

Abstract
Hypothermic preservation of solid allografts causes profound damage of vascular endothelial cells. This, in turn, might activate innate immunity. In the present study we employed an in vitro model to study to what extent supernatants of damaged endothelial cells are able to activate innate immunity and to study the nature of these signals. The expression of high mobility group box 1 (HMGB1) and adhesion molecules on human umbilical vein endothelial cell was studied by immunofluorescence, fluorescence activated cell sorter and Western blotting. Cytokine production was performed by enzyme-linked immunosorbent assay. HMGB1 expression was lost completely in endothelial cells after hypothermic preservation. This was associated with cell damage as it occurred only in untreated endothelial cell but not in cells rendered resistant to hypothermia-mediated damage by dopamine treatment. Only supernatants from hypothermia susceptible cells up-regulated the expression of interleukin (IL)-8 and adhesion molecules in cultured endothelial cells in an HMGB1-dependent manner. In whole blood assays, both supernatants of hypothermia susceptible and resistant cells inhibited tumour necrosis factor (TNF)-α production concomitantly with an increased IL-10 secretion. The activity of the supernatants was already found after 6 h of hypothermic preservation, and paralleled the decrease in intracellular adenosine triphosphate (ATP) levels. Modulation of TNF-α and IL-10 production by these supernatants was abrogated completely by prior treatment with adenosine deaminase and was similar to the response of an A2R agonist. Our study demonstrates that both HMGB1 and adenosine are released during hypothermic preservation. While release of HMGB1 is caused by cell damage, release of adenosine seems to be related to ATP hydrolysis, occurring in both susceptible and resistant cells.
Keywords: adenosine, dopamine, endothelium, HMGB1, hyothermia
Introduction
Allocation of organ allografts has become possible by adequate improvements in organ preservation. In transplantation of solid organs, static cold storage is the most widely used modality for organ preservation. However, acute shortage of donor organs and the inevitable pressure to use suboptimal donors has made this method questionable to prevent deterioration of organ quality [1,2]. Prolonged hypothermic preservation remains a significant cause of pretransplantation injury of allografts. Because pretransplantation injury affects short- and long-term organ function significantly [3–5], cold ischaemia time is a critically important factor for transplantation outcome [3,4]. Tissue injury caused by hypothermia is mediated largely by oxidative stress [6–8]. Injury to endothelial cells may lead to impaired control of vascular permeability [9], thrombosis [10] and inflammation [10,11].
According to the danger hypothesis [12], tissue damage might evoke immune activation and therefore tissue damage seems to increase tissue immunogenicity of the allograft. Although this theory has now been accepted widely, the signals that initiate immune activation and modulate innate immunity are not defined clearly in organ transplantation.
High mobility group box chromosomal protein 1 (HMGB1) is a highly conserved chromatin binding protein which is abundantly expressed in mammalian tissues [13]. In addition to its crucial roles in stabilizing nucleosome formation, facilitating gene transcription and modulating steroid hormone receptors, its role in innate immunity has been appreciated widely [13–15]. HMGB1 can be released passively by necrotic cells [16] or secreted actively by monocytes, macrophages and dendritic cells via a non-classical vesicle-mediated secretory pathway [17–19]. The presence of extracellular HMGB1 is not only a indicator that a cell or tissue has suffered from damage, but also provides ‘danger’ signals to a variety of cells that constitute the innate immune system [14,20,21]. Binding of HMGB1 to the receptor for advanced glycation end products (RAGE) on endothelial cells and monocytes increases the expression of adhesion molecules and stimulates the production of an array of proinflammatory cytokines [22,23]. The involvement of Toll-like receptors (TLR)-2 and -4 in cell activation by HMGB1 has also been demonstrated [24].
We and others have shown previously that cold preservation of endothelial cells results in a rapid loss of endothelial barrier function [9] and induces necrosis of these cells [25–27]. Therefore, cold preservation of allografts might be accompanied by the release of HMGB1 from endothelial cells and hence might contribute to activation of the innate immune system and subsequently to inflammation [13–15,22,23].
Apart from signals that activate innate immunity, other factors might be present that down-regulate inflammation. In particular adenosine, a purine nucleoside that is released extracellularly, has a potent endogenous anti-inflammatory potential [28]. Under circumstances such as ischaemia, a rapid and massive degradation of intracellular adenosine triphosphate (ATP) leads eventually to accumulation of adenosine 5′-phosphate which, in turn, is converted to adenosine [29,30]. Also, during cold preservation of organ allografts, intracellular ATP is depleted within hours, an eligible condition for extracellular release of adenosine. It is therefore plausible that adenosine release during hypothermic preservation of allografts is another pivotal modulator of the immune response in the recipient.
In the present study we used an in vitro model to investigate if hypothermic preservation of endothelial cells is associated with the release of HMGB1 and adenosine. In addition, the functional relevance of these factors in terms of immune activation was studied.
Materials and methods
Reagents
Reagents used were as follows: endothelial cell growth MedKIT, phenol-red free (PRF) medium (Promocell, Heidelberg, Germany), phosphate-buffered saline (PBS), RPMI-1640 (Gibco invitrogen, NY, USA), fetal bovine serum (FBS) Gold (PAA Laboratories GmbH, Pasching, Austria), trypsin/ethylenediamine tetraacetic acid solution, lipopolysaccharide (LPS) from Escherichia coli, CGS 21680, DMSO, TritonX-100 (Sigma, St Louis, MO, USA), bovine serum albumin (BSA) (Serva, Heidelberg, Germany), 37% formaldehyde (Mallinckrodt Baker, Deventer, the Netherlands), Ficoll-paqueTM plus (Amersham Biosciences AB, Uppsala, Sweden), peptidoglycan Bacillus subtilis (PGN-BS) (B. subtilis), Pam3CysSerLys4 (Pam3CSK4), Poly (I:C), flagellin (B. subtilis), CL087, ssRNA40/LyoVec, ODN2006 (Invitrogen, San Diego, CA, USA), ZM241385 (Tocris Cookson Inc., Ellisville, MO, USA) and high mobility group protein 1 (Proteinone, Bethesda, MD, USA).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated from fresh umbilical cords as described previously [26]. The cells were grown in basal endothelial medium supplemented with 10% FBS and essential growth factors until they formed a confluent monolayer. Cold preservation was performed essentially as described [26]. In brief, 2 h before cold preservation was started HUVECs were either treated with dopamine (DA, 30 μM) or left untreated. The cells were stored at 4°C in PRF cell-culture medium or preservation solution (University of Wisconsin, UW) depending on the specific experiment.
Immunohistochemical staining
High mobility group box chromosomal protein 1 staining was performed on HUVECs and peripheral blood mononuclear cells (PBMC). HUVECs were cultured on glass coverslips until confluence. The cells were treated for 2 h with DA (30 μM) or left untreated and stored for 24 h at 4°C in UW solution. Cells that were cultured for the same period of time at 37°C were included in each experiment. PBMC were isolated by Ficoll-Paque plus density-gradient centrifugation and mounted on glass by cytospin centrifugation. All cells were fixed in 4% freshly made formaldehyde for 1 h at room temperature followed by permeabilization with ice-cold 0·5% Triton X-100 for 5 min. The cells were then incubated for 30 min at room temperature with PBS containing 2% BSA to block unspecific background staining. A polyclonal rabbit anti-HMGB1 antibody (Biomol, Lake Placid, NY, USA) was added overnight to the samples at 4°C. After washing extensively with PBS/BSA the samples were incubated for 1 h at room temperature with a Texas red-conjugated goat anti-rabbit immunoglobulin G (IgG) (Molecular Probes, Leiden, the Netherlands) or fluorescein isothiocyanate (FITC)-conjugated swine anti-rabbit IgG (Dako, Hamburg, Germany). Cells were incubated with 4,6-diamidino-2-phenylindole or TOTO3 (Molecular Probes, Leiden, the Netherlands) for 15 min at room temperature to visualize nuclei. Fluorescence signals were analysed by fluorescence and confocal microscopy.
Assessment of intracellular ATP
Confluent HUVEC monolayers were treated for 2 h with 30 μM DA or left untreated. Subsequently the cells were washed and stored at 4°C. Intracellular ATP was extracted at serial time-points and measured by luciferase-driven bioluminescence using the ATP Bioluminescence Assay Kit CLS II (Roche Diagnostics, Mannheim, Germany).
Activation of endothelial cells by supernatants
To test if the supernatants of endothelial cells that were stored for 24 h at 4°C contain biological activity, normal cultured HUVEC were incubated for 24 h at 37°C with the supernatants in a 1:1 dilution. Hereafter interleukin (IL)-8 production was measured by enzyme-linked immunosorbent assay (ELISA), as recommended by the manufacturer (Beckman Coulter, Marseille Cedex, France). HMGB1 specificity in the supernatants was tested by immune-absorption. To this end, HMGB1 by adding a polyclonal rabbit anti-HMGB1 antibody (Biomol) or normal rabbit IgG was added for 1 h followed by the addition of protein A/G (1 h) (Santa Cruz, Heidelberg, Germany). The supernatants were centrifuged and used for further experiments. Absorption of HMGB1 was demonstrated by Western blot (data not shown). Apart from IL-8 production, activation of HUVEC by the supernatants was also tested by fluorescence activated cell sorter (FACS) analysis as described below.
Fluorescence activated cell sorter analysis
Human umbilical vein endothelial cells were cultured in medium supplemented with supernatants (dilution 1 : 1) from endothelial cells that were subjected to cold preservation. FACS analysis was performed with 2 × 106 cells using the following FITC-conjugated monoclonal antibodies; anti-human intracellular adhesion molecule 1 (ICAM-1), anti-human vascular adhesion molecule 1 (VCAM-1) and anti-human E-selectin (all from Becton Dickinson, Heidelberg, Germany). Antibodies were added for 30 min at 4°C followed by washing extensively with PBS. FACS analysis was performed on a FACScalibur equipped with the CELLQuest software (Becton Dickinson). The data were analysed by Windows Multiple Document Interface (WinMDI) software (version 2·8).
Whole blood assay
Blood was obtained from healthy adult donors with informed consent and diluted 1 : 1 with PRF medium or with supernatants obtained from HUVECs that were stored for 24 h at 4°C in PRF medium. HUVECs were either left untreated or treated with DA (30 μM) for 2 h prior to cold storage, as described above. The diluted blood was plated subsequently in 24-well plates containing either culture medium or one of the following stimuli: 20 μg/ml PGN-BS (B. subtilis), 10 μg/ml Pam3CSK4, 50 μg/ml Poly(I:C), 0·5 μg/ml LPS, 1 μg/ml flagellin (B. subtilis), 1 μg/ml CL087, 1 μg/ml ssRNA40/LyoVec, 1 μM ODN2006 (Invivogen). In some experiments, recombinant HMGB1 (0·1 μg/ml) (Proteinone) or the adenosine receptor A2A agonist CGS 21680 (5 μM) (Sigma) was added. The concentrations of different TLR ligands were based on a previous publication [31] and product data sheet. The plates were incubated for 24 h at 37°C (5% CO2/95% humidity) after which supernatants were harvested and assessed for tumour necrosis factor (TNF)-α and IL-10 production. TNF-α and IL-10 concentrations of the supernatant after whole blood stimulation were measured by ELISA (both from Beckman Coulter), according to the manufacturer's instructions.
Western blot analysis
Supernatants from HUVECs that were subjected to cold preservation for 24 h were analysed for the presence of HMGB1 by Western blot. The supernatant were 50× concentrated by means of centricon columns (Millipore, Billerica, MA, USA). The presence of HMGB1 in cell nuclei isolated from the same HUVEC was also analysed. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting was performed essentially similarly, as described [26]. Briefly, samples were resolved on 10% SDS-PAGE and electrotransferred onto polyvinylidene difluoride filters. Hereafter the filters were incubated overnight with 5% non-fat dry milk powder in PBS to block unspecific background staining. A polyclonal rabbit anti-HMGB1 antibody (Biomol) followed by a horseradish peroxidise-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) (both dissolved in PBS/0·2% Tween-20/5% non-fat dry milk powder) were added subsequently for 60 min. Visualization of immunoreactive bands was performed by chemiluminescence reagent (PerkinElmer LAS Inc., Boston, MA, USA), according to the manufacturer's instructions.
Statistical analysis
Data were presented as mean ± standard deviation for the indicated number of separate experiments. All analyses were based on more than three separate experiments. Differences between groups were determined by Student's t-test. A P-value less than 0·05 was considered statistically significant.
Results
Release of HMGB1 during cold preservation
The expression of HMGB1 was investigated by means of immunefluorescence. In cultured HUVEC, HMGB1 expression was found both in the nuclei and in an extranuclear compartment in close vicinity to the nucleus (Fig. 1, upper panel, left). By using confocal immunofluorescence microscopy extranuclear HMGB1 expression could be confirmed (Fig. 1a, upper panel, middle). Extranuclear HMGB1 expression was found only in HUVEC and not in PBMC (Fig. 1a, upper panel, right). In untreated HUVEC that were subjected to 24 h of cold preservation, the extranuclear compartment was lost completely, while nuclear staining of HMGB1 was diminished strongly (Fig. 1a, lower panel, left). Cold-preservation of untreated HUVEC was characterized by release of lactate dehydrogenase, which was abrogated completely by 2 h of DA treatment prior to cold preservation (data not shown). Under the latter condition, loss of HMGB1 expression was not observed (Fig. 1a, lower panel, right). Western blot analysis of isolated nuclei and supernatants confirmed the loss of HMGB1 in nuclei of untreated cells concomitantly with the appearance of the protein in the supernatant (Fig. 1b).
Fig. 1.
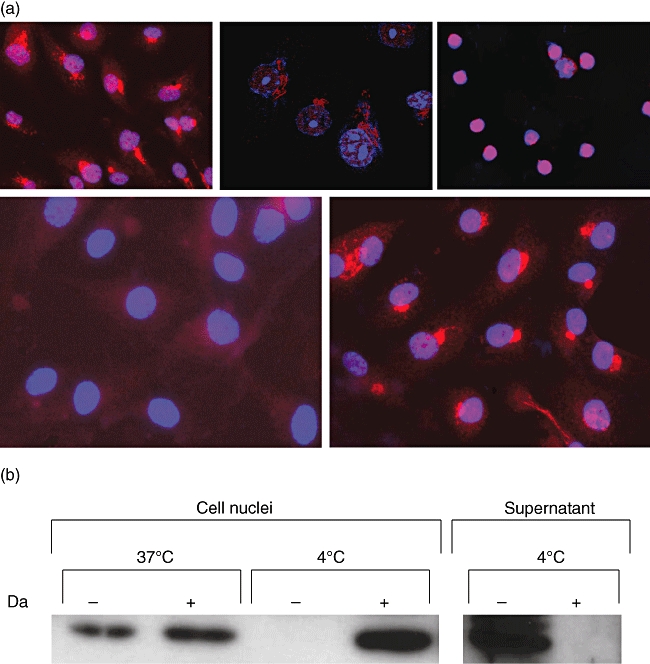
(a) High mobility group box 1 (HMGB1) expression in endothelial cells. Cultured human umbilical vein endothelial cells (HUVEC) (upper two panels, left) and freshly isolated peripheral blood mononuclear cells (upper panel, right) were stained for HMGB1 as described in the Materials and methods section. Analysis of HMGB1 was performed by means of immunofluorescence (upper panels, left and right) and by confocal microscopy. Original magnification: 200× and 400× respectively. The lower panels show HMGB1 expression in untreated HUVEC (left) and dopamine-treated HUVEC (right) after 24 h of cold storage. Original magnification: 400×. (b) Western blot analysis of HMGB1 in cell nuclei from HUVEC before (37°C) and after cold storage (4°C). Note the disappearance of HMGB1 from the nuclei after cold storage of untreated HUVEC (–) and its appearance the supernatant of these cells. Da: dopamine 30 μM.
Modulation of adhesion molecules and IL-8 expression in cultured endothelial cells
To test if the release of HMGB1 from unprotected HUVEC has functional significance, cultured endothelial cells were stimulated with supernatants of untreated or DA-treated HUVEC for 24 h, after which the expression of ICAM-1, E-selectin and VCAM-1 was determined. The expression of all three adhesion molecules was up-regulated by supernatants obtained from untreated HUVEC (Fig. 2a). Similarly, IL-8 production was two times higher in endothelial cell cultures stimulated with supernatants of untreated HUVEC. This was due to the release of HMGB1, as depletion of HMGB1 from these supernatants by means of immune-absorption completely normalized IL-8 production (Fig. 2b).
Fig. 2.
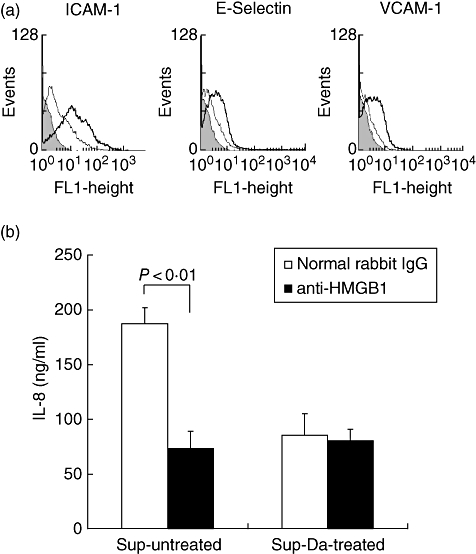
Up-regulation of adhesion molecules and interleukin (IL)-8 production by supernatants. (a) Normal cultured endothelial cell were incubated for 24 h with supernatants obtained from untreated (bold line) and dopamine-treated (thin line) human umbilical vein endothelial cells (HUVEC) that were subjected to 24 h of cold storage. Note that supernatants from untreated HUVEC up-regulates the expression of intercellular adhesion molecule type 1, E-selectin and vascular cell adhesion molecule type 1 compared with supernatants from dopamine treated HUVEC. Filled histogram represents the negative control. The results from a representative experiment (n = 5) is depicted. (b) Normal cultured endothelial cells were incubated for 24 h with supernatants obtained from untreated (Sup-untreated) and dopamine-treated (Sup-Da-treated) HUVEC that were subjected to 24 h of cold storage. The supernatants were immune-absorbed with anti-high mobility group box 1 (HMGB1) (filled bars) or normal rabbit immunoglobulin G, as described in the Materials and methods section. IL-8 production was measured by enzyme-linked immunosorbent assay using triplicates for each condition. The results are expressed as mean IL-8 production (ng/ml) ± standard deviation from four different experiments. ICAM, intracellular adhesion molecule; VCAM, vascular adhesion molecule.
Modulation of TNF-α and IL-10 production
We next investigated if supernatants obtained from HUVEC after cold storage are able to modulate LPS-mediated TNF-α and IL-10 production in whole blood assays. Both supernatants from untreated and DA-treated HUVEC (n = 6) suppressed TNF-α production consistently, while the production of IL-10 was increased. This was unlikely to be mediated by HMGB1, as addition of recombinant HMGB1 augmented both TNF-α and IL-10 production (Fig. 3).
Fig. 3.
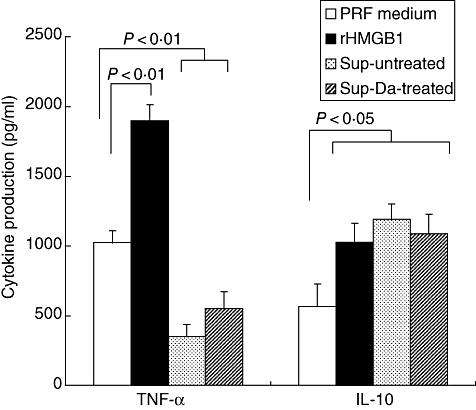
Lipopolysaccharide (LPS)-mediated tumour necrosis factor (TNF)-α and interleukin (IL)-10 production in whole blood assays. Whole blood was diluted 1 : 1 in normal phenol-red free (PRF) medium to which either nothing (open bars) or 0·1 μg/ml recombinant high mobility group box 1 (HMGB1) (filled bars) was added. In addition, whole blood was diluted 1 : 1 with supernatants obtained after cold storage (24 h) of untreated human umbilical vein endothelial cells (HUVEC) (Sup-untreated; dotted bars) or dopamine-treated (Sup-Da-treated; hatched bars). To each condition 0·5 μg/ml of LPS was added. TNF-α and IL-10 production was assessed after 24 h by enzyme-linked immunosorbent assay, using triplicates for each condition. A total of six experiments were performed with different blood donors and different HUVEC supernatants. The result of a representative experiment is depicted.
Although up-regulation in TNF-α and IL-10 production was also observed when other TLR ligands were added to the whole blood assay, inhibition of TNF-α production by supernatants obtained from HUVEC after cold storage occurred only in a TLR-4-dependent fashion (Fig. 4). This was also found for the up-regulation in IL-10 production. Supernatants from DA-treated and untreated HUVEC behaved similarly in this regard (data not shown).
Fig. 4.
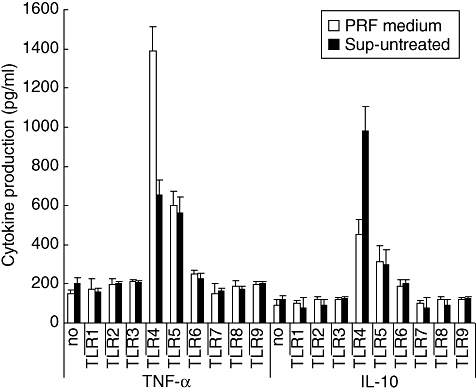
Modulation of Toll-like receptor (TLR)-mediated tumour necrosis factor (TNF)-α and interleukin (IL)-10 production. Whole blood was diluted 1 : 1 in normal phenol-red free (PRF) medium (open bars) or in supernatants of untreated human umbilical vein endothelial cells (Sup-untreated), obtained directly after 24 h of cold storage. To the wells were added either nothing (no) or different TLR ligands (TLR-1–9). The different ligands that were used were as follows: peptidoglycan Bacillus subtilis (PGN-BS) (20 μg/ml) (TLR-1), Pam3CysSerLys4 (10 μg/ml) (TLR-2), Poly (I:C) (50 μg/ml) (TLR-3), LPS 500 ng/ml (TLR-4), flagellin (1 μg/ml) (TLR-5), CL087 (1 μg/ml) (TLR-6), ssRNA (1 μg/ml) (TLR-8) and ODN2006 (1 μM) (TLR-9). TNF-α and IL-10 production was assessed as described in Fig. 3. A total of four different experiments were performed. The result of a representative experiment is expressed as mean cytokine production (pg/ml) ± standard deviation.
The modulatory activity found in the supernatants was already present 6 h after cold storage and coincided with loss of intracellular ATP levels (Fig. 5). Although intracellular ATP levels decreased further in time during cold preservation, modulation of TNF-α and IL-10 production was maximal after 6 h. In subsequent experiments the involvement of adenosine, a product of ATP hydrolysis, in modulation of cytokine production was studied. When whole blood assays were performed in normal PRF medium, addition of the non-specific adenosine A2A and A2B receptor agonist 5′-N-ethylcarboxamidoadenosine (NECA) inhibited TNF-α production significantly, while IL-10 production was up-regulated significantly to a similar extent as supernatants from HUVEC after cold storage (Fig. 6a). The specific adenosine A2A receptor agonist CGS 21680 did not differ from NECA in this regard (data not shown). Addition of NECA HUVEC supernatants did not influence TNF-α or IL-10 production (Fig. 6a). In contrast, addition of adenosine deaminase (ADA) abrogated completely the modulatory effect of the supernatants, while addition of ADA to normal PRF medium did not influence the production of TNF-α or IL-10 in whole blood assays (Fig. 6b).
Fig. 5.
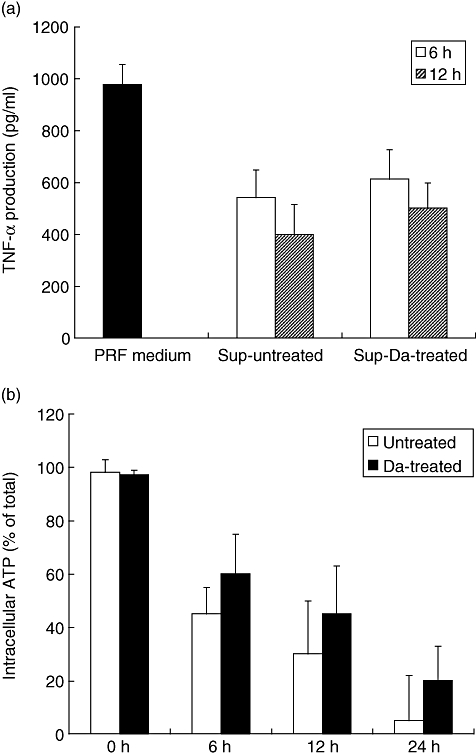
Acquirement of tumour necrosis factor (TNF)-α suppressing activity in supernatants and intracellular adenosine triphosphate (ATP) depletion in time. (a) Whole blood was diluted 1:1 in normal phenol-red free (PRF) medium (filled bars) and in supernatants of untreated human umbilical vein endothelial cells (HUVEC) (Sup-untreated) or in supernatants of dopamine-treated HUVEC (Sup-Da-treated), obtained directly after 6 h (open bars) or 12 h (hatched bars) of cold storage. To each condition 0·5 μg/ml of lipopolysaccharide was added. TNF-α production was assessed as described in Fig. 3. A total of three different experiments were performed. The result of a representative experiment is expressed as mean TNF-α production (pg/ml) ± standard deviation (s.d.). (b) Untreated (open bars) and dopamine-treated (filled bars) HUVEC were stored at 4°C for different periods of time. Hereafter, intracellular ATP was measured as described in Materials and methods. The total amount of ATP before cold storage was taken as 100% for each condition. The result of a representative experiment is depicted and expressed as mean percentage of total ATP ± s.d. A total number of three experiments were performed.
Fig. 6.
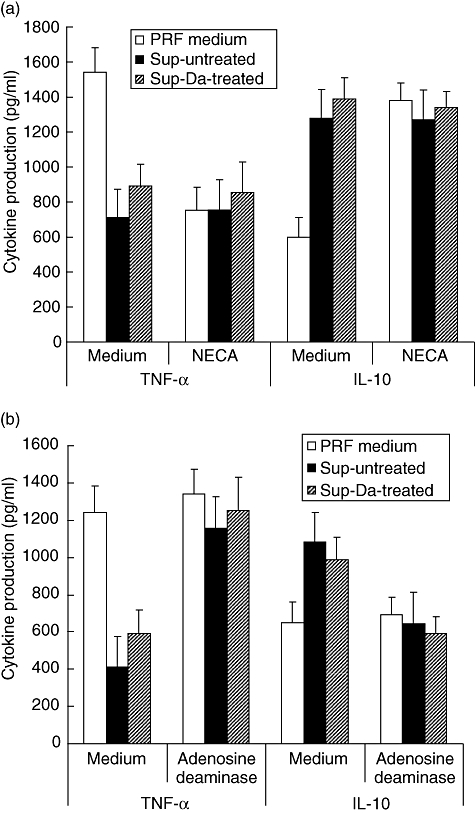
Modulation of lipopolysaccharide (LPS)-induced cytokine production is mediated by adenosine. (a) Whole blood was diluted 1:1 in normal phenol-red free (PRF) medium (open bars) and in supernatants of untreated human umbilical vein endothelial cells (HUVEC) (filled bars; Sup-untreated) or in supernatants of dopamine treated HUVEC (hatched bars; Sup-Da-treated), obtained directly after 24 h of cold storage. To each condition 0·5 μg/ml of LPS was added. The whole blood assay was performed in the absence (medium) or presence of 5′-N-ethylcarboxamidoadenosine (10 μM). (b) Similar to (a), but the supernatants were not treated (medium) or treated with adenosine deaminase (7 U/ml) before initiation of the experiments. In (a) and (b), tumour necrosis factor (TNF)-α and interleukin (IL)-10 production were assessed as described in Fig. 3. A total of four different experiments were performed. The result of a representative experiment is depicted and expressed as mean cytokine production (pg/ml) ± standard deviation.
Discussion
Long-term graft survival of renal allografts obtained from living donors is generally better than that obtained from post-mortem donors, despite better human leucocyte antigen-matching of the latter [32]. According to the danger hypothesis [12], this might be explained by the fact that immune activation following transplantation will be much stronger in allografts that have suffered from severe damage during the transplantation process. Compared with allografts from living donors, pretransplantation injury in allografts from post-mortem donors is expected to be larger as a consequence of brain death [33,34] and prolonged hypothermic preservation [3]. While brain death influences microcirculation and organ perfusion [35] and is associated with inflammation in end-organs [33,34], hypothermic preservation causes cell necrosis in a time-dependent fashion [3,8,26]. How hypothermic preservation might cause immune activation, however, is not yet clear. In the present study we therefore investigated if hypothermic preservation of endothelial cells is associated with the release of HMGB1 and adenosine, two important molecules known to modulate innate immunity. The major findings of this study are, first, that HMGB1 expression is lost completely in endothelial cells that underwent necrosis during hypothermic preservation. This is not observed in endothelial cells rendered resistant to cold preservation damage by DA pretreatment. Secondly, HMGB1 released in the supernatant of damage cells is biologically active as it up-regulates adhesion molecules and IL-8 production in viable endothelial cells. Thirdly, during hypothermic preservation adenosine is released independently of cell damage. Adenosine modulates TNF-α and IL-10 production in monocytes in a TLR-4-dependent fashion.
To our knowledge, extranuclear HMGB1 expression in viable endothelial cells has not been reported thus far. Although Mullins et al. [36] have reported that in LPS or TNF-α stimulated HUVEC relocation of HMGB1 to the cytoplasm can occur, they did not mention relocation of HMGB1 to a specific intracellular compartment. Our findings on the extranuclear compartment of HMGB1 in HUVEC was unlikely because of a fixation or staining artefact, as HMGB1 expression in PBMC that were processed similarly to HUVEC did not reveal an extranuclear HMGB1 compartment. Based on the HMGB1 staining pattern we speculate that this compartment might be related to the endoplasmatic reticulum (ER); however, this must be confirmed in future studies. In support of ER localization is a recent report from Ivanov et al. [37], who showed co-localization of HMGB1 with markers of the ER, the ER Golgi intermediate compartment and the Golgi in bone marrow-derived dendritic cells and macrophages.
Because prolonged hypothermic preservation leads to tissue necrosis, the finding that HMGB1 was lost during cold preservation was not surprising. Cold preservation injury can be prevented by appropriate pretreatment of cells with a number of compounds, e.g. catecholamines [8,9] and iron scavengers [26,38]. Prevention of the loss of HMGB1 expression in DA-treated HUVEC demonstrates that hypothermia does not cause the release of HMGB1 per se but that this reflects cell necrosis.
Although we did not address if HMGB1 expression was also reduced in organ allografts after hypothermic preservation, a number of studies have demonstrated unambiguously the occurrence of tissue necrosis under this condition [25,39]. Therefore, it is likely that HMGB1 is indeed released during cold preservation of whole organs. The presence of HMGB1 in organ allografts might influence neutrophil infiltration [40], endothelial production of proinflammatory cytokines [23], recruitment and proliferation of smooth muscle cells [41] and homing of endothelial progenitor cells [42].
Apart from HMGB1, adenosine could also be demonstrated in supernatants of HUVEC after cold storage. This might explain why supernatants of damaged cells down-regulated TNF-α production in whole blood assay, despite the presence of HMGB1. Adenosine release was associated with ATP hydrolysis and occurred independently of cell necrosis. Extracellular ATP can be converted to adenosine by a cascade of ectonucleotidases, including CD39 and CD73, both of which are expressed on endothelial cells [43,44]. Because ATP hydrolysis also occurs intracellularly, adenosine can be released alternatively through nucleoside transporters [44].
We have previously demonstrated in vitro that endothelial barrier dysfunction is restored after cold preservation by DA pretreatment [9]. Because adenosine enhances barrier function [43–46], this might underlie the beneficial effect of DA on barrier function. Although adenosine was also released during cold storage in untreated cells, it cannot restore barrier dysfunction in these cells as they become necrotic with increasing cold preservation time [26]. The involvement of adenosine on barrier function, however, has not been addressed directly in the present study.
Because in vitro HMGB1 and adenosine are released in the supernatants and organ allografts are flushed before implantation, it can be argued that release of these factors do not have any significance in organ transplantation. Endothelial cells express RAGE [47], a putative receptor for HMGB1, which might already bind its ligand during cold preservation. Subsequent warm reperfusion then can cause receptor activation leading to phenotypic and functional changes in endothelial cells. The relevance of endogenous release of adenosine during hypothermia is indeed questionable, as adenosine is added to some preservation solution, e.g. UW.
In conclusion, our study identified two mediators, i.e. HMGB1 and adenosine, that are released during cold preservation and that are able to influence innate immunity. While the release of HMGB1 is associated with tissue necrosis and activation of innate immunity, adenosine release is independent of cell damage and suppresses activation of the innate immune system in a TLR-4-dependent fashion. Prevention of cold preservation injury by adequate donor treatment might thus prevent release of HMGB1 and preserve the action of adenosine on endothelial barrier function, thereby improving immediate graft function and long-term graft survival after transplantation of organ allografts from post-mortem donors.
Acknowledgments
This study was supported by a grant of the German Research Foundation (DFG, European graduate school, GRK 880).
References
- 1.Maathuis MH, Leuvenink HJ, Ploeg RJ. Perspectives in organ preservation. Transplantation. 2007;83:1289–98. doi: 10.1097/01.tp.0000265586.66475.cc. [DOI] [PubMed] [Google Scholar]
- 2.Fuller BJ, Lee CY. Hypothermic perfusion preservation: the future of organ preservation revisited? Cryobiology. 2007;54:129–45. doi: 10.1016/j.cryobiol.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Salahudeen AK, Haider N, May W. Cold ischemia and the reduced long-term survival of cadaveric renal allografts. Kidney Int. 2004;65:713–18. doi: 10.1111/j.1523-1755.2004.00416.x. [DOI] [PubMed] [Google Scholar]
- 4.Salahudeen AK. Consequences of cold ischemic injury of kidneys in clinical transplantation. J Invest Med. 2004;52:296–8. doi: 10.1136/jim-52-05-28. [DOI] [PubMed] [Google Scholar]
- 5.Kieran NE, Rabb H. Immune responses in kidney preservation and reperfusion injury. J Invest Med. 2004;52:310–14. doi: 10.1136/jim-52-05-30. [DOI] [PubMed] [Google Scholar]
- 6.Zieger MA, Gupta MP. Endothelial cell preservation at 10 degrees C minimizes catalytic iron, oxidative stress, and cold-induced injury. Cell Transplant. 2006;15:499–510. doi: 10.3727/000000006783981756. [DOI] [PubMed] [Google Scholar]
- 7.Vairetti M, Griffini P, Pietrocola G, Richelmi P, Freitas I. Cold-induced apoptosis in isolated rat hepatocytes: protective role of glutathione. Free Radic Biol Med. 2001;31:954–61. doi: 10.1016/s0891-5849(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 8.Bartels-Stringer M, Kramers C, Wetzels JF, Russel FG, Groot H, Rauen U. Hypothermia causes a marked injury to rat proximal tubular cells that is aggravated by all currently used preservation solutions. Cryobiology. 2003;47:82–91. doi: 10.1016/s0011-2240(03)00071-3. [DOI] [PubMed] [Google Scholar]
- 9.Brinkkoetter PT, Beck GC, Gottmann U, et al. Hypothermia-induced loss of endothelial barrier function is restored after dopamine pre-treatment: role of p42/p44 activation. Transplantation. 2006;82:534–42. doi: 10.1097/01.tp.0000229396.34362.e2. [DOI] [PubMed] [Google Scholar]
- 10.Verrier E. The microvascular cell and ischemia-reperfusion injury. J Cardiovasc Pharmacol. 1996;27(Suppl. 1):S26–30. doi: 10.1097/00005344-199600001-00007. [DOI] [PubMed] [Google Scholar]
- 11.Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-KappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation. 2001;103:2296–302. doi: 10.1161/01.cir.103.18.2296. [DOI] [PubMed] [Google Scholar]
- 12.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 13.Mueller S, Scaffidi P, Degryse B, et al. New EMBO member's review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 2001;20:4337–40. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumitriu IE, Barau HP, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–7. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Popovic PJ, DeMarco L, Lotze MT, et al. High mobility group B1 protein suppresses the human plasmacytoid dendritic cell response to TLR9 agonists. J Immunol. 2006;177:8701–7. doi: 10.4049/jimmunol.177.12.8701. [DOI] [PubMed] [Google Scholar]
- 16.Scaffidi P, Misteli T, Bianchi ME. Relaese of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 18.Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Report. 2002;10:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi: 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- 20.Harris HE, Raucci A. Alarming news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–8. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeMarco RA, Fink MP, Lotze MP. Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol Immunol. 2005;42:433–44. doi: 10.1016/j.molimm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 22.Adersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG-1) stimulates pro-inflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 24.Park JS, Svetkauskaite D, He QB, et al. Involvement of Toll like receptor 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 25.Huet PM, Nagaoka MR, Desbiens G, et al. Sinusoidal endothelial cell and hepatocyte death following cold ischemia–warm reperfusion of the rat liver. Hepatology. 2004;39:1110–19. doi: 10.1002/hep.20157. [DOI] [PubMed] [Google Scholar]
- 26.Yard B, Beck G, Schnuelle P, et al. Prevention of cold-preservation injury of cultured endothelial cells by catecholamines and related compounds. Am J Transplant. 2004;4:22–30. doi: 10.1046/j.1600-6143.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 27.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia–reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 28.Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- 29.Van Belle H, Goossens F, Wynants J. Formation and release of purine catabolites during hypoperfusion, anoxia and ischemia. Am J Physiol. 1987;252:H886–93. doi: 10.1152/ajpheart.1987.252.5.H886. [DOI] [PubMed] [Google Scholar]
- 30.Matherne GP, Headrick JP, Coleman SD, Berne RM. Interstitial transudate purines in normoxic and hypoxic immature and mature rabbit hearts. Pediatr Res. 1990;28:348–53. doi: 10.1203/00006450-199010000-00010. [DOI] [PubMed] [Google Scholar]
- 31.Morris GE, Parker LC, Ward JR, et al. Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 2006;20:1539–49. doi: 10.1096/fj.06-5910fje. [DOI] [PubMed] [Google Scholar]
- 32.Terasaki PI, Cecka JM, Gjetson DW, Takemoto S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med. 1995;333:333–6. doi: 10.1056/NEJM199508103330601. [DOI] [PubMed] [Google Scholar]
- 33.Weiss S, Kotsch K, Francuski M, et al. Brain death activates donor organs and is associated with a worse I/R injury after liver transplantation. Am J Transplant. 2007;7:1584–93. doi: 10.1111/j.1600-6143.2007.01799.x. [DOI] [PubMed] [Google Scholar]
- 34.Pratscke J, Wilhelm MJ, Laskowski I, et al. Influence of donor brain death on chronic rejection of renal transplants in rats. J Am Soc Nephrol. 2001;12:2474–81. doi: 10.1681/ASN.V12112474. [DOI] [PubMed] [Google Scholar]
- 35.Herijgers P, Leunens V, Tjandra-Maga TB, Mubagwa K, Flameng W. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation. 1996;62:330–5. doi: 10.1097/00007890-199608150-00005. [DOI] [PubMed] [Google Scholar]
- 36.Mullins GE, Suden-Gullberg J, Johansson AS, et al. Activation of human umbilical vein endothelial cells leads to relocation and release of high-mobility group box chromosomal protein 1. Scand J Immunol. 2004;60:566–73. doi: 10.1111/j.0300-9475.2004.01518.x. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov S, Dragoi A-M, Wang X, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–81. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rauen U, Kerkweg U, de Groot H. Iron-dependent vs. iron-independent cold-induced injury to cultured rat hepatocytes: a comparative study in physiological media and organ preservation solutions. Cryobiology. 2007;54:77–86. doi: 10.1016/j.cryobiol.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Reddy SP, Battacharjya S, Maniakin N, et al. Preservation of porcine non-heart-beating donor livers by sequential cold storage and warm perfusion. Transplantation. 2004;77:1328–32. doi: 10.1097/01.tp.0000119206.63326.56. [DOI] [PubMed] [Google Scholar]
- 40.Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–39. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porto A, Palumbo R, Pieroni M, et al. Smooth muscle cells in human atherosclerotic plaques secrete and proliferate in response to high mobility group box 1 protein. FASEB J. 2006;20:2565–6. doi: 10.1096/fj.06-5867fje. [DOI] [PubMed] [Google Scholar]
- 42.Chavakis E, Hain A, Vincini M, et al. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100:204–12. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 43.Eltzschig HK, Iba JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–96. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eltzschig HK, Weissmuller T, Mager A, Eckle T. Nucleotide metabolism and cell–cell interactions. Methods Mol Biol. 2006;341:73–87. doi: 10.1385/1-59745-113-4:73. [DOI] [PubMed] [Google Scholar]
- 45.Satpathy M, Gallagher P, Jin Y, Srinivas SP. Extracellular ATP opposes thrombin-induced myosin light chain phosphorylation and loss of barrier integrity in corneal endothelial cells. Exp Eye Res. 2005;81:183–92. doi: 10.1016/j.exer.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 46.Eltzschig HK, Eckle T, Mager A, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–8. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 47.Yonekura H, Yamamoto Y, Sakurai S, Watanabe T, Yamamoto H. Roles of the receptor for advanced glycation endproducts in diabetes-induced vascular injury. J Pharmacol Sci. 2005;97:305–11. doi: 10.1254/jphs.cpj04005x. [DOI] [PubMed] [Google Scholar]