

Abstract
Recently we identified galectin-3 (gal-3), which is secreted by colonic epithelial cells (CEC), to be a strong activator of colonic lamina propria fibroblasts (CLPF). Modulation of CLPF function may play a role during stricture and fistula formation in inflammatory bowel disease (IBD). Therefore, we investigated further the expression of gal-3 and effects on CLPF. The aim of this study is to perform a direct comparison of gal-3 between tissue from healthy controls and from patients with either Crohn's disease (CD) or ulcerative colitis (UC). CEC, CLPF and intestinal macrophages (IMAC) were isolated from control and IBD colonic tissue. Interleukin-8 secretion as a readout of CLPF activation was quantified by enzyme-linked immunosorbent assay. Gal-3 in cell cultures and tissue samples was evaluated by Western blot, immunofluorescence and immunohistochemistry. CLPF-migration was assayed in the 48-well modified Boyden chamber. Gal-3 expression was found in all segments of the colon. In the terminal ileum, less gal-3 was found compared with the colon. Immunohistochemistry and immunofluorescence revealed a homogenous distribution of gal-3 in CEC and IMAC of control mucosa and UC. However, significantly less gal-3 was found in IMAC from CD patients. In CD fistulae and stenoses, gal-3 expression was reduced significantly and barely detectable. In co-incubation studies lactose reduced significantly the CLPF-stimulatory potential of gal-3, indicating that the C-terminal domain of gal-3 is responsible for CLPF activation. Gal-3 stimulated CLPF migration in CLPF derived from fistulae. In conclusion, gal-3 expression is down-regulated in CD-fistulae and stenoses as well as in IMAC in CD patients. Gal-3 induces migration of CLPF derived from fistulae. Its role for stricture and fistula formation warrants further investigation.
Keywords: Crohn's disease, epithelial cells, fibroblasts, fistula, galectin-3
Introduction
Recently, we identified galectin-3 (gal-3) as a new colonic lamina propria fibroblasts (CLPF) activating factor isolated from colonic epithelial cells (CEC) [1]. Supernatants of cultured primary human CEC strongly activated primary colonic fibroblast cultures or the intestinal fibroblast cell line CCD-18Co, as seen with nuclear factor kappa B activation and interleukin (IL)-8 secretion. Using a classical biochemical approach we isolated the major protein of the activating fractions from the CEC supernatants by fast performance liquid chromatography. Using matrix-assisted-laser desorption/ionization time-of-flight-mass-spectrometer we identified the protein to be gal-3. Immunoprecipitation confirmed that gal-3 is part of the CEC-conditioned media and the major activator of CLPF. Stimulation of CLPF with recombinant gal-3 resulted in a high increase of CLPF IL-8 secretion.
Galectin-3 is a member of a group of β-galactoside binding proteins with 14 mammalian members known [2–4]. Three distinct groups of galectins are defined by their structures and sequences. Galectins contain a unique domain of 130 amino acids which is responsible for the carbohydrate-binding activity, the so-called carbohydrate recognition domain (CRD). Galectins are expressed by a variety of cell types and are present both intracellularly and extracellularly. By binding to and cross-linking of glycoconjugates on cell surfaces, galectins can modulate processes such as apoptosis, cytokine secretion, cell adhesion and migration [5]. These proteins have been implicated in the regulation, activation, differentiation and survival of T cells and thus may play critical roles in the modulation of chronic inflammatory disorders such as inflammatory bowel diseases (IBD) and other autoimmune diseases.
Galectin-3 is the only member of the so-called ‘chimera group’. It consists of an unusual prolin- and glycine-rich N-terminal domain attached to the C-terminal CRD domain. The N-terminus was shown to lack carbohydrate-binding activity, but is essential for full biological activity of gal-3 [6]. The N-terminal domain has been implicated in the secretion of gal-3 [7]. Gal-3 contains an extended binding site for longer oligosaccharides such as polylactosaminoglycan [8]. The C-terminal domain includes a sugar binding site known to be blocked by lactose [6,9]. Gal-3 is expressed in a large number of tissues and cell types such as epithelial cells, fibroblasts, keratinocytes, osteoclasts, Langerhans cells, dendritic cells, monocytes and macrophages [10–12]. It is localized in the nucleus, in the cytoplasm and at the cell surface [13,14]. Gal-3 is released from cells by an unorthodox secretory mechanism that uses the endoplasmic reticulum and the Golgi. The function of gal-3 is pleiotropic, including cell proliferation, adhesion and survival [15–18]. A gal-3 knock-out model showed attenuation of liver fibrosis, probably because of reduced procollagen expression [19].
The gal-3 protein has gained increasing interest during the last years. A role for stromal reactions in different kinds of cancer, such as thyroid carcinoma, colon carcinoma or gastric cancer, and an involvement in metastasis formation has been postulated [20–25]. In colon cancer, reduction of gal-3 expression in the colon is associated with a reduced risk of liver metastases [24]. However, the role of gal-3 as a prognostic factor in cancer is still discussed controversially [20,26,27]. Gal-3 has been shown to interact with bcl-2, a known suppressor of apoptosis with some sequence similarity with gal-3 thereby influencing and regulating apoptosis [28,29]. In addition, gal-3 protects against the formation of reactive oxygen products [30]. Gal-3-transfected T cells showed higher and faster growth rates compared with controls. Gal-3 also may be involved in nuclear splicing of pre-mRNA. It plays a role in wound healing and accelerating re-epithelialization [31].
In contrast to these mainly anti-inflammatory features, gal-3 may also act as an inducer of inflammation in some tissues. Gal-3-deficient mice developed less inflammatory cell infiltrates in the peritoneal cavity after thioglycollate instillation compared with control mice [32].
These known features, and our observation that gal-3 is a major activator of CLPF, make it an interesting factor whose functions in the intestinal immune system and for the pathophysiology of IBD need to be evaluated. Proliferation of the muscularis mucosa and muscularis propria occurs in Crohn's disease (CD) but not in ulcerative colitis (UC). This contributes to the development of strictures and intestinal obstruction. Fistulae and stenoses are found frequently in patients with CD. The exact mechanisms leading to stenoses- and fistula-formation are unknown, limiting conservative treatment options; however, involvement of CLPF is postulated by most experts.
The persistence of fibroblast activation beyond an inflammatory insult may be a risk factor for scarring and pathological remodelling of the tissue. Fibroblast migration plays an important role in tissue formation and wound healing and is altered in CD mucosa [33–36].
To date, the potential involvement of gal-3 in IBD pathophysiology has not been investigated in greater detail. In active CD gal-3 expression seems to be decreased in the inflamed epithelium [37]. Gal-3 expression could be reduced by tumour necrosis factor (TNF) incubation in the colonic adenocarcinoma cell line HCT-8. A substantially higher percentage of sera from CD patients contained anti-gal-3 immunoglobulin G (IgG) autoantibodies than from those with UC and controls. In CD patients the titre of autoantibodies correlated negatively with disease activity [38]. This result was confirmed recently and gal-3 was shown to modulate T cell function in IBD [39].
Because of this fragmentary information and the important role of gal-3 which we found for CLPF activation, we further investigated functions of gal-3 in the intestinal immune systems under physiological conditions and its potential role during IBD.
Materials and methods
Isolation and culture of primary CEC
Colonic tissue was obtained from patients undergoing surgical resection for colorectal carcinoma or diverticulitis. The isolation of CEC was performed as described [40]. Using our protocol, purity of the cells can be given as 90–98% [40]. The isolation of CEC from biopsies and surgical specimens from IBD patients and healthy controls was approved by the University of Regensburg Ethics Committee. All patients gave informed consent. Table 1 gives details for the patients and materials involved in this study.
Table 1.
Characteristics of tissue obtained from patients undergoing surgical resection or biopsies. The material was used for reverse transcription–polymerase chain reaction, immunohistochemical staining and cell isolation (epithelial cells, macrophages, fibroblasts).
| Patient | Localization | Sex | Age | Diagnosis | Medication | Isolated tissue | Grade of inflammation |
|---|---|---|---|---|---|---|---|
| 1 | Colon | M | 72 | Carcinoma | No medication | CEC IMAC | No inflammation |
| 2 | Sigma | M | 59 | Carcinoma | No medication | CEC CLPF | No inflammation |
| 3 | Sigma | F | 55 | Carcinoma | No medication | CEC | No inflammation |
| 4 | Colon | M | 63 | Carcinoma | No medication | CEC | No inflammation |
| 5 | Colon | F | 69 | Carcinoma | No medication | CEC IMAC | No inflammation |
| 6 | Sigma | F | 80 | Carcinoma | No medication | CEC | No inflammation |
| 7 | Sigma | M | 45 | Carcinoma | No medication | CEC CLPF | No inflammation |
| 8 | Rectum | F | 48 | Carcinoma | No medication | CEC IMAC | No inflammation |
| 9 | Rectum | M | 68 | Carcinoma | No medication | CEC | No inflammation |
| 10 | Rectum | M | 69 | Carcinoma | No medication | CEC | No inflammation |
| 11 | Colon | F | 69 | Carcinoma | No medication | IMAC | No inflammation |
| 12 | Rectum | F | 54 | Carcinoma | No medication | CEC | No inflammation |
| 13 | Ileum | M | 66 | Carcinoma | No medication | CEC IMAC | No inflammation |
| 14 | Colon | M | 36 | CD stenosis | Azathioprin, mesalazine, prednisolone 10 mg | CEC CLPF | Mild–severe inflammation |
| 15 | Colon | M | 53 | CD stenosis | No medication | CLPF | Mild–severe inflammation |
| 16 | Colon | F | 31 | CD stenosis | Prednisolone 5 mg | CEC CLPF | Mild inflammation |
| 17 | Colon | F | 37 | CD stenosis | Prednisolone 20 mg | CLPF | Severe inflammation |
| 18 | Colon | M | 69 | CD | Unknown | CLPF | Mild–severe inflammation |
| 19 | Ileocoecal | M | 23 | CD | methotrexate 15 mg | CEC CLPF | Mild inflammation |
| 20 | Ileum | M | 39 | CD | Prednisolone 7·5 mg | CLPF | Mild inflammation |
| 21 | Ileocoecal | F | 53 | CD | Budesonide | CEC CLPF | Mild inflammation |
| 22 | Colon | M | 38 | CD | Sulfasalazine | CEC CLPF | No − mild inflammation |
| 23 | Colon | F | 17 | CD | Budenoside, sulfasalazine, | CLPF | Mild inflammation |
| 24 | Colon | F | 42 | CD | Prednisolone 60 mg | CEC CLPF | Severe inflammation |
| 25 | Colon | F | 40 | CD stenosis | No medication | CLPF | Mild inflammation |
| 26 | Ileum | M | 43 | CD stenosis | Azathioprine, budesonide, sulfasalazine | CLPF | Mild inflammation |
| 27 | Colon | M | 27 | CD | Budesonide | CLPF IMAC | Mild |
| 28 | Colon | F | 31 | CD | No medication | CEC CLPF | No inflammation |
| 29 | Ileum | M | 39 | CD | Sulfasalazine | CEC | Mild inflammation |
| 30 | Fistula | M | 69 | CD | Unknown | CLPF | Unknown |
| 31 | Fistula | F | 47 | CD | Azathioprine | CLPF | Unknown |
| 32 | Fistula | F | 40 | CD | Sulfasalazine | CLPF | Unknown |
| 33 | Fistula | F | 40 | CD | Azathioprine | CLPF | Unknown |
| 34 | Fistula | M | 53 | CD | No medication | CLPF | Unknown |
| 35 | Colon | F | 39 | UC | Prednisolone 7·5 mg, budesonide | CEC CLPF | Mild inflammation |
| 36 | Colon | M | 42 | UC | Prednisolone 10 mg, azathioprine | CEC CLPF | Severe inflammation |
| 37 | Colon | M | 46 | UC | Sulfasalazine | CEC CLPF | No − mild inflammation |
| 38 | Colon | M | 58 | UC | No medication | CEC CLPF | Mild inflammation |
| 39 | Sigma | M | 37 | UC | Mesalazine | CEC | Mild |
| 40 | Colon | F | 46 | UC | Prednisolone 60 mg, sulfasalazine | CEC CLPF | Severe inflammation |
| 41 | Colon | M | 48 | UC | No medication | CEC IMAC | No inflammation |
| 42 | Sigma | F | 65 | Diverticulitis | No medication | CEC | No inflammation |
| 43 | Sigma | M | 48 | Diverticulitis | No medication | CEC | Mild inflammation |
| 44 | Sigma | M | 39 | Diverticulitis | No medication | CEC | Mild–severe inflammation |
CD, Crohn's disease; CEC, colonic epithelial cells; CLPF, colonic lamina propria fibroblasts; IMAC, intestinal macrophages; UC, ulcerative colitis.
Isolation and purification of human intestinal macrophages
Lamina propria mononuclear cells isolated from normal and IBD mucosa specimens were incubated with MicroBeads armed with a monoclonal mouse anti-human macrophage CD33 antibody (Miltenyi Biotec, Bergisch Gladbach, Germany) and purified twice with AS separation columns (Miltenyi Biotec), as described [41]. The magnetically labelled cells were retained in the column. After removal of the column from the magnetic field, the retained fraction was eluted. Eluted cells were passed through a second AS separation column to increase the purity of intestinal macrophages (IMAC). The final purity of > 95% IMAC was confirmed by fluorescence activated cell sorter (FACS) analysis using phycoerythrin-conjugated goat anti-mouse IgG antibody (Caltag, Medac, Hamburg, Germany).
Conditioned medium from CEC
Colonic epithelial cells were isolated and cultured on collagen A-coated (Biochrom, Berlin, Germany) Millicell CM filters (Millipore Corporation, Bedford, MA, USA) in CEC-medium (MEM-Earle/Biochrom) (penicillin/streptomycin and gentamycin; PAA Laboratories GmbH, Linz, Austria) as described [40]. After 24 h of incubation the medium was collected, centrifuged to remove cell debris and stored at −20°C for up to 3 months.
Cell lines
HT-29 cells and CaCo-2 cells were obtained from the European Collection of Cell Culture and cultured in Dulbecco's modified Eagle's medium (DMEM) (PAA Laboratories) supplemented with 10% fetal calf serum (FCS) (Pan Biotech, Aidenbach, Germany), 1% sodium-pyruvate (PAA Laboratories), 1% non-essential amino acids (PAA Laboratories) and 1% penicillin/streptomycin under standard tissue-culture conditions. CCD-18Co cells represent an immortalized fibroblast cell line from human colon (American Type Culture Collection, Manassas, VA, USA). Cells were cultured at 37°C in a 10% CO2 atmosphere in DMEM supplemented with 20% FCS, 1% sodium-pyruvate, 1% non-essential amino acids and 1% penicillin/streptomycin.
Interleukin-8 enzyme-linked immunosorbent assay
IL-8 protein was quantified by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (Biozol, Eching, Germany).
Immunofluorescence and immunohistochemistry
Paraffin-embedded specimens were cut at 4 μm and mounted on glass slides. Slides were washed twice in phosphate-buffered saline (PBS) for 5 min, air-dried and incubated for 10 min in acetone at −20°C. Subsequently, slides were incubated in medium containing 10% FCS for 30 min. Then, 50 μl of anti-gal-3 antibody (BD Biosciences, Pharmingen; clone B2C10, or mouse IgG1 isotype control mouse IgG1) antibody as isotype control in dilutions of 1:250 and 1:200 respectively, were added. The slides were placed in a humid box for 1 h. Immunohistochemical staining was performed with a Vectastain ABC-elite standard system (Vector Laboratories, Burlingame, CA, USA). Immunofluorescence was incubated with a secondary antibody (goat anti-mouse 1:150; Molecular Probes Eugene, OR, USA) for 30 min at 37°C. Fluorescent mounting medium (DakoCytomation, Hamburg, Germany) was added, and the slides were covered and stored at 4°C.
Co-incubation studies
The fibroblast cell line CCD-18Co was incubated with recombinant gal-3 (10 μg/ml; R&D Systems, Wiesbaden, Germany) and lactose in different concentrations for 24 h. IL-8 was quantified in the supernatant by ELISA. Protein was measured with the bicinchoninic acid test (Sigma); 400 000 cells/per well were used in a six-well plate.
Western blot
Thirty μg protein were loaded per lane. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Equal loading of the gels was controlled by Ponceau S staining. The gels were blotted onto nitrocellulose membranes (Schleicher/Schull, Dassel, Germany) for 30 min. The membranes were incubated subsequently in blocking buffer containing PBS, 0·2% Triton-X (Sigma) and 5% milk powder, followed by washing with Triton-X and PBS. Anti-gal-3 antibody in 5 ml of blocking buffer was added at a concentration of 5 μg/ml for 1 h, followed by washing. Anti-biotin antibody with streptavidin in concentrations of 1:1000 and 1:4000 was added for 30 min. After washing, peroxidase was visualized by enhanced chemiluminescence development (ECL Plus Western Blotting Detection System, Amersham, UK) and exposured to X-ray film (Curix 60, Agfa, Mortsel, Belgium).
Quantitative reverse transcription–polymerase chain reaction
Colonic mucosa was used for quantitative reverse transcription–polymerase chain reaction (RT–PCR) and transferred to ice-cold RNAlater solution (Ambion, Huntingdon, UK). RNA was extracted using the RNeasy-kit (Qiagen, Hilden, Germany) and the Qiagen shredder-kit, following the manufacturer's recommendations. RNA was transcribed using the Promega (Mannheim, Germany) reverse transcription system, as recommended by the manufacturer. Quantification of cytokine mRNA was performed using Taqman PCR, as described previously [42] (gal-3 forward GCG GAA AAT GGC AGA CAA TT, gal-3 reverse CAT CCT TGA GGG TTT GGG TTT). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression was measured using an appropriate GAPDH kit (Applied Biosystems, Darmstadt, Germany) and served as reference.
Isolation and culture of CLPF
Human CLPF were isolated and cultured as described previously [36]. The mucosa from surgical specimens was cut into 1-mm pieces, whereas biopsy specimens were used directly for the isolation of CLPF. Epithelial cells were separated in Hank's balanced salt solution without Ca2+ and Mg2+ (PAA Laboratories GmbH, Linz, Austria) with 2 mM ethylenediamine tetraacetic acid (Sigma, Deisenhofen, Germany). The remaining tissue was rinsed and digested for 30 min at 37°C with 1 mg/ml collagenase 1 (Sigma), 0·3 mg/ml DNase I (Boehringer Ingelheim, Ingelheim, Germany) and 2 mg/ml hyaluronidase (Sigma) in PBS (Gibco, Karlsruhe, Germany). The isolated cells were cultured in 25 cm2 cell culture flasks (Costar, Bodenheim, Germany) with DMEM containing 10% FCS, penicillin (PAA Laboratories), streptomycin 10 mg/ml (PAA Laboratories), ciprofloxacin 2 mg/ml (Bayer, Leverkusen, Germany), gentamycin 50 mg/ml (PAA Laboratories) and amphotericin B 1 mg/ml (Biochrom, Berlin, Germany). Non-adherent cells were removed by subsequent changes of medium. The cultured cells were used between passages 3 and 8.
Migration assays
All migration assays were performed in the modified 48-well Boyden chamber, as described previously [36]. A polycarbonate filter (8-μm pore size, Gerbu Biotechnik, Gaiberg, Germany) divided the chamber into an upper and a lower compartment. Each test substance was placed into the wells of the lower compartment in replicates. A total of 20 000 CLPF/well were seeded into the wells of the upper compartment of the Boyden chamber. The Boyden chamber was incubated at 37°C in 10% CO2 atmosphere for 6 h. The filter was removed from the chamber and the non-migrated cells on the upper side of the filter were scraped off with a rubber policeman. The migrated cells on the lower side of the filter were fixed and stained with the Hemacolor staining kit (Merck, Darmstadt, Germany) and counted at a 400-fold magnification.
Migration assays were performed with fibronectin as positive control and unconditioned media as negative control for 6 h. Recombinant gal-3 was applied in concentrations of 1 and 10 μg/ml. Fibronectin was used at a concentration of 25 μg/ml. The extent of migration was quantified as cells/microscopic field. Each experiment was repeated eight to 12 times per patient.
Statistics
Statistical analyses were performed using the Student's t-test and Mann–Whitney rank sum test. Differences were considered significant at a P-value < 0·05.
Results
Galectin-3 is expressed strongly in the colon, sigma and rectum and less expressed in the terminal ileum
Galectin-3 mRNA was quantified in epithelial cells isolated from colon, sigma and rectum of controls by real-time PCR. Similar amounts of gal-3 mRNA were found in rectum, sigma and colon (Fig. 1a).
Fig. 1.
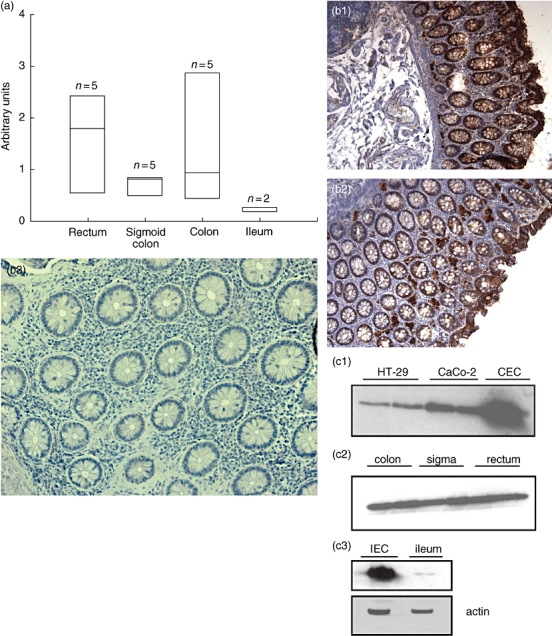
(a) Quantification of galectin-3 (gal-3) expression in the colon and terminal ileum by real-time polymerase chain reaction (PCR). Gal-3 mRNA expression is given as arbitrary units. Gal-3 is expressed homogeneously in the sigma, rectum and the colon and found mainly in epithelial cells; n = 5 per group, no significant differences between all groups. (b) Immunohistochemical staining of gal-3 colon and ileum of controls. Gal-3 staining brown. Gal-3 is expressed homogeneously in tissue of controls (1 and 2). Isotype control 3 (non-inflamed mucosa, control). Gal-3 is found predominantly in the epithelial cell barrier, whereas almost no gal-3 can be found in the lamina propria. (c) Western blot analysis of gal-3 expression in epithelial cell lines, rectum, sigma, colon (epithelial cells) and ileum (epithelial cells and biopsies). Compared with colonic epithelial cells (CEC), gal-3 is expressed less in both epithelial cell lines HT-29 and CaCo-2. Gal-3 is expressed homogeneously in the sigma, rectum and colon. In the terminal ileum gal-3 is expressed less compared with rectum, colon and sigma and found predominantly in the epithelial cells.
Sequential specimens of the colon (rectum, sigma and colon) were investigated for gal-3 protein expression by immunohistochemistry (Fig. 1b). Gal-3 protein was found homogeneously in the sigma, rectum and colon. Gal-3 positive cells were identified mainly as epithelial cells. In the lamina propria, gal-3 protein was rarely detectable. Increasing gal-3 staining was seen in the epithelial layer towards the gut lumen.
Galectin-3 protein expression was investigated by Western blot analyses. In epithelial cell lines (HT-29 and CaCo-2) little gal-3 was found compared with CEC (Fig. 1c). In rectum, sigma and colon equal amounts of gal-3 protein were detectable. In isolated intestinal epithelial cells (IEC) and biopsies from the terminal ileum less gal-3 was found (Fig. 1c).
Reduced gal-3 mRNA expression in CD fistulae, stenoses and IMAC
The expression of gal-3 mRNA was characterized further in CD fistulae and stenoses. As IMAC are both important players in IBD and potential source of gal-3, we also investigated gal-3 mRNA expression in IMAC isolated from patients with IBD and controls. Gal-3 expression in CD, CD fistulae and CD stenoses was compared with controls and UC tissue.
In non-inflamed colonic specimens of UC patients, gal-3 mRNA expression quantified by real-time PCR tended to be lower compared with controls, but this difference did not reach statistical significance because of high variability. In inflamed UC inflamed tissue gal-3 mRNA expression was comparable with specimens from UC patients without inflammation. In colonic tissue from CD patients gal-3 mRNA expression was lower compared with controls. Inflamed mucosa of CD patients showed lower gal-3 mRNA expression than non-inflamed tissue. Gal-3 expression in CD was lower than in controls (no significance) and in UC colonic tissue (no significance). In CD stenoses gal-3 expression was lower compared with controls (P < 0·04). The same was found in CD fistulae (P < 0·001).
In IMAC derived from colonic tissue of IBD patients, gal-3 was lower compared with IMAC from controls (P < 0·012) (Fig. 2).
Fig. 2.
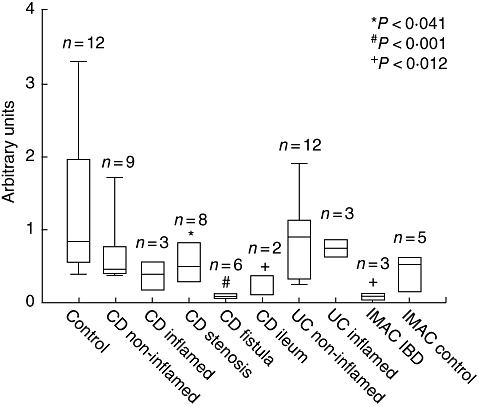
Taqman-polymerase chain reaction for galectin-3 (gal-3) mRNA expression in controls, ulcerative colitis, Crohn's disease (CD), CD-stenoses and CD-fistulae. In CD colonic tissue gal-3 expression was lower compared with controls (no significance). In CD stenoses gal-3 expression was low compared with controls (P < 0·041), whereas in CD fistulae gal-3 expression was also significantly low (P < 0·001). In macrophages derived from colonic tissue of inflammatory bowel disease gal-3 was low compared with controls (P < 0·012) and macrophages from controls.
In UC and CD, gal-3 expression was low compared with control tissue. In CD stenoses and fistulae, as well as in IMAC isolated from IBD tissue, gal-3 expression was significantly lower compared with controls.
Reduced gal-3 protein expression in CD fistulae and macrophages
In controls and in UC patients, gal-3 protein was found to be expressed mainly in epithelial cells, with no difference between both groups. Gal-3 was found predominantly in the bowel wall next to the lumen in epithelial cells (Fig. 3a,c). In IMAC from controls and UC patients, gal-3 was expressed similarly (Fig. 3b,d). In CD, gal-3 expression was reduced as described [37]. However, in CD fistulae minimal or no gal-3 staining was seen (Fig. 3e).Additionally, only low gal-3 was detected in macrophages from CD (Fig. 3f).
Fig. 3.
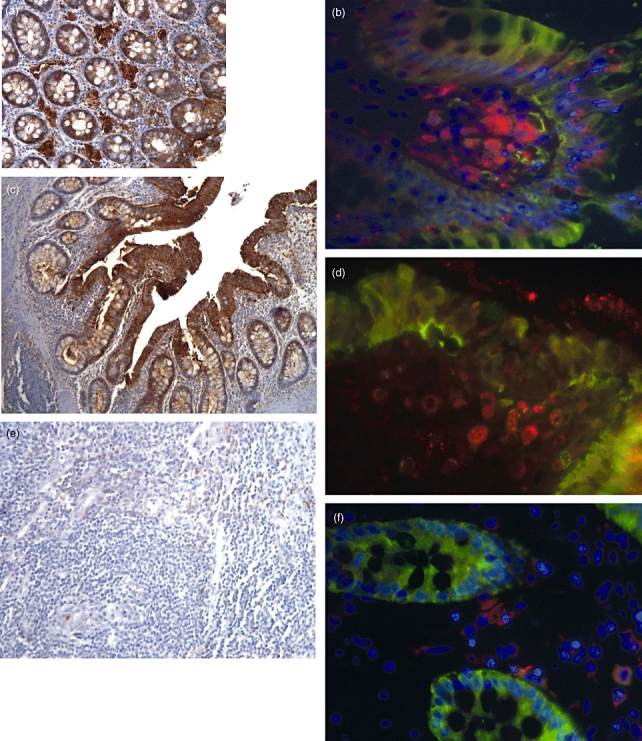
Immunofluorescence and immunohistochemical staining of galectin-3 (gal-3) in inflammatory bowel disease (IBD) colonic tissue, macrophages and controls. Three to five patients per group were investitgated. Representative staining of each group is shown in this figure. Gal-3 staining brown, CD 68 (macrophages) red, gal-3 and CD 68 orange. (a) Gal-3 staining in colon tissue of controls (brown, 200×). Gal-3 is expressed homogenously in the epithelial cell layer. (b) Gal-3 expression in macrophages (fluorescein isothiocyanate-tr-dapi, CD 68 red). (c) Gal-3 in ulcerative colitis (UC) colon (100×). (d) Gal-3 was found equally in UC macrophages. (e) Gal-3 expression in fistulae of Crohn's disease (CD) (200×). No gal-3 staining was found in this area. (f) Gal-3 in CD macrophages. Compared with control and UC patients there was no gal-3 expression in CD fistulae and a reduced expression in CD macrophages.
Galectin-3-induced fibroblast activation is inhibited by lactose
Galectin-3 is composed of two domains, the N-terminal domain with a consensus sequence element and the C-terminal CRD domain with distinct functional properties. The C-terminal domain includes the sugar binding site known to be blocked by lactose [6]. We therefore aimed to elucidate whether one or both domains are essential for the fibroblast activation described recently. CCD-18Co fibroblasts were incubated with recombinant human gal-3 (10 μg/ml) for 24 h. Additionally, increasing concentrations of lactose were added. As readout of fibroblast activation, IL-8 secretion into the supernatant was quantified. Gal-3 alone induced a strong induction of IL-8 secretion, as described previously (6843·0 ± 884·7 pg/ml). The addition of lactose in low concentrations reduced the stimulatory effect of gal-3 slightly (lactose 50 mM: 5413·7 ± 1693·9 pg/ml), but this effect was not significant. Lactose in higher concentrations abrogated the fibroblast stimulating effect of gal-3 (Fig. 4) indicating that the CRD-domain is responsible for CLPF activation.
Fig. 4.
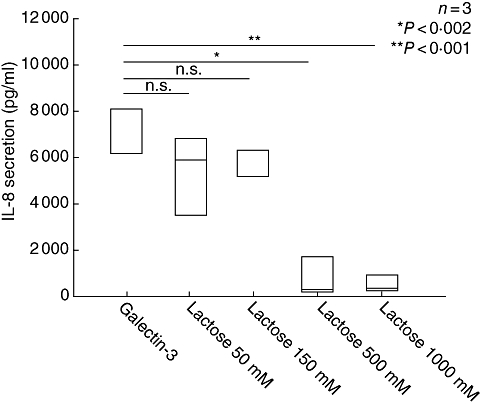
The fibroblast cell line CCD-18Co was incubated with galectin-3 (gal-3) (10 μg/ml) and different concentrations of lactose (50–500 mM) for 24 h. Interleukin (IL)-8 secretion was measured by enzyme-linked immunosorbent assay in the supernatant of the cells and is given as pg/ml. Lactose inhibited significantly the gal-3 inducing IL-8 secretion in a concentration-dependent manner; n = 3.
Galectin-3 inhibits CLPF migration at high concentrations, but induces migration at low concentrations
Because we found low expression of gal-3 RNA and protein in CD fistulae and stenoses and because disturbed fibroblast migration is implicated to play a role in the formation of both fistulae and stenoses, we investigated the influence of recombinant gal-3 on colonic fibroblast migration. Migration of CLPF from CD tissue, fistulae and stenoses were compared with control and UC tissue. Migration assays of CLPF were performed for 6 h with CEC-conditioned media; fibronectin was used as positive control and unconditioned media as negative control. Recombinant gal-3 (with or without addition of fibronectin) was added in concentrations of 1 and 10 μg/ml. Fibronectin was used at 25 μg/ml.
In CLPF from controls, UC, CD and CD stenoses gal-3 inhibited fibroblast migration significantly compared with fibronectin in high concentrations (Fig. 5a–d, gal-3 10 μg/ml versus control: P < 0·001 CLPF control, P < 0·029 CLPF UC, P < 0·001 CLPF CD, P < 0·009 CLPF CD stenosis).
Fig. 5.
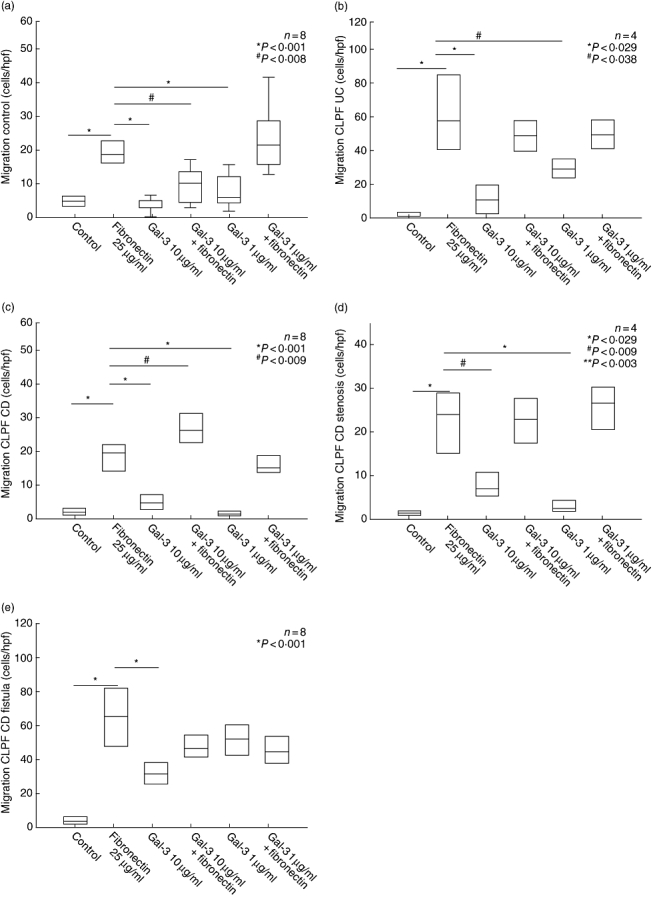
Colonic lamina propria fibroblasts (CLPF) migration assay in the 48-well Boyden chamber. Unconditioned medium was used as negative control, fibronectin as positive control. The influence of galectin-3 (gal-3) with or without addition of fibronectin in different concentrations on migration was investigated. (a, b) In CLPF from controls and ulcerative colitis gal-3 inhibited fibroblast migration (gal-3 10 μg/ml). (c, d) In CLPF from Crohn's disease (CD) and CD stenoses gal-3 also inhibited the migratory effect. (e) Gal-3 stimulated CLPF migration in CD fistulae comparable with the effect of fibronectin (gal-3 at a concentration of 1 μg/ml).
Exclusively in CLPF from CD fistulae, gal-3 induced a migration comparable with the level of fibronectin alone (gal-3 1 μg/ml 51·1 ± 9·9 cells/high power field, fibronectin 25 μg/ml 64·8 ± 18·5 cells/high power field) (Fig. 5e).
Discussion
As we have found recently that gal-3 is a strong activator of CLPF, we have now investigated the expression and functional role of gal-3 in intestinal mucosa from controls and IBD tissue. We found gal-3 to be expressed homogeneously in all parts of the colon, but a clearly lower expression in the terminal ileum. Recently we had found a low gal-3 expression in the CEC lines HT-29 and CaCo-2 gal-3, whereas in primary CEC a strong expression had been detected [1], corresponding with data presented here. Our study on gal-3 expression confirms a recent report on decreased gal-3 expression in CD mucosa [37]. In this report, the expression of gal-3 in IEC from CD patients, ileum adjacent to resected colon carcinoma, unspecific bowel inflammation, diverticulosis, UC and healthy jejunum was investigated. It was shown that gal-3 is distributed homogeneously in epithelial cells from control patients. However, according to our findings, gal-3 was decreased in CD epithelial cells.
We could demonstrate in our study that in CD stenoses and fistulae gal-3 mRNA and protein expression was reduced strongly or virtually absent, whereas gal-3 was expressed homogeneously and found predominantly in the epithelium of controls and UC patients.
Tumour necrosis factor is known to play an important role in fistulae formation. Anti-TNF strategies in the therapy of fistulae in CD have evolved rapidly during recent years and improved the clinical outcome of the patients [43,44]. Concerning gal-3, the incubation of intestinal biopsies with TNF leads to a down-regulation of gal-3 mRNA in vitro[37,39]. We found a strong epithelial expression of gal-3 in controls. In inflammatory conditions such as CD, as well as fistulae and strictures, gal-3 is barely or not detectable, as we could demonstrate clearly in vivo. It is likely that increased TNF levels are responsible for the down-regulation of gal-3 expression in affected tissue of IBD patients as well as CD stenoses and fistulae. Because loss of gal-3 in fistulae and stenoses seems to be correlated with disturbed immune functions and prolonged activation of fibroblasts, this may point to a potential role of gal-3 as a protective agent in inflammatory conditions such as IBD.
The expression level of gal-3 in IMAC from controls and UC patients was similar. However, gal-3 expression was clearly down-regulated in IMAC isolated from CD patients on protein and RNA levels. In inflamed IBD mucosa proinflammatory cytokines secreted by activated effector T cells stimulate macrophages to secrete large amounts of TNF-α, IL-1 and IL-6 [45]. These cytokines contribute to a up-regulated expression of adhesion molecule ligands on the vascular endothelium of the mucosal blood vessels followed by leucocyte adhesion and extravasation into the tissue and inflammation [46,47]. A loss of gal-3 expression in IMAC from CD patients − as we have shown in this study − might be correlated with consecutive disturbed gal-3 function and contribute to a prolonged and dysbalanced immune response of IMAC, particularly, in IBD.
However, peritoneal macrophages from gal-3−/− mice were more prone to undergo apoptosis than those from gal-3+/+ mice when treated with apoptotic stimuli, suggesting that expression of gal-3 in inflammatory cells may lead to longer cell survival, thus prolonging inflammation. These results support strongly a role for gal-3 as a proinflammatory mediator in the peritoneal cavity [32]. Gal-3 also plays a central role in neutrophil extravasation [16] and is a migratory factor for monocytes and macrophages [48], again indicating a role as proinflammatory mediator because of these investigations.
Further studies on the role of gal-3 loss in IMAC of IBD patients are warranted to clarify the influence of gal-3 on immune functions in IBD. The exact regulation of gal-3 function still remains unclear. Gal-3 is regulated and secreted by a pathway that excludes the ER and Golgi apparatus [14]. In human neutrophils CD66a and CD66b have been identified as functional receptors of gal-3 [49]. Known binding partners of gal-3 are β-catenin, fibronectin, beta1 integrins, collagens and laminin [50–53]. Incubation of gal-3 with different concentrations of lactose reduced significantly the stimulatory potential of gal-3 on CLPF. We could show, therefore, that the induction of IL-8 production of CLPF by gal-3 was mediated mainly by the CRD-domain of gal-3. Co-incubation with lactose, a natural inhibitor of gal-3 which binds to the CRD domain, inhibited the CLPF activating effect of gal-3. However, the detailed mechanism of signal transduction and receptor binding of gal-3 in CLPF activation, especially in CLPF from CD fistulae and migration, needs to be elucidated further.
In CD fistulae, CLPF migration by gal-3 was increased compared with controls and UC. In contrast, in CLPF derived from controls, UC, CD and CD stenoses, in high concentrations gal-3 inhibited CLPF migration significantly.
Recently we demonstrated that fistulae in CD differ markedly from non-CD fistulae with regard to their cellular composition. CD-fistulae had a lining of flattened intestinal epithelium or a narrow squamous epithelium. Non-epithelialized fistulae were covered by a thin layer of fibroblast-like cells, focally forming a new basement membrane. We could demonstrate that these fibroblast-like cells originate from epithelial cells via epithelial–mesenchymal transition (EMT). It could be speculated that a reduced ability of fibroblasts to migrate and close wounds could be associated with the necessity for epithelial cells to step in and facilitate wound closure via EMT. As gal-3 is able to induce fibroblast migration, a reduced gal-3 expression could be associated with impaired wound closure. This would explain why EMT is necessary for wound repair leading finally to fistula formation. However, this hypothesis warrants further experimental support.
A participation of gal-3 in fibrosis of the lung has been described. Increased synthesis and secretion of gal-3 during irradiation-induced lung injury was found. Gal-3 seems to have a modulating effect in pulmonary alveolar epithelial expansion and differentiation during injury and repair [54]. Additionally, the influence of gal-3 in the development of tissue fibrosis in chronic pancreatitis has been reported [55].
Other members of the galectin family have been only barely investigated in IBD. Gal-1 has been shown to play a role in the activation of pancreatic stellate cells [56]. However, in HCT-15, LoVo and CoLo201 cells, gal-1 induced reduced cell migration [57]. In an experimental model of colitis induced by administration of 2,4,6-trinitrobenzene sulphonic acid (TNBS), gal-1 showed a protective and immunomodulatory role in the colon [58].
Galectin-3 is a protein with increasing interest in IBD. We could show in this paper that gal-3 is little expressed (or not) in fistulae and stenoses as well as in IMAC in CD. In the terminal ileum, gal-3 expression is reduced compared with controls. Additionally, gal-3 induces increased fibroblast migration in CLPF from CD fistulae. Its role for fistula and stricture formation in IBD is shown in Fig. 6 and has to be clarified in further studies to develop protective targets to disrupt the perpetuation of inflammation and fibroblast activation.
Fig. 6.
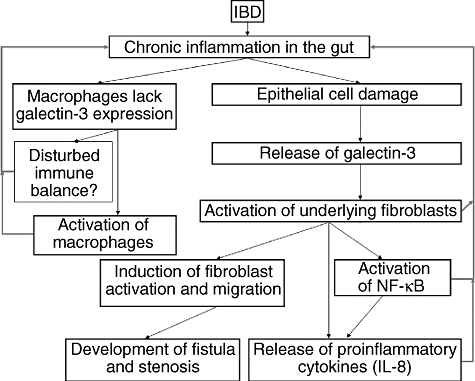
Galectin-3 (gal-3) and its potential role in inflammatory bowel disease (IBD). Chronic inflammation is found in IBD, especially in Crohn's disease (CD). These inflammatory conditions cause damage of the epithelial cell layer. Gal-3 is contained in epithelial cells of the colon, as stated in this paper. Damaged epithelial cells might release gal-3. Released gal-3 could then lead to an activation of underlying lamina propria cells, such as colonic lamina propria fibroblasts. Especially in CD, this activation of fibroblast might contribute to the formation of fistulae and stenosis as well as an increased migration of fibroblasts. In this paper, we demonstrate that intestinal macrophages (IMAC) from CD patients do not express gal-3. In general, chronic inflammatory conditions might contribute to a reduced expression of gal-3 in IMAC. This reduced expression might therefore contribute to a disturbed and prolonged immune reaction. Further investigations will contribute to a better understanding of the interaction of gal-3 between epithelial cells and IMAC.
Acknowledgments
This work was supported by the Bundesministerium für Bildung und Forschung, BMBF (Kompetenznetz CED) and the Deutsche Forschungsgemeinschaft (SFB 585). The authors thank the surgeons and pathologists of the University of Regensburg for providing us with colonic specimens. We are grateful for the patients' contribution of the tissue samples.
References
- 1.Lippert E, Falk W, Bataille F, et al. Soluble galectin-3 is a strong, colonic epithelial-cell derived, lamina propria fibroblast-stimulating factor. Gut. 2007;56:43–51. doi: 10.1136/gut.2005.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins. Structure and function of a large family of animal lectins. J Biol Chem. 1994;269:20807–10. [PubMed] [Google Scholar]
- 3.Barondes SH, Castronovo V, Cooper DN, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76:597–8. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovich GA, Baum LG, Tinari N, et al. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 2002;23:313–20. doi: 10.1016/s1471-4906(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 5.Rubinstein N, Ilarregui JM, Toscano MA, Rabinovich GA. The role of galectins in the initiation, amplification and resolution of the inflammatory response. Tissue Antigens. 2004;64:1–12. doi: 10.1111/j.0001-2815.2004.00278.x. [DOI] [PubMed] [Google Scholar]
- 6.Seetharaman J, Kanigsberg A, Slaaby R, Leffler H, Barondes SH, Rini JM. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J Biol Chem. 1998;273:13047–52. doi: 10.1074/jbc.273.21.13047. [DOI] [PubMed] [Google Scholar]
- 7.Menon RP, Hughes RC. Determinants in the N-terminal domains of galectin-3 for secretion by a novel pathway circumventing the endoplasmic reticulum–Golgi complex. Eur J Biochem. 1999;264:569–76. doi: 10.1046/j.1432-1327.1999.00671.x. [DOI] [PubMed] [Google Scholar]
- 8.Cowles EA, Agrwal N, Anderson RL, Wang JL. Carbohydrate-binding protein 35. Isoelectric points of the polypeptide and a phosphorylated derivative. J Biol Chem. 1990;265:17706–12. [PubMed] [Google Scholar]
- 9.Henrick K, Bawumia S, Barboni EA, Mehul B, Hughes RC. Evidence for subsites in the galectins involved in sugar binding atthe nonreducing end of the central galactose of oligosaccharide ligands: sequence analysis, homology modeling and mutagenesis studies of hamster galectin-3. Glycobiology. 1998;8:45–57. doi: 10.1093/glycob/8.1.45. [DOI] [PubMed] [Google Scholar]
- 10.Kim K, Mayer EP, Nachtigal M. Galectin-3 expression in macrophages is signaled by Ras/MAP kinase pathway and up-regulated by modified lipoproteins. Biochim Biophys Acta. 2003;1641:13–23. doi: 10.1016/s0167-4889(03)00045-4. [DOI] [PubMed] [Google Scholar]
- 11.Moutsatsos IK, Wade M, Schindler M, Wang JL. Endogenous lectins from cultured cells: nuclear localization of carbohydrate-binding protein 35 in proliferating 3T3 fibroblasts. Proc Natl Acad Sci USA. 1987;84:6452–6. doi: 10.1073/pnas.84.18.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flotte TJ, Springer TA, Thorbecke GJ. Dendritic cell and macrophage staining by monoclonal antibodies in tissue sections and epidermal sheets. Am J Pathol. 1983;111:112–24. [PMC free article] [PubMed] [Google Scholar]
- 13.Davidson PJ, Davis MJ, Patterson RJ, Ripoche MA, Poirier F, Wang JL. Shuttling of galectin-3 between the nucleus and cytoplasm. Glycobiology. 2002;12:329–37. doi: 10.1093/glycob/12.5.329. [DOI] [PubMed] [Google Scholar]
- 14.Mehul B, Hughes RC. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J Cell Sci. 1997;110:1169–78. doi: 10.1242/jcs.110.10.1169. [DOI] [PubMed] [Google Scholar]
- 15.Maeda N, Kawada N, Seki S, et al. Stimulation of proliferation of rat hepatic stellate cells by galectin-1 and galectin-3 through different intracellular signaling pathways. J Biol Chem. 2003;278:18938–44. doi: 10.1074/jbc.M209673200. [DOI] [PubMed] [Google Scholar]
- 16.Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. 2002;168:1813–22. doi: 10.4049/jimmunol.168.4.1813. [DOI] [PubMed] [Google Scholar]
- 17.Akahani S, Nangia-Makker P, Inohara H, Kim HR, Raz A. Galectin-3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 1997;57:5272–6. [PubMed] [Google Scholar]
- 18.Inohara H, Akahani S, Raz A. Galectin-3 stimulates cell proliferation. Exp Cell Res. 1998;245:294–302. doi: 10.1006/excr.1998.4253. [DOI] [PubMed] [Google Scholar]
- 19.Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. 2006;103:5060–5. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad ML, Pellegata NS, Huang Y, de la Nagaraja HNCA, Kloos RT. Galectin-3, fibronectin-1, CITED-1, HBME1 and cytokeratin-19 immunohistochemistry is useful for the differential diagnosis of thyroid tumors. Mod Pathol. 2005;18:48–57. doi: 10.1038/modpathol.3800235. [DOI] [PubMed] [Google Scholar]
- 21.Saggiorato E, Aversa S, Deandreis D, et al. Galectin-3: presurgical marker of thyroid follicular epithelial cell-derived carcinomas. J Endocrinol Invest. 2004;27:311–7. doi: 10.1007/BF03351054. [DOI] [PubMed] [Google Scholar]
- 22.Oestreicher-Kedem Y, Halpern M, Roizman P, et al. Diagnostic value of galectin-3 as a marker for malignancy in follicular patterned thyroid lesions. Head Neck. 2004;26:960–6. doi: 10.1002/hed.20087. [DOI] [PubMed] [Google Scholar]
- 23.Schoeppner HL, Raz A, Ho SB, Bresalier RS. Expression of an endogenous galactose-binding lectin correlates with neoplastic progression in the colon. Cancer. 1995;75:2818–26. doi: 10.1002/1097-0142(19950615)75:12<2818::aid-cncr2820751206>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 24.Bresalier RS, Mazurek N, Sternberg LR, et al. Metastasis of human colon cancer is altered by modifying expression of the beta-galactoside-binding protein galectin 3. Gastroenterology. 1998;115:287–96. doi: 10.1016/s0016-5085(98)70195-7. [DOI] [PubMed] [Google Scholar]
- 25.Woo HJ, Joo HG, Song SW, Sohn YS, Chae C. Immunohistochemical detection of galectin-3 in canine gastric carcinomas. J Comp Pathol. 2001;124:216–18. doi: 10.1053/jcpa.2000.0442. [DOI] [PubMed] [Google Scholar]
- 26.Okada K, Shimura T, Suehiro T, Mochiki E, Kuwano H. Reduced galectin-3 expression is an indicator of unfavorable prognosis in gastric cancer. Anticancer Res. 2006;26:1369–76. [PubMed] [Google Scholar]
- 27.O'Driscoll L, Linehan R, Kennedy M, et al. Lack of prognostic significance of survivin, survivin-deltaEx3, survivin-2B, galectin-3, bag-1, bax-alpha and MRP-1 mRNAs in breast cancer. Cancer Lett. 2003;201:225–36. doi: 10.1016/s0304-3835(03)00518-4. [DOI] [PubMed] [Google Scholar]
- 28.Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci USA. 1996;93:6737–42. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshii T, Fukumori T, Honjo Y, Inohara H, Kim HR, Raz A. Galectin-3 phosphorylation is required for its anti-apoptotic function and cell cycle arrest. J Biol Chem. 2002;277:6852–7. doi: 10.1074/jbc.M107668200. [DOI] [PubMed] [Google Scholar]
- 30.Matarrese P, Tinari N, Semeraro ML, Natoli C, Iacobelli S, Malorni W. Galectin-3 overexpression protects from cell damage and death by influencing mitochondrial homeostasis. FEBS Lett. 2000;473:311–15. doi: 10.1016/s0014-5793(00)01547-7. [DOI] [PubMed] [Google Scholar]
- 31.Dagher SF, Wang JL, Patterson RJ. Identification of galectin-3 as a factor in pre-mRNA splicing. Proc Natl Acad Sci USA. 1995;92:1213–17. doi: 10.1073/pnas.92.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu DK, Yang RY, Pan Z, et al. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol. 2000;156:1073–83. doi: 10.1016/S0002-9440(10)64975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leeb SN, Vogl D, Gunckel M, et al. Reduced migration of fibroblasts in inflammatory bowel disease: role of inflammatory mediators and focal adhesion kinase. Gastroenterology. 2003;125:1341–54. doi: 10.1016/j.gastro.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Leeb SN, Vogl D, Grossmann J, et al. Autocrine fibronectin-induced migration of human colonic fibroblasts. Am J Gastroenterol. 2004;99:335–40. doi: 10.1111/j.1572-0241.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- 35.Badid C, Mounier N, Costa AM, Desmouliere A. Role of myofibroblasts during normal tissue repair and excessive scarring: interest of their assessment in nephropathies. Histol Histopathol. 2000;15:269–80. doi: 10.14670/HH-15.269. [DOI] [PubMed] [Google Scholar]
- 36.Leeb SN, Vogl D, Falk W, Scholmerich J, Rogler G, Gelbmann CM. Regulation of migration of human colonic myofibroblasts. Growth Factors. 2002;20:81–91. doi: 10.1080/08977190290031941. [DOI] [PubMed] [Google Scholar]
- 37.Jensen-Jarolim E, Gscheidlinger R, Oberhuber G, et al. The constitutive expression of galectin-3 is downregulated in the intestinal epithelia of Crohn's disease patients, and tumour necrosis factor alpha decreases the level of galectin-3-specific mRNA in HCT-8 cells. Eur J Gastroenterol Hepatol. 2002;14:145–52. doi: 10.1097/00042737-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Jensen-Jarolim E, Neumann C, Oberhuber G, et al. Anti-galectin-3 IgG autoantibodies in patients with Crohn's disease characterized by means of phage display peptide libraries. J Clin Immunol. 2001;21:348–56. doi: 10.1023/a:1012240719801. [DOI] [PubMed] [Google Scholar]
- 39.Muller S, Schaffer T, Flogerzi B, et al. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm Bowel Dis. 2006;12:588–97. doi: 10.1097/01.MIB.0000225341.37226.7c. [DOI] [PubMed] [Google Scholar]
- 40.Rogler G, Daig R, Aschenbrenner E, et al. Establishment of long-term primary cultures of human small and large intestinal epithelial cells. Lab Invest. 1998;78:889–90. [PubMed] [Google Scholar]
- 41.Rogler G, Hausmann M, Vogl D, et al. Isolation and phenotypic characterization of colonic macrophages. Clin Exp Immunol. 1998;112:205–15. doi: 10.1046/j.1365-2249.1998.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brenmoehl J, Herfarth H, Gluck T, et al. Genetic variants in the NOD2/CARD15 gene are associated with early mortality in sepsis patients. Intens Care Med. 2007;33:1541–8. doi: 10.1007/s00134-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 43.Osterman MT, Lichtenstein GR. Infliximab in fistulizing Crohn's disease. Gastroenterol Clin North Am. 2006;35:795–820. doi: 10.1016/j.gtc.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–33. doi: 10.1053/j.gastro.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 45.Fuss IJ, Becker C, Yang Z, et al. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. doi: 10.1097/01.mib.0000194183.92671.b6. [DOI] [PubMed] [Google Scholar]
- 46.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–21. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 47.Goebel S, Huang M, Davis WC, et al. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2006;290:G648–G654. doi: 10.1152/ajpgi.00466.2005. [DOI] [PubMed] [Google Scholar]
- 48.Sano H, Hsu DK, Yu L, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–64. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 49.Feuk-Lagerstedt E, Jordan ET, Leffler H, Dahlgren C, Karlsson A. Identification of CD66a and CD66b as the major galectin-3 receptor candidates in human neutrophils. J Immunol. 1999;163:5592–8. [PubMed] [Google Scholar]
- 50.Kuwabara I, Liu FT. Galectin-3 promotes adhesion of human neutrophils to laminin. J Immunol. 1996;156:3939–44. [PubMed] [Google Scholar]
- 51.Sato S, Hughes RC. Binding specificity of a baby hamster kidney lectin for H type I and II chains, polylactosamine glycans, and appropriately glycosylated forms of laminin and fibronectin. J Biol Chem. 1992;267:6983–90. [PubMed] [Google Scholar]
- 52.Ochieng J, Leite-Browning ML, Warfield P. Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochem Biophys Res Commun. 1998;246:788–91. doi: 10.1006/bbrc.1998.8708. [DOI] [PubMed] [Google Scholar]
- 53.Shimura T, Takenaka Y, Tsutsumi S, Hogan V, Kikuchi A, Raz A. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004;64:6363–7. doi: 10.1158/0008-5472.CAN-04-1816. [DOI] [PubMed] [Google Scholar]
- 54.Kasper M, Hughes RC. Immunocytochemical evidence for a modulation of galectin 3 (Mac-2), a carbohydrate binding protein, in pulmonary fibrosis. J Pathol. 1996;179:309–16. doi: 10.1002/(SICI)1096-9896(199607)179:3<309::AID-PATH572>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, Friess H, Zhu Z, et al. Galectin-1 and galectin-3 in chronic pancreatitis. Lab Invest. 2000;80:1233–41. doi: 10.1038/labinvest.3780131. [DOI] [PubMed] [Google Scholar]
- 56.Masamune A, Satoh M, Hirabayashi J, Kasai K, Satoh K, Shimosegawa T. Galectin-1 induces chemokine production and proliferation in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G729–G736. doi: 10.1152/ajpgi.00511.2005. [DOI] [PubMed] [Google Scholar]
- 57.Hittelet A, Legendre H, Nagy N, et al. Upregulation of galectins-1 and -3 in human colon cancer and their role in regulating cell migration. Int J Cancer. 2003;103:370–9. doi: 10.1002/ijc.10843. [DOI] [PubMed] [Google Scholar]
- 58.Santucci L, Fiorucci S, Rubinstein N, et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology. 2003;124:1381–94. doi: 10.1016/s0016-5085(03)00267-1. [DOI] [PubMed] [Google Scholar]