

Abstract
Lidocaine is a commonly used local anaesthetic agent which has also been found to possess anti-inflammatory activity in several disorders. However, the mechanism of this effect has been little explored. The aim of this study was to investigate the effect of lidocaine on stimulated human T cells. The effect of lidocaine on Jurkat T cells was examined by enzyme-linked immunosorbent assay (ELISA) to determine secretion of interleukin (IL)-2, and by the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] viability assay. Tumour necrosis factor (TNF)-α and IL-2 mRNA expression was determined by reverse transcription–polymerase chain reaction. In addition, the effect of lidocaine on the proliferation of freshly isolated peripheral blood (PB) CD3+ T cells was examined by carboxyfluorescein succinimidyl ester dilution. Apoptosis induction and cytokine production and secretion were determined by annexin V/PI assay, intracellular immunostaining and ELISA respectively. The results showed that lidocaine exerts a dose-dependent inhibition of IL-2 and TNF-α secretion by Jurkat T cells at the protein and mRNA levels. Moreover, lidocaine reduced nuclear factor-κB (NF-κB) signalling in clinically relevant concentrations. Similarly, proliferation of anti-CD3 stimulated PB T cells was abrogated significantly by lidocaine, and the percentage of interferon-γ- and TNF-α-producing T cells was diminished after culture with this agent. In both experimental systems, lidocaine's effect was not mediated by cytotoxic mechanism, as no significant apoptosis or necrosis was demonstrated following co-culture of T cells with this drug. In conclusion, lidocaine's anti-inflammatory effect may be mediated by a drug-induced abrogation of T cell proliferation and cytokine secretion independent of cell death. These effects are mediated at least partly by inhibition of NF-κB signalling.
Keywords: cytokine, inflammation, lidocaine, NF-κB, T cell
Introduction
Lidocaine, a commonly used local anaesthetic agent, has been known to possess an anti-inflammatory effect [1]. Both systemic administration and local application of the drug have been shown to ameliorate murine experimental colitis [2,3]. In addition, distal ulcerative colitis has responded to topical lidocaine in some clinical studies [4,5]. The mechanisms responsible for the anti-inflammatory effect of lidocaine have been little investigated. We have found previously that lidocaine prevents tumour necrosis factor (TNF)-α-induced secretion of the proinflammatory mediators interleukin (IL)-8 and IL-1β from intestinal epithelial cell lines [6]. Moreover, others have demonstrated that lidocaine down-regulates the immune functions of several cell lineages. Thus, lidocaine and lidocaine-related local anaesthetics were shown to inhibit lymphocyte maturation and proliferation and the migration of macrophages into tissues [7], expression of CD11b/CD18 by polymorphonuclear cells [8], adhesion of leucocytes to injured vessels [8–10], the lipopolysaccharide-stimulated secretion of leukotriene B4 and IL-1α from peripheral blood mononuclear cells (PBMC) [11], histamine release from mast cells [12] and the generation of oxygen derivates by neutrophils [13]. However, the specific effects of this drug on the T cell lineage have been little explored [14].
Because T cells play a paramount role in many chronic immune-driven inflammatory disorders, the aim of this study was to evaluate the effects of lidocaine on this subset, and delineate mechanisms that may be instrumental in suppressing T cell-driven inflammation. Our results suggest that lidocaine abrogates T cell proliferation and suppresses expression of the T cell-derived proinflammatory cytokines IL-2, TNF-α and interferon (IFN)-γ via nuclear factor-κB (NF-κB)-mediated inhibition of mRNA expression.
Materials and methods
Human subjects
The study was approved by the Institutional Ethics Committee at the Chaim Sheba Medical Center. Peripheral blood (PB) samples were obtained from healthy donors after their informed consent was obtained.
Reagents
IL-2 was purchased from Hoffman-La Roche Inc. (Nutley, NJ, USA). Lidocaine was purchased from MP Biomedicals, Inc. (Solon, OH, USA), solubilized in phosphate-buffered saline (PBS) from Sigma-Aldrich (St Louis, MO, USA) to the stock concentrations of 0·5 M, and then diluted in RPMI-1640 (Invitrogen/Gibco, Paisley, UK). Concentrations used were within the therapeutic range [4]. Phytohaemagglutinin (PHA) and phorbol myristate acetate (PMA) were also from Sigma-Aldrich.
Cell line and cultures
Fresh PBMC were resuspended in RPMI-1640 medium supplemented with 10% fetal bovine serum (FCS) (Gibco/Invitrogen Life Technologies, CA, USA), 2 mM L-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml) (Gibco/Invitrogen Life Technologies). Cells were placed in 96-well flat-bottomed tissue culture plates (Corning Inc., Corning, NY, USA) at 3 × 105 cells/well containing 200 μl of medium and stimulated immediately with anti-CD3 (OKT3) at 2 μg/ml. Cells were cultured for the designated time in a humidified 37°C, 5% CO2 incubator, and the medium was supplemented on day 3 with 20 U/ml of recombinant IL-2 (Hoffman-La Roche Inc.).
The human T cell line Jurkat was purchased from the American Type Culture Collection TIB-152 (Manassas, VA, USA). The cell line was cultured in RPMI- 1640 supplemented by 10% FBS (Gibco/Invitrogen Life Technologies) and 1% penicillin–streptomycin (Gibco/Invitrogen Life Technologies) at 37°C, 5% CO2 incubation, 100% humidity incubator. Cells were tested routinely for mycoplasma infection by the Mycotrim TC kit (Irvine Scientific, Santa Ana, CA, USA), according to the manufacturer's instructions. On the day of the experiment cells at the concentration of 2 × 106/ml were transferred from tissue culture flasks (Nunc, Brand Products, Roskilde, Denmark) to non-tissue culture 24-well plates with fresh medium (1 ml/well). Cells were triggered by PHA (2·5 μg/ml) and PMA (50 ng/ml) in the absence or presence of the indicated concentrations of lidocaine, or cultured in media alone as a negative control. Cells were incubated for the indicated time-points for the different assays detailed below.
Antibodies and fluorescence activated cell sorter analysis
The various fluorescent-conjugated anti-CD3, anti-CD4, anti-CD8, anti-CD14, anti-IFN-γ, anti-TNF-α and the corresponding fluorochrome-conjugated mouse immunoglobulin G isotype controls were all purchased from PharMingen (San Diego, CA, USA). Stained cells were analysed on a BD FACSCaliburTM (Becton Dickinson, NJ, USA) after gating on the viable lymphocyte population in forward-/side-scatter dot plots.
Enzyme-linked immunosorbent assays of cytokine secretion
Supernatants were collected at the designated time-points by centrifugation of the medium-containing cells for 7 min at 180 g. The concentration of IL-2 in the medium was assessed by enzyme-linked immunosorbent assays (ELISA), as per the manufacturer's instructions (IL-2 Duoset; R&D Systems, MN, USA).
Quantitative real-time polymerase chain reaction analysis
Total RNA was extracted from the Jurkat cells incubated for 2 h after stimulation as above. RNA extraction was performed using the TriReagent kit (Molecular Research Centre, Cincinnati, OH, USA) according to the manufacturer's instructions. cDNA was synthesized from 5 μg RNA at 37°C for 1 h using 200 μM deoxynucleotides (Sigma-Aldrich), 5 μM random hexamers (Amersham Pharmacia Biotech, Piscataway, NJ, USA), 20 U RNAguard (Amersham) and 200 U/μl Moloney murine leukaemia virus (MMLV–RT) Quant (US Biomedical, Cleveland, OH, USA).
Interleukin-2 and TNF-α mRNA expression levels were determined by quantitive real-time polymerase chain reaction (PCR) using the TaqMan assay on-demand kit with the ABI-Prism 7900HT (Applied Biosystems, Foster City, CA, USA). Analysis was carried out in triplicate in 20 μl for 2 min at 50°C, for 10 min at 95°C using a total of 40 cycles, each of 15 s at 95°C, and for 1 min at 60°C. The comparative threshold cycle method was used for TNF-α and IL-2 and the endogenous reference gene 18 S mRNA. In each experiment the non-stimulated RNA sample was used as a calibrate to allow comparison of relative quantity between the samples.
Nuclear localization of NF-κB
Jurkat cells were stimulated as above and incubated for 7 min. The nuclear fraction was then collected from cells using the Active Motif nuclear extract kit (cat. 40010) (Active Motif North America, Carlsbad, CA, USA) according to the manufacturer's instructions. NF-κB levels in the nuclear fraction were measured using the Active Motif NF-κB family kit (cat. 43296) according to the manufacturer's instructions.
Cell viability assay
The viability of the Jurkat cells was assessed by a [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay kit (Hoffman-La Roche), after stimulation and incubation of Jurkat cells for 24 h as above. MTT was added at the end of the 24-h incubation period and compared with the experimental control (cells without lidocaine).
Analysis of T cell proliferation
The flow cytometric-based analysis of PBMC proliferation by serial halving of the fluorescence intensity of the vital dye, carboxyfluorescein succinimidyl ester (CFSE), was employed. Briefly, PBMC at 107/ml were suspended in 1 ml RPMI-1640 and then pulsed with 4 μM CFSE (Molecular Probes Inc., Eugene, OR, USA) for 15 min at 37°C with constant shaking. Subsequently, the cells were washed twice with large volumes of RPMI-1640 + 10% FCS medium for quenching the CFSE. The cells were then triggered with anti-CD3 (OKT3, 2 μg/ml) in the absence or presence of lidocaine at the designated concentrations, or incubated in medium alone as a negative control. Cells were harvested on day 5 of culture and analysed for CFSE content (generation number) by flow cytometry (FACSCaliber, BD, San Jose, CA, USA).
Analysis of cell apoptosis
Apoptotic cells were detected by staining with annexin V and propidium iodide using the annexin V FITC apoptosis detection kit I (PharMingen) after 5 days of incubation in the absence or presence of lidocaine at the designated concentrations. PBMC were washed twice in PBS, and the pellet was resuspended in annexin V binding buffer (PharMingen) at a concentration of 106 cells/ml. Annexin V FITC and propidium iodide were added (5 μl of each per 105 cells). Samples were mixed gently and incubated for 15 min at room temperature in the dark before fluorescence activated cell sorter (FACS) analysis.
Intracellular cytokine staining
Peripheral blood mononuclear cells were activated with 20 ng/ml of phorbol 12,13-dibutyrate and 0·8 μM of ionomycin (Sigma-Aldrich) in the presence of 2 μg/ml of monensin (GolgiStop; PharMingen) for 5 h at 37°C. Following activation, cells were harvested, fixed and permeabilized using the Cytofix/Cytoperm Plus kit and then immunostained by anti-CD3 and for intracellular IFN-γ or TNF-α production, all in accordance with the manufacturer's recommendations (PharMingen). All samples were then analysed on a FACScan cytofluorograph.
Statistical analysis
All values are presented as means ± standard deviation. Repeated-measures analysis of variance was used for statistical analysis of the data. The Tukey–Kramer multiple comparison test or Wilcoxon's rank test was used to evaluate significance between experimental groups, as appropriate. P < 0·05 was considered significant.
Results
Lidocaine inhibits IL-2 production by the Jurkat T cell line
To evaluate the effects of lidocaine on the secretion of IL-2 from Jurkat cells, cells were stimulated by the combination of the potent activators PMA + PHA and then cultured in the absence or presence of graded concentrations of lidocaine. The supernatant was collected at 24 h and analysed for the content of IL-2. We found that the addition of lidocaine inhibited significantly the amount of IL-2 secreted to the supernatant in a dose-dependent manner (Fig. 1).
Fig. 1.
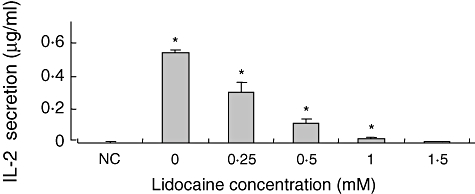
The effect of lidocaine on phorbol myristate acetate + phytohaemagglutitin (PMA + PHA)-stimulated interleukin (IL)-2 secretion from the Jurkat cell line. Jurkat cells were stimulated by PMA + PHA, and then cultured in the absence or presence of graded concentrations of lidocaine. The IL-2 level in the supernatant was determined at 24 h by enzyme-linked immunosorbent assay. The values represent the mean and standard deviation obtained in three different experiments, each performed in duplicate. *P < 0·05. NC: negative control (non-stimulated cells).
Lidocaine does not affect Jurkat T cell viability
The above results, indicating an inhibiting effect of lidocaine on IL-2 secretion, could result potentially from reducedcellular function, or alternatively from drug-mediated cytotoxicity. In order to gain further insight into the responsible mechanism for the observed effect, we next aimed to determine the effect of lidocaine on Jurkat cell viability. Thus, cells were stimulated as above, and cellular death was determined by an MTT assay after 24 h of culture with the designated concentrations of lidocaine. We find that increasing concentrations of lidocaine had only a slight and non-significant effect on T cell viability (Fig. 2). This suggests that the mechanism for reduced IL-2 production under lidocaine treatment is mediated through non-cytotoxic effect.
Fig. 2.
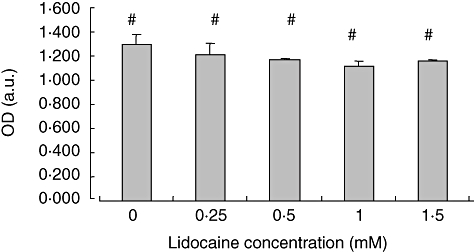
Lidocaine does not affect cell viability of the Jurkat cell line. Jurkat cells were incubated for 24 h with graded concentrations of lidocaine or in media alone. Cell viability was assessed at the end of culture by an [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay kit. The values represent the mean and standard deviation obtained in three different experiments. OD (a.u.): Optical density (arbitrary units). #P = not significant.
Lidocaine reduces IL-2 and TNF-α mRNA expression in Jurkat T cells
Because the above results indicated that down-regulation of cytokine production was not related to induction of cell death, we next aimed to test the effect of lidocaine on cytokine transcription. Thus, Jurkat cells were stimulated and cultured as above, RNA was extracted (see the Methods section) and TNF-α and IL-2 mRNA levels were determined by quantitative reverse transcription–PCR. We find that under these experimental conditions lidocaine suppresses significantly the expression of TNF-α and IL-2 mRNA in a dose-dependent manner (Fig. 3a and b). These data indicate that lidocaine abrogates cytokine secretion at the transcriptional level, via inhibition of mRNA expression.
Fig. 3.
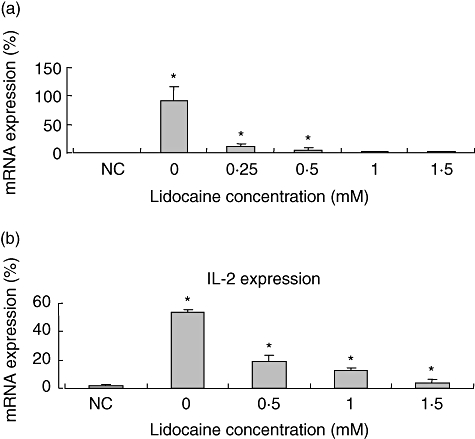
The effect of lidocaine on phorbol myristate acetate + phytohaemagglutitin (PMA + PHA)-stimulated tumour necrosis factor (TNF)-α and interleukin (IL)-2 transcription in Jurkat cells. Lidocaine (0·25 mM−1·5 mM) was added for 2 h to Jurkat cells that were prestimulated with PMA + PHA. Thereafter, RNA was extracted and assayed for TNF-α (a) and IL-2 (b) mRNA expression by reverse transcription–polymerase chain reaction. The values represent the mean and standard deviation obtained in three different experiments, each performed in duplicate. *P < 0·05. NC: negative control (non-stimulated cells).
Nuclear fraction NF-κB binding is reduced by lidocaine treatment
Because lidocaine was shown to inhibit mRNA expression of the proinflammatory mediators IL-2 and TNF-α, we next evaluated whether the upstream signal transduction pathway of NF-κB was also affected. Thus, Jurkat cells were stimulated and cultured as above, and NF-κB translocation was measured as detailed in the Methods section. We find that lidocaine lowers NF-κB translocation of Jurkat cells significantly cultured with the drug compared with cells cultured in medium alone. Moreover, this inhibitory effect was dose-dependent at all doses tested, except for the difference between 0·5 mM and 1 mM of lidocaine that did not reach statistical significance (Fig. 4). Collectively, these data suggest that lidocaine suppresses proinflammatory cytokine production by Jurkat T cells at the transcriptional level through a mechanism that involves, at least partly, the NF-κB-signal transduction pathway.
Fig. 4.
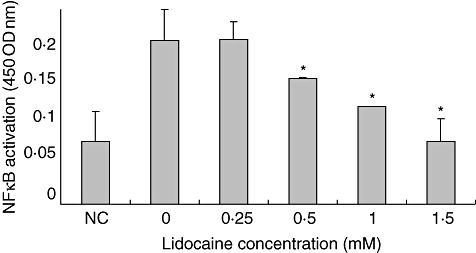
Lidocaine reduces nuclear factor (NF-κB) nuclear localization in a dose-dependent manner. Jurkat cells were stimulated and incubated as above in the presence or absence of lidocaine at the designated concentration. The nuclear fraction was collected and NF-κB levels in the nuclear fraction were measured using the Active Motif NF-κB family kit (see the Methods section for details). The values represent the mean and standard deviation obtained in three different experiments, each performed in duplicate. *P < 0·05. NC: negative control (non-stimulated cells).
Lidocaine inhibits IL-2 production by activated PB T cells
Because T cells are implicated in a multitude of human chronic inflammatory disorders the above results, observed in a transformed T cell line, motivated us to examine the effects of lidocaine on freshly isolated and activated PB T cells. To evaluate the effects of lidocaine on the secretion of IL-2 from T cells, cells were stimulated by PMA + PHA, and then cultured in the absence or the presence of graded concentrations of lidocaine. The supernatant was collected at 24 h, and analysed for the content of IL-2 by ELISA. We found that the addition of lidocaine inhibited significantly the amount of IL-2 secreted to the supernatant in a dose-dependent manner. Results obtained from six different donors are shown in Fig. 5.
Fig. 5.

The effect of lidocaine on phorbol myristate acetate + phytohaemagglutitin (PMA + PHA)-stimulated interleukin (IL)-2 secretion from peripheral blood (PB) T cells. PB T cells obtained from healthy donors were stimulated by PMA + PHA, and then cultured in the absence or presence of graded concentrations of lidocaine. IL-2 level in the supernatant was determined at 24 h by enzyme-linked immunosorbent assay. Results obtained from six different donors are shown. IL-2 secretion was reduced significantly between all different lidocaine concentrations at P = 0·03 for trend, using the Wilcoxon rank test. NC: negative control (non-stimulated cells).
Lidocaine reduces production of TNF-α and IFN-γ by activated PB T cells
In order to assess further the effect of lidocaine on T cells at the single cell level, and as TNF-α and IFN-γ are known to play a major role in driving many immune disorders, we aimed to examine the effect of lidocaine on the production of these proinflammatory cytokines. Hence, PBMC were isolated from healthy donors, stimulated by OKT3 and cultured for 3 days in the absence or the presence of the indicated concentrations of lidocaine. Cells were harvested at day 3, activated briefly by PMA and ionomycin in the presence of Golgi-apparatus inhibitor, and gated permealized CD3+ cells were analysed for TNF-α and IFN-γ production by FACS analysis, following intracellular immunostaining by the appropriate monoclonal antibodies. As shown in a representative experiment depicted in Fig. 6, lidocaine reduces the number of T cells producing both mediators in a dose-dependent manner.
Fig. 6.
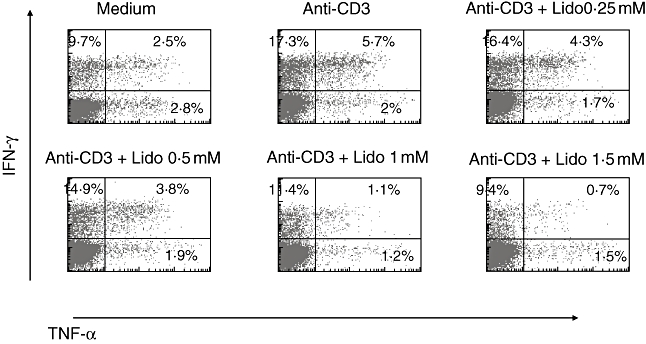
Lidocaine reduces the expansion of tumour necrosis factor (TNF)-α- and interferon (IFN)-γ-producing CD3+ T cells. Peripheral blood lymphocytes obtained from healthy donors were stimulated by anti-CD3 in the presence or absence of the indicated drug. Intracellular cytokine production by gated CD3+ T cells was determined after immunostaining by fluorescence activated cell sorter (FACScan) cytofluorograph analysis. The percentage of positive cells is indicated in the corresponding quadrants of the dot plots. Results are representative of three separate experiments.
T cell proliferation is inhibited by lidocaine
In order to assess further the inhibitory effect of lidocaine on activated PB T cells, we next addressed its effect on cellular proliferation following T cell receptor (TCR) ligation. Thus, after loading with CFSE freshly isolated PBMC were stimulated and cultured as above. The divisions of the gated CD3+ population were determined on day 5 by CFSE content. We find that addition of lidocaine reduces significantly the proliferation of T cells following TCR stimulation (Fig. 7). These results indicate that lidocaine may suppress the inflammatory reaction not only by suppressing cytokine production, but also by inhibiting T cell proliferation.
Fig. 7.
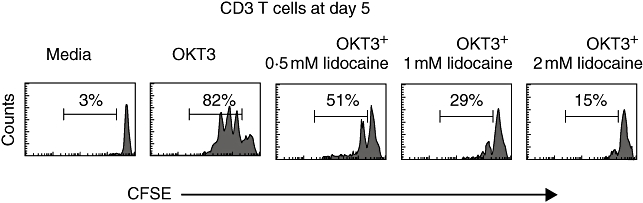
Lidocaine inhibits T cell proliferation. Peripheral blood mononuclear cells from healthy donors were pulsed with carboxyfluorescein succinimidyl ester (CFSE). The cells were then triggered with anti-CD3 in the absence or presence of lidocaine at the designated concentrations, or incubated in medium alone as a negative control. Cells were harvested on day 5 of culture and analysed for CFSE content. The percentage of dividing cells is indicated. Results are representative of three different experiments.
Lidocaine induces only marginal T cell apoptosis
In order to elucidate further lidocaine's anti-inflammatory effect, its pro-apoptotic potential was examined on human PB T cells. Thus, freshly isolated PBMC were stimulated and cultured as above, and cell death of CD3+ gated cells was determined by annexin V/PI content at the time-points indicated. We find that lidocaine exerted only a marginal pro-apoptotic effect on activated T cells at 72 h post-stimulation (right lower quadrants), and that no clear dose–response pattern was discernable (Fig. 8). Similar results were obtained when cells were examined after 24 h and 5 days of culture (data not shown). These results suggest that the lidocaine anti-inflammatory effect is mediated only minimally, if at all, by induction of cellular death.
Fig. 8.
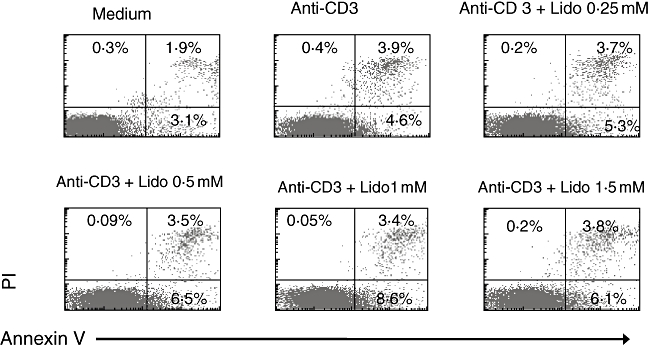
Lidocaine has only a modest pro-apoptotic effect on stimulated peripheral blood (PB) CD3+ T cells. PB cells from healthy donors were stimulated as above, and harvested at day 3. Apoptosis was assessed by annexin V/PI staining on a flow cytometer. The percentage of positive cells is indicated in the corresponding quadrants of the dot plots. Results are representative of three experiments.
Discussion
The results of this study indicate that lidocaine inhibits the production of three major T cell inflammatory cytokines: IL-2, TNF-α and IFN-γ. This down-regulation was shown to involve inhibition at the mRNA expression level and reduction of NF-κB translocation to the nucleus. Furthermore, we demonstrate that the effect of lidocaine was not associated with induction of significant cell death, but arrested cellular proliferation.
The current findings are of importance for understanding the anti-inflammatory effects of lidocaine, observed in both animal models and in human disease. The inhibition of TNF-α synthesis is of particular interest in light of its major role in irritable bowel disease (IBD) pathogenesis, which is supported both by studies demonstrating increased TNF-α levels in intestinal tissue and PB of IBD patients [15,16] and by the therapeutic effects of anti-TNF-α agents in Crohn's disease and ulcerative colitis [17,18]. The fact that lidocaine exerted its therapeutic effects after local application is of particular interest, because of the possibility to block TNF-α locally and thus to avoid the deleterious effects of systemic continuous therapy with anti-TNF-α agents. Design of appropriate formulations may allow exploration of this therapeutic approach more efficiently.
That lidocaine inhibited the expression of a number of proinflammatory mediators suggests regulation of a common proinflammatory signalling pathway. Previous studies have demonstrated that activation of NF-κB was necessary for the expression of IL-2 [19], IFN-γ[20] and TNF-α[21]. Our observations indicate that lidocaine inhibited signalling through the NF-κB signal transduction pathway in a dose-dependent manner, and its application to the cells led to lower cellular mRNA levels of relevant inflammatory mediators. However, the upstream events leading to this inhibition were not identified in this study. Previous studies have demonstrated that amide local anaesthetics such as lidocaine inhibit K+ channels [22]. In turn, blocking K+ channels was shown to inhibit NF-κB signalling [23]. Although the specific K+ channels blocked by lidocaine and their possible effect on cellular signalling remains to be explored, these observations may provide a plausible explanation for lidocaine's effect. None the less, other signalling pathways were not assessed and the effects of lidocaine could also be mediated through additional cellular mechanisms. Of relevance in this respect, TNF-α expression was reduced significantly at minimal lidocaine concentrations (0·25 mM) that were below the threshold for lidocaine-mediated NF-κB inhibition, which became apparent at slightly higher concentrations (Figs 3 and 4). These results may indicate that other signal transduction pathways may play a role in conferring the anti-inflammatory effects of lidocaine at the cellular level.
Our results have also demonstrated that lidocaine inhibited cellular proliferation, yet induced only mild apoptosis. This type of effect may explain the modest therapeutic outcome observed with lidocaine therapy only. None the less, arrest of cellular proliferation was also demonstrated with potent therapeutic agents such as azathioprine and methotrexate [24,25]. This may give rise to a notion whereby anti-inflammatory therapy could be initiated with apoptosis-inducing agents, and lidocaine may be employed in adjunct subsequently to maintain remission. This is reminiscent of the commonly accepted treatment regimens with immunosuppressive agents, albeit without the risk of bone marrow suppression. The fact that local anaesthetics undergo extensive hepatic metabolism contribute further to the attractiveness of this therapeutic approach. Our results, which demonstrate that lidocaine does not affect T cell viability, differ from those obtained by Boselli et al. [26]. However, in our study we used lidocaine concentrations ranging from 10- to 1·5-fold lower than that used by Boselli et al. This range of concentrations corresponds to previously reported therapeutic serum concentrations in patients [4]. Furthermore, while we used an MTT assay in order to assess Jurkat cell viability, the methods used by Boselli et al. were annexin V and propidium iodide staining. Thus, it is possible that higher lidocaine concentrations and a different cell viability assessment method might have caused that dissimilarity. The fact that our results were verified in PB T cells using the annexin V method (Fig. 7) supports our data further.
Our study had a number of limitations. Admittedly, the relevance of our study to IBD was limited by the fact that we used PB-derived cells rather than intestinal-derived cells. Future studies with a similar design, employing intestinally derived T cells, will be necessary to corroborate the validity of our findings for these cell populations as well. Furthermore, our study was conducted using both Jurkat cells and PB T cells. The purpose of the studies in Jurkat cells was to study the effect of lidocaine in a homogeneous population of cells, which would also allow for the study of the NF-κB signal transduction pathway in a convenient experimental system. However, because of the transformed nature of the Jurkat cells, we confirmed the functional implications of our findings in cells obtained from normal PB.
In conclusion, our findings shed light on the anti-inflammatory effects of local anaesthetics and suggest novel mechanisms mediating these actions. Greater understanding of local anaesthetic anti-inflammatory mechanisms may prove beneficial for designing drug combinations with conventional therapeutic agents rationally which may, in turn, pave the way for further improvement in the therapy of inflammatory disorders.
References
- 1.Hollmann MW, Durieux ME. Local anesthetics and the inflammatory response: a new therapeutic indication? Anesthesiology. 2000;93:858–75. doi: 10.1097/00000542-200009000-00038. [DOI] [PubMed] [Google Scholar]
- 2.Bjorck S, Jennische E, Dahlstrom A, et al. Influence of topical rectal application of drugs on dextran sulfate-induced cilitis in rats. Dig Dis Sci. 1997;42:824–32. doi: 10.1023/a:1018880501437. [DOI] [PubMed] [Google Scholar]
- 3.McCafferty DM, Sharkey KA, Wallace JL. Beneficial effects of local or systemic lodocaine in experimental colitis. Am J Physiol. 1994;266:G560–7. doi: 10.1152/ajpgi.1994.266.4.G560. [DOI] [PubMed] [Google Scholar]
- 4.Bjocrk S, Dahlstrom A, Ahlman H. Treatment of distal colitis with local anaesthetic agents. Pharmacol Toxicol. 2002;90:173–80. doi: 10.1034/j.1600-0773.2002.900401.x. [DOI] [PubMed] [Google Scholar]
- 5.Bjocrk S, Dahlstrom A, Johansson L, et al. Treatment of the mucosa with local anaesthetics in ulcerative colitis. Agents Actions. 1992;Spec No:C60–72. [PubMed] [Google Scholar]
- 6.lahav M, Levite M, Bassani L, et al. Lidocaine inhibits secretion of IL-8 and IL-1beta and stimulates secretion of IL-1 receptor antagonist by epithelial cells. Clin Exp Immunol. 2002;127:226–33. doi: 10.1046/j.1365-2249.2002.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickstein R, Kiremidjian-Schumacher L, Stotzky G. Effects of lidocaine on the function of immunocompetent cells. I. In vitro exposure of mouse spleen lymphocytes and peritoneal macrophages. Immunopharmacology. 1985;9:117–25. doi: 10.1016/0162-3109(85)90007-4. [DOI] [PubMed] [Google Scholar]
- 8.Martinsson T, Oda T, Fernvik E, et al. Ropivacaine inhibits leukocyte rolling, adhesion and CD11b/CD18 expression. J Pharmacol Exp Ther. 1997;283:59–65. [PubMed] [Google Scholar]
- 9.Giddon DB, Lindhe J. In vivo quantitation of local anesthetic suppression of leukocyte adherence. Am J Pathol. 1972;68:327–38. [PMC free article] [PubMed] [Google Scholar]
- 10.MacGregor RR, Thorner RE, Wright DM. Lidocaine inhibits granulocyte adherence and prevents granulocyte delivery to inflammatory sites. Blood. 1980;56:203–9. [PubMed] [Google Scholar]
- 11.Sinclair R, Eriksson AS, Gretzer C, et al. Inhibitory effects of amide local anaesthetics on stimulus-induced human leukocyte metabolic activation, LTB4 release and IL-1 secretion in vitro. Acta Anaesthesiol Scand. 1993;37:159–65. doi: 10.1111/j.1399-6576.1993.tb03693.x. [DOI] [PubMed] [Google Scholar]
- 12.Yanagi H, Sankawa H, Saito H, et al. Effect of lidocaine on histamine release and Ca2+ mobilization from mast cells and basophils. Acta Anaesthesiol Scand. 1996;40:1138–44. doi: 10.1111/j.1399-6576.1996.tb05577.x. [DOI] [PubMed] [Google Scholar]
- 13.Ohsaka A, Saionji K, Sato N, et al. Local anesthetic lidocaine inhibits the effect of granulocyte colony-stimulating factor on human neutrophil functions. Exp Hematol. 1994;22:460–6. [PubMed] [Google Scholar]
- 14.Tanaka A, Minoguchi K, Oda N, et al. Inhibitory effect of lidocaine on T cells from patients with allergic asthma. J Allergy Clin Immunol. 2002;109:485–90. doi: 10.1067/mai.2002.122155. [DOI] [PubMed] [Google Scholar]
- 15.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–81. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elsässer-Beile U, von Kleist S, Gerlach S, et al. Cytokine production in whole blood cell cultures of patients with Crohn's disease and ulcerative colitis. J Clin Lab Anal. 1994;8:447–51. doi: 10.1002/jcla.1860080618. [DOI] [PubMed] [Google Scholar]
- 17.Baert FJ, D'Haens GR, Peeters M, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn's ileocolitis. Gastroenterology. 1999;116:22–8. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 18.Gisbert JP, González-Lama Y, Maté J. Systematic review: infliximab therapy in ulcerative colitis. Aliment Pharmacol Ther. 2007;25:19–37. doi: 10.1111/j.1365-2036.2006.03131.x. [DOI] [PubMed] [Google Scholar]
- 19.Lindstein T, June CH, Ledbetter JA, et al. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–43. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 20.Sica A, Dorman L, Viggiano V, et al. Interaction of NF-κB and NFAT with the interferon-promoter. J Biol Chem. 1997;272:30412–20. doi: 10.1074/jbc.272.48.30412. [DOI] [PubMed] [Google Scholar]
- 21.Elewaut DJA, Didonato JM, Kim F, et al. NF-κB is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol. 1999;158:3285–92. [PubMed] [Google Scholar]
- 22.Kindler CH, Paul M, Zou H, et al. Amide local anesthetics potently inhibit the human tandem pore domain background K+ channel TASK-2 (KCNK5) J Pharmacol Exp Ther. 2003;306:84–92. doi: 10.1124/jpet.103.049809. [DOI] [PubMed] [Google Scholar]
- 23.Papavlassopoulos M, Stamme C, Thon L, et al. axiK blockade selectively inhibits the lipopolysaccharide-induced I kappa B-alpha/NF-kappa B signaling pathway in macrophages. J Immunol. 2006;177:4086–93. doi: 10.4049/jimmunol.177.6.4086. [DOI] [PubMed] [Google Scholar]
- 24.Schroll S, Sarlette A, Ahrens K, et al. Effects of azathioprine and its metabolites on repair mechanisms of the intestinal epithelium in vitro. Regul Pept. 2005;131:1–11. doi: 10.1016/j.regpep.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Stromhaug A, Slordal L, Warren DJ. Differential effects of the antifolates methotrexate, aminopterin and trimetrexate on murine haemopoietic progenitor cells. Br J Haematol. 1996;92:514–20. doi: 10.1046/j.1365-2141.1996.d01-50.x. [DOI] [PubMed] [Google Scholar]
- 26.Boselli E, Duflo F, Debon R, et al. The induction of apoptosis by local anesthetics: a comparison between lidocaine and ropivacaine. Anesth Analg. 2003;96:755–6. doi: 10.1213/01.ANE.0000047201.85815.9D. [DOI] [PubMed] [Google Scholar]