYQC and LYPB haplotypes, causing MBL deficiency in the homozygous state or in association with the LXPA haplotype, nevertheless reach frequencies as high as the fully functional haplotypes in African and South Amerindian populations [18].
Numerous investigations have found association between the MBL2*LXPA, LYPB, LYQC and HYPD haplotypes and predisposition and/or severity of various immunodeficiencies, autoimmune and infectious diseases in children and adults (for a review, see [19,20]). The high worldwide frequency of MBL deficiency has sparked hypotheses postulating that (1) MBL might be harmful in certain conditions of exacerbated inflammation and/or could be highjacked as a vehicle for pathogens to gain access to the intracellular space, and (2) the MBL2 polymorphism might be under balancing selection. Some investigators have presented results in support of the first hypothesis [21–27] while other investigations, based on epidemiological and molecular evolution analyses, failed to corroborate the second hypothesis [18,28,29].
The reason why some people and not others develop severe HCV disease and why some achieve sustained response to therapy (cure), whereas others do not, may be determined by the effect of a large number of both host and viral factors that can influence disease progression. For example, while the genotypes of HCV are related to treatment responses and carriers of 2 and 3 genotypes usually respond better to therapy than those with genotype 1, they are not related to the severity of liver fibrosis [2].
In order to help to clarify these questions, we analysed the association of MBL2 gene polymorphisms with the development of chronic HCV infection per se, as well as with the severity of liver fibrosis and with the response to interferon (IFN) therapy in a sample of Euro–Brazilian patients from Southern Brazil.
Materials and methods
Subjects and samples
A total of 102 Euro–Brazilian patients (65 men and 37 women) were studied with a mean age of 51 [standard deviation + 9·5] years from Curitiba, southern Brazil. The patients were defined as being from European ascendence based on physical characteristics. It is known from former studies that the genotype distribution of MBL2 haplotypes of Euro–Brazilians of Southern Brazil, with ascendence defined in the same manner, is homogeneous with the MBL2 genotype distribution of most European populations [18,30]. All patients had positive anti-HCV by enzyme-linked immunosorbent assay (ELISA) and positive viral RNA using reverse transcriptase–polymerase chain reaction Amplicor® HCV, version 2.0 (Roche, Branchburg, NJ, USA). The HCV genotypes were analysed by VERSANT™HCV Genotype Assay (Bayer, Tarrytown, NY, USA). The diagnosis of chronic hepatitis C was confirmed further by positive liver biopsy. Only patients older than 18 years and with stage F2, or higher, liver fibrosis according to the Metavir classification [31] were included. Other exclusion criteria included: (1) alcohol usage (> 20 g daily for women and 40 g for men); (2) co-infection with hepatitis B virus or human immunodeficiency virus (HIV); (3) the presence of other liver or systemic disease; and (4) refusal to participate in this study. HCV genotypes were known for 95 of the 102 patients: 54% were 3 (45/51 were 3a), 41% were 1 (at least 18/39 were 1b and 10/39, 1a) and 1% were 2, which is in accordance with the viral distribution pattern as seen in South Brazil [32]. The study was approved by the Ethics Committee of Human Research of the Clinical Hospital, Federal University of Paraná, Brazil. All patients and controls read and signed an informed consent form prior to their inclusion in the study.
Among the cases analysed, 21 (20·6%) patients had stage F2 liver fibrosis, 36 (35·3%) had stage F3 and 45 (44·1%), stage F4 liver fibrosis or cirrhosis. The histopathological analyses were assessed by a pathologist and then confirmed in turn by another. The control group included 102 healthy Euro–Brazilian individuals matched with the patients according to gender and age. All controls were negative for anti-HCV, anti-hepatitis B core antigen and anti-HIV-1 and -2 antibodies.
Most patients were treated either with conventional IFN-α or with pegylated-IFN-α (pgIFN) associated with ribavirin. They were classified as sustained responders (SR) or non-responders according to their response to the treatment. Among the 61 patients treated with IFN, 27 (44%) were SR and 34 (56%) were non-responders. Among the 38 patients treated with pgIFN associated with ribavirin, 13 (34%) were SR and 25 (66%) were non-responders. At follow-up after 6 months of therapy, all SR patients were negative for HCV RNA and had normal alanine aminotransferase levels. Because patients submitted to a second treatment with pgIFN usually present a lower SR than treatment-naive patients who initiated their therapy with pgIFN, we conducted their analysis on the association of MBL2 genotypes separately.
Blood was collected with anti-coagulant ethylenediamine tetraacetic acid. The plasma was frozen and DNA was extracted from the peripheral blood mononuclear cells through standard salting-out and phenol/chloroform/isoamyl alcohol methods.
MBL2 genotyping and MBL serology
Six SNPs located at positions g.273C > G (MBL2*L,H), g.602G > C (MBL2*Y,X), g.826C > T (MBL2*P,Q), g.1045C > T (p.Arg52Cys, MBL2*A,D), g.1052G > A (p.Gly54Asp, MBL2*A,B) and g.1061G > A (p.Gly57Glu, MBL2*A,C) were analysed using real-time polymerase chain reaction with fluorescent hybridization probes, as described previously [33]. Nucleotide positions were determined according to official recommendations [34], taking Y16577 as the reference sequence. The phase between the variants could be inferred based on the strong linkage disequilibrium between them.
The concentration of MBL was estimated by ELISA in 82 patients and in 50 controls with available plasma using wells coated with mannan and developed with monoclonal anti-MBL antibody, 131-1 [35]. This assay has a cut-off of 10 ng/ml and detects only high-order MBL oligomers.
Statistical analysis
Genotype and allele frequencies were obtained by direct counting. Deviations from Hardy–Weinberg equilibrium were tested using the approach described by Guo and Thompson [36]. The allele frequency distributions of the patient and control populations and of other populations of European descent were compared by applying the exact test of population differentiation of Raymond and Rousset [37]. Both analyses were performed using the software package Arlequin version 3.1 [38].
Possible associations between MBL2 genotypes or alleles and susceptibility to severe disease were analysed with χ2 test. The parametric Student's t-test and the non-parametric Kruskal–Wallis or Mann–Whitney tests were used with quantitative data. The software ‘Primer of Biostatistics’ was used for comparisons between two proportions, and χ2 was performed using the software Epi-Info. Bonferroni correction was applied when several dependent or indepent statistical tests were performed simultaneously. The value of P < 0·05 was adopted as significance level.
Results
All groups assessed in this study were found to be in Hardy–Weinberg equilibrium. The haplotype and genotype distribution was homogeneous with those of other European and Euro-Brazilian populations [16–18]. A rare eighth haplotype (LYPD) was observed in one control subject (Table 1). The distribution of MBL2 haplotypes (Table 1) and the overall circulating levels of MBL showed no significant difference between HCV patients and controls [mean 1360·5 versus 1272·9 ng/ml, P = not significant (n.s.)]. A significant association of MBL concentration with the different genotypes was observed for both HCV patients and controls (P < 0·0001 and P < 0·004, respectively, with Bonferroni correction PB < 0·02), Fig. 1.
Table 1.
MBL2 haplotypes in hepatitis C virus (HCV) patients and controls.
| MBL2 haplotypes | HCV patients | Controls | ||
|---|---|---|---|---|
| n = 204 | % | n = 204 | % | |
| HYPA | 57 | 27·9 | 54 | 26·5 |
| LYQA | 37 | 18·1 | 43 | 21·1 |
| LYPA | 21 | 10·3 | 19 | 9·3 |
| LXPA | 44 | 21·6 | 43 | 21·1 |
| LYPB | 29 | 14·2 | 31 | 15·2 |
| HYPD | 10 | 4·9 | 10 | 4·9 |
| LYQC | 6 | 2·9 | 3 | 1·5 |
| LYPD | 0 | 0 | 1 | 0·5 |
n, number of chromosomes.
Fig. 1.
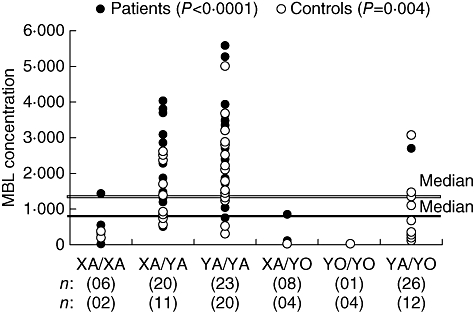
MBL2 genotypes in relation to the concentration of mannose-binding lectin (MBL) in hepatitis C virus patients and controls. Double line: controls; single line: patients. The statistical analysis refers to the comparison between the XA/YA, YA/YA and YA/YO genotypes (Kruskal–Wallis test; significant for both groups, P < 0·017, with Bonferroni correction).
The diplotypes including X/Y of the promoter region and A/O of exon 1 were analysed according to their influence on the MBL levels [18]. MBL concentrations were low in all the cases with XA/XA and XA/YO genotypes, and very low in all the cases with YO/YO genotype (<10 ng/ml). The YA/YO genotype was associated with intermediate levels of MBL (Table 2).
Table 2.
MBL2 genotypes and protein concentration in hepatitis C virus (HCV) patients and controls.
| MBL2 genotype | HCV patients | Controls | ||||||
|---|---|---|---|---|---|---|---|---|
| n1 = 102 | % | n2 = 82 | (MBL ng/ml) | n1 = 102 | % | n2 = 50 | (MBL ng/ml) | |
| YA/YA | 30 | 29·4 | 23 | 2413 (481–5594) | 39 | 38·2 | 20 | 2093·5 (280–5023) |
| XA/YA | 23 | 22·5 | 20 | 1579 (479–4059) | 20 | 19·6 | 11 | 915 (513–2600) |
| YA/YO | 32 | 31·4 | 26 | 370·5 (107–2713) | 17 | 16·7 | 12 | 520 (97–3100) |
| XA/XA | 6 | 5·9 | 6 | 423 (36–1388) | 6 | 5·9 | 2 | 279 (195–363) |
| XA/YO | 9 | 8·8 | 6 | 71·5 (11–835) | 12 | 11·8 | 1 | 47 |
| YO/YO | 2 | 2 | 1 | < 10 | 8 | 7·8 | 4 | < 10 |
The medians of mannose-binding lactose (MBL) levels on admission are given, followed by the range of values in parentheses. Bold type, significant difference (P = 0·022, odds ratio 2·29, confidence interval 95% 1·17–4·46). n1, number of genotyped individuals, n2, number of serologically quantified individuals.
The frequency of the YA/YO genotype was significantly higher in the hepatitis C patients compared with the controls [31·4% versus 16·7%, P = 0·022, PB = n.s. odds ratio (OR) 2·29, confidence interval (CI) 95% 1·17–4·46] (Table 2). The frequencies of the genotypes associated with low levels of MBL (XA/XA, XA/YO and YO/YO) were lower in the patients with severe fibrosis stage F4 than in the patients with moderate fibrosis stage F2 (4/41, 8·9% versus 6/15, 28·6% P = 0·04, PB = 0·08 OR 0·24 CI 95% 0·06–0·99) (Table 3), and also when compared with the control group (4/41, 8·9% versus 26/102, 25·5%, P = 0·011, PB = 0·022 OR 0·29, CI 95% 0·09–0·87) (not shown).
Table 3.
MBL2 genotypes and severity of fibrosis in hepatitis C virus patients.
| Stages of liver fibrosis | ||||||
|---|---|---|---|---|---|---|
| Stage F2 | Stage F3 | Stage F4 | ||||
| MBL2 genotype | (n = 21) | % | (n = 36) | % | (n = 45) | % |
| YA/YA | 7 | 33·3 | 10 | 27·8 | 13 | 28·9 |
| XA/YA | 4 | 19·1 | 8 | 22·2 | 11 | 24·4 |
| YA/YA + XA/YA | 11 | 52·4 | 18 | 50·0 | 24 | 53·3 |
| YA/YO | 4 | 19·1 | 11 | 30·6 | 17 | 37·8 |
| XA/XA | 2 | 9·5 | 3 | 8·3 | 1 | 2·2 |
| XA/YO | 3 | 14·3 | 3 | 8·3 | 3 | 6·7 |
| YO/YO | 1 | 4·7 | 1 | 2·8 | 0 | 0 |
| XA/XA + XA/YO + YO/YO | 6 | 28·6 | 7 | 19·4 | 4 | 8·9 |
n, number of individuals. Bold type, significant difference (P = 0·04, odds ratio 0·24 confidence interval 95% 0·06–0·99).
For the analysis of the response to treatment, the patients were divided into two subgroups according to Matsushita et al. [39]: one comprising those homozygous for the promoter and exon 1 common alleles (YA/YA) and the other group those heterozygous or homozygous to promoter and exon 1 mutated alleles (YA/YO, XA/XA, XA/YO, YO/YO). The X or O mutations were found less frequently in patients who were responsive to pgIFN treatment than in patients who did not respond to this treatment (5/13, 38·5% versus 19/25, 75%, P = 0·023, PB = 0·046 OR 5·01 CI 95% 1·19–21·51). Seven patients were non-responders of the two treatments, first with IFN and ribavirin and afterwards with pgIFN and ribavirin. Five of those non-responders patients (5/7, 71%) had the X or O mutations. In addition, MBL concentrations were higher in the pgIFN responders than in non-responder patients (mean: 2312 mg/ml × 1498 mg/ml, P = n.s.).
No association was found between HCV genotypes and the degree of liver fibrosis or MBL2 genotypes.
Discussion
Data on MBL2 polymorphisms in European HCV patients are scarce [9]. Most of the studies assessing the MBL2 gene polymorphism in HCV infection were performed in Asian patients. Asians present only one common SNP in exon 1, at codon 54, i.e. the B allele [18,40]. A recent Brazilian study was conducted on an ethnically highly mixed population presenting a viral genotype distribution significantly different from that of the present study (51% 1b, 29% 3a and 7% 1a versus 18% 1b, 44% 3a and 10% 1a) [41].
Furthermore, previous studies on MBL and hepatitis C have comprised patients with different clinical forms of HCV infection: patients with no liver involvement, patients with cirrhosis and others cured spontaneously, and with diagnosis based only on serology. This compromises the analysis regarding the severity of liver commitment [39,41–44]. These were the grounds for studying patients based on the result of their liver biopsy, as this is the most reliable measure for grading the disease.
In this study, the MBL2 polymorphisms known to have large effects on the concentration and functional capacity of MBL were assessed in Euro–Brazilian HCV patients with biopsy-proven liver fibrosis and controls. The observed MBL2 haplotype and genotype frequencies are in agreement with previous studies in the Euro–Brazilian population [18] and did not differ significantly from the distribution reported for western European populations [16,17]. The limited number of haplotypes observed (n = 8) is due to linkage disequilibrium between the SNPs of the promoter region and the exon 1 of the MBL2 gene [18]. The LYPD haplotype, found in one of the controls, has been described previously in the Euro–Brazilian and in other European populations with an allelic frequency of approximately 1% [30,45,46]. It is believed to be originated from a recent intragene recombination between LYPA and HYPD or between LYPB and HYPD[30].
Based on the results, we suggest that the MBL2*YA/YO genotype, associated with intermediate plasma levels of MBL, is involved in the development of chronic hepatitis C. The frequency of the LYA/YO genotype was also significantly higher in Iranian HCV-infected patients compared with controls [44]. Recently, Segat et al. showed that in addition to MBL2 O/O, HCV patients from North-east Brazil also presented a highly significant increase of the A/O genotype in comparison with ethnically matched healthy controls. According to some authors [8,23,47], the MBL levels associated with A/O genotypes in the present study could be considered as MBL insufficiency. Taken together, these data suggest that MBL2 genotypes associated with low or intermediate levels of circulating protein are associated with the susceptibility of HCV infection, and that MBL might have an important anti-viral effect both in acute as well in chronic disease. The secretion of proinflammatory cytokines is MBL dose-dependent [48]. Thus, our results suggest that intermediate concentrations of MBL have a dose-response effect on the production of proinflammatory cytokines by the live Kupfer cells, contributing to the chronification of the virus C hepatitis. Conversely, the YO/YO, XA/YO and XA/XA genotypes, associated with low levels of MBL, were found to be negatively associated with the severity of liver fibrosis, and low levels of MBL might thus reduce the risk of chronic hepatitis C. This is in accordance with recent data indicating increased complement activation and increased activity of the MBL/MASP1 complex in HCV-induced hepatic fibrosis [49–51]. A protective role for these genotypes in chronic diseases has been reported by several other authors [22–24,52]. These data therefore corroborate the proinflammatory role of MBL in chronic disease. However, it's important to note that the exclusion of patients with no or low degree of fibrosis from this study, we probably missed important additional information to evaluate the association between disease progression and MBL2 genotype.
In addition, MBL2 genotypes containing X or O mutations were found to be associated with non-responsiveness to pgIFN and ribavirin treatment. In another report, the authors also found that Japanese HCV patients with MBL2 genotypes containing X or O alleles were significantly less likely to respond to therapy with IFN than similar patients presenting haplotypes associated with higher amounts of MBL [42]. Taken together, these data suggest that MBL may play an immunomodulatory role during treatment with IFN, as it is known that MBL regulates the release of different cytokines from immune cells in response to infection [53]. This is potentially an important finding as the expense, adverse side effects and long duration of anti-viral therapy make screening for potential non-responders desirable. In this context, the identification of MBL2 genotypes may be useful in predicting the response to IFN treatment in HCV patients.
In conclusion, MBL2 polymorphisms may therefore be associated not only with the development of chronic hepatitis C, but also with its outcome and response to pgIFN and ribavirin treatment.
Acknowledgments
This study was supported partially by grants from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) to Iara Messias-Reason and by grants from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) to Lilian Pereira-Ferrari.
References
- 1.McHutchison JG. Understanding hepatitis C. Am J Manag Care. 2004;10:S21–S29. [PubMed] [Google Scholar]
- 2.Poupon R. Hepatitis C: epidemiology, management and treatment. Bull Acad Natl Med. 2005;189:375–84. [PubMed] [Google Scholar]
- 3.Keller BC, Johnson CL, Erickson AK, Gale M., Jr Innate immune evasion by hepatitis C virus and West Nile virus. Cytokine Growth Factor Rev. 2007;18:535–44. doi: 10.1016/j.cytogfr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 5.Grakoui A. Hepatitis C virus infection. How does the host respond? Minerva Gastroenterol Dietol. 2004;50:21–8. [PubMed] [Google Scholar]
- 6.Gremion C, Cerny A. Hepatitis C virus and the immune system: a concise review. Rev Med Virol. 2005;15:235–68. doi: 10.1002/rmv.466. [DOI] [PubMed] [Google Scholar]
- 7.Datz C, Cramp M, Haas T, et al. The natural course of hepatitis C virus infection 18 years after an epidemic outbreak of non-A, non-B hepatitis in a plasmapheresis centre. Gut. 1999;44:563–7. doi: 10.1136/gut.44.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiel S, Frederiksen PD, Jensenius JC. Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol. 2006;43:86–96. doi: 10.1016/j.molimm.2005.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown KS, Ryder SD, Irving WL, Sim RB, Hickling TP. Mannan binding lectin and viral hepatitis. Immunol Lett. 2007;108:34–44. doi: 10.1016/j.imlet.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Garred P, Larsen F, Madsen HO, Koch C. Mannose-binding lectin deficiency − revisited. Mol Immunol. 2003;40:73–84. doi: 10.1016/s0161-5890(03)00104-4. [DOI] [PubMed] [Google Scholar]
- 11.Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40:423–9. doi: 10.1016/s0161-5890(03)00155-x. [DOI] [PubMed] [Google Scholar]
- 12.Larsen F, Madsen HO, Sim RB, Koch C, Garred P. Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J Biol Chem. 2004;279:21302–11. doi: 10.1074/jbc.M400520200. [DOI] [PubMed] [Google Scholar]
- 13.Terai I, Kobayashi K, Matsushita M, Miyakawa H, Mafune N, Kikuta H. Relationship between gene polymorphisms of mannose-binding lectin (MBL) and two molecular forms of MBL. Eur J Immunol. 2003;33:2755–63. doi: 10.1002/eji.200323955. [DOI] [PubMed] [Google Scholar]
- 14.Juliger S, Luckner D, Mordmuller B, et al. Promoter variants of the human mannose-binding lectin gene show different binding. Biochem Biophys Res Commun. 2000;275:617–22. doi: 10.1006/bbrc.2000.3343. [DOI] [PubMed] [Google Scholar]
- 15.Madsen HO, Garred P, Thiel S, et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol. 1995;155:3013–20. [PubMed] [Google Scholar]
- 16.Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol. 1998;161:3169–75. [PubMed] [Google Scholar]
- 17.Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC. Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods. 2000;241:33–42. doi: 10.1016/s0022-1759(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 18.Boldt AB, Culpi L, Tsuneto LT, de Souza IR, Kun JF, Petzl-Erler ML. Diversity of the MBL2 gene in various Brazilian populations and the case of selection at the mannose-binding lectin locus. Hum Immunol. 2006;67:722–34. doi: 10.1016/j.humimm.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Eisen DP, Minchinton RM. Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin Infect Dis. 2003;37:1496–505. doi: 10.1086/379324. [DOI] [PubMed] [Google Scholar]
- 20.Kilpatrick DC. Mannan-binding lectin and its role in innate immunity. Transfus Med. 2002;12:335–52. doi: 10.1046/j.1365-3148.2002.00408.x. [DOI] [PubMed] [Google Scholar]
- 21.Dornelles LN, Pereira-Ferrari L, Messias-Reason I. Mannan-binding lectin plasma levels in leprosy: deficiency confers protection against the lepromatous but not the tuberculoid forms. Clin Exp Immunol. 2006;145:463–8. doi: 10.1111/j.1365-2249.2006.03161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garred P, Harboe M, Oettinger T, Koch C, Svejgaard A. Dual role of mannan-binding protein in infections: another case of heterosis? Eur J Immunogenet. 1994;21:125–31. doi: 10.1111/j.1744-313x.1994.tb00183.x. [DOI] [PubMed] [Google Scholar]
- 23.de Messias-Reason IJ, Boldt AB, Moraes Braga AC, et al. The association of mannan-binding lectin gene polymorphism with clinical leprosy: new insight into an old paradigm. J Infect Dis. 2007;196:1379–85. doi: 10.1086/521627. [DOI] [PubMed] [Google Scholar]
- 24.Messias RI, Schafranski MD, Jensenius JC, Steffensen R. The association between mannose-binding lectin gene polymorphism and rheumatic heart disease. Hum Immunol. 2006;67:991–8. doi: 10.1016/j.humimm.2006.08.296. [DOI] [PubMed] [Google Scholar]
- 25.Schafranski MD, Stier A, Nisihara R, Messias-Reason IJ. Significantly increased levels of mannose-binding lectin (MBL) in rheumatic heart disease: a beneficial role for MBL deficiency. Clin Exp Immunol. 2004;138:521–5. doi: 10.1111/j.1365-2249.2004.02645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos IK, Costa CH, Krieger H, et al. Mannan-binding lectin enhances susceptibility to visceral leishmaniasis. Infect Immun. 2001;69:5212–5. doi: 10.1128/IAI.69.8.5212-5215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mombo LE, Lu CY, Ossari S, et al. Mannose-binding lectin alleles in sub-Saharan Africans and relation with susceptibility to infections. Genes Immun. 2003;4:362–7. doi: 10.1038/sj.gene.6363979. [DOI] [PubMed] [Google Scholar]
- 28.Dahl M, Tybjaerg-Hansen A, Schnohr P, Nordestgaard BG. A population-based study of morbidity and mortality in mannose-binding lectin deficiency. J Exp Med. 2004;199:1391–9. doi: 10.1084/jem.20040111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdu P, Barreiro LB, Patin E, et al. Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum Mol Genet. 2006;15:2650–8. doi: 10.1093/hmg/ddl193. [DOI] [PubMed] [Google Scholar]
- 30.Boldt AB, Petzl-Erler ML. A new strategy for mannose-binding lectin gene haplotyping. Hum Mutat. 2002;19:296–306. doi: 10.1002/humu.10051. [DOI] [PubMed] [Google Scholar]
- 31.Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24:289–93. doi: 10.1002/hep.510240201. [DOI] [PubMed] [Google Scholar]
- 32.Campiotto S, Pinho JR, Carrilho FJ, et al. Geographic distribution of hepatitis C virus genotypes in Brazil. Braz J Med Biol Res. 2005;38:41–9. doi: 10.1590/s0100-879x2005000100007. [DOI] [PubMed] [Google Scholar]
- 33.Steffensen R, Hoffmann K, Varming K. Rapid genotyping of MBL2 gene mutations using real-time PCR with fluorescent hybridisation probes. J Immunol Methods. 2003;278:191–9. doi: 10.1016/s0022-1759(03)00190-x. [DOI] [PubMed] [Google Scholar]
- 34.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 35.Petersen SV, Thiel S, Jensen L, Steffensen R, Jensenius JC. An assay for the mannan-binding lectin pathway of complement activation. J Immunol Methods. 2001;257:107–16. doi: 10.1016/s0022-1759(01)00453-7. [DOI] [PubMed] [Google Scholar]
- 36.Guo SW, Thompson EA. Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–72. [PubMed] [Google Scholar]
- 37.Raymond M, Rousset F. An exact test for population differentiation. Evolution. 1995;49:1280–3. doi: 10.1111/j.1558-5646.1995.tb04456.x. [DOI] [PubMed] [Google Scholar]
- 38.Schneider S, Roessli D, Excoffier L. Arlequin: a software for population genetic data analysis. Geneva: Genetics and Biometry Laboratory, University of Geneva; 2000. [computer program] [Google Scholar]
- 39.Matsushita M, Hijikata M, Ohta Y, et al. Hepatitis C virus infection and mutations of mannose-binding lectin gene MBL. Arch Virol. 1998;143:645–51. doi: 10.1007/s007050050320. [DOI] [PubMed] [Google Scholar]
- 40.Lipscombe RJ, Beatty DW, Ganczakowski M, et al. Mutations in the human mannose-binding protein gene: frequencies in several population groups. Eur J Hum Genet. 1996;4:13–9. doi: 10.1159/000472164. [DOI] [PubMed] [Google Scholar]
- 41.Segat L, Silva Vasconcelos LR, Montenegro de Melo F, et al. Association of polymorphisms in the first exon of mannose binding lectin gene (MBL2) in Brazilian patients with HCV infection. Clin Immunol. 2007;124:13–7. doi: 10.1016/j.clim.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Matsushita M, Hijikata M, Matsushita M, Ohta Y, Mishiro S. Association of mannose-binding lectin gene haplotype LXPA and LYPB with interferon-resistant hepatitis C virus infection in Japanese patients. J Hepatol. 1998;29:695–700. doi: 10.1016/s0168-8278(98)80248-1. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki K, Tsutsumi A, Wakamiya N, et al. Mannose-binding lectin polymorphisms in patients with hepatitis C virus infection. Scand J Gastroenterol. 2000;35:960–5. doi: 10.1080/003655200750023039. [DOI] [PubMed] [Google Scholar]
- 44.Somi MH, Farhang S, Asgharzadeh M, Estakhry R, Pouri AA. Mannose binding lectin gene haplotype in Iranian patients with hepatitis C infection. Hep Mon. 2007;7:21–6. [Google Scholar]
- 45.Cedzynski M, Szemraj J, Swierzko AS, et al. Mannan-binding lectin insufficiency in children with recurrent infections of the respiratory system. Clin Exp Immunol. 2004;136:304–11. doi: 10.1111/j.1365-2249.2004.02453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skalnikova H, Freiberger T, Chumchalova J, Grombirikova H, Sediva A. Cost-effective genotyping of human MBL2 gene mutations using multiplex PCR. J Immunol Methods. 2004;295:139–47. doi: 10.1016/j.jim.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 47.Valdimarsson H, Vikingsdottir T, Bang P, et al. Human plasma-derived mannose-binding lectin: a phase I safety and pharmacokineticstudy. Scand J Immunol. 2004;59:97–102. doi: 10.1111/j.0300-9475.2004.01357.x. [DOI] [PubMed] [Google Scholar]
- 48.Jack DL, Read RC, Tenner AJ, Frosch M, Turner MW, Klein NJ. Mannose-binding lectin regulates the inflammatory response of human professional phagocytes to Neisseria meningitidis Serogroup B. J Infect Dis. 2001;1184:1152–62. doi: 10.1086/323803. [DOI] [PubMed] [Google Scholar]
- 49.Brown KS, Keogh MJ, Tagiuri N, et al. Severe fibrosis in hepatitis C virus-infected patients is associated with increased activity of the mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex. Clin Exp Immunol. 2007;147:90–8. doi: 10.1111/j.1365-2249.2006.03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gangadharan B, Antrobus R, Dwek RA, Zitzmann N. Novel serum biomarker candidates for liver fibrosis in hepatitis C patients. Clin Chem. 2007;53:1792–9. doi: 10.1373/clinchem.2007.089144. [DOI] [PubMed] [Google Scholar]
- 51.Lee IN, Chen CH, Sheu JC, et al. Identification of complement C3a as a candidate biomarker in human chronic hepatitis C and HCV-related hepatocellular carcinoma using a proteomics approach. Proteomics. 2006;6:2865–73. doi: 10.1002/pmic.200500488. [DOI] [PubMed] [Google Scholar]
- 52.Soborg C, Madsen HO, Andersen AB, Lillebaek T, Kok-Jensen A, Garred P. Mannose-binding lectin polymorphisms in clinical tuberculosis. J Infect Dis. 2003;188:777–82. doi: 10.1086/377183. [DOI] [PubMed] [Google Scholar]
- 53.Jack DL, Turner MW. Anti-microbial activities of mannose-binding lectin. Biochem Soc Trans. 2003;31:753–7. doi: 10.1042/bst0310753. [DOI] [PubMed] [Google Scholar]