

Abstract
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine that has been demonstrated to regulate the apoptosis of several cell types. Dysregulated apoptosis of fibroblasts has been implicated in a variety of fibrotic diseases, including systemic sclerosis (SSc). In this study, we investigated the role of MIF in the apoptosis of dermal fibroblasts. The concentrations of MIF were measured in sera and in culture supernatants of peripheral blood mononuclear cells (PBMCs) and dermal fibroblasts by enzyme-linked immunosorbent assay. The degree of apoptosis was determined by colorimetric assay, and signalling pathways were examined by Western blot. The results showed that serum levels of MIF were significantly higher in patients with SSc (n = 47) than in healthy controls (n = 56). Stimulation of PBMCs by anti-CD3 and anti-CD28 increased the production of MIF by fourfold over the constitutive levels. SSc dermal fibroblasts produced higher amounts of MIF than normal dermal fibroblasts. When treated with sodium nitroprusside (SNP), SSc dermal fibroblasts showed a lower degree of apoptosis compared with normal dermal fibroblasts. Exogenous MIF (1–100 ng/ml) inhibited SNP-induced apoptosis of dermal fibroblasts dose-dependently. Both extracellular regulated kinase (ERK) inhibitor (PD98059) and protein kinase B (Akt) inhibitor (LY294002) almost completely blocked the inhibitory effect of MIF on apoptosis. Furthermore, MIF increased the expression of Bcl-2, phospho-ERK and phospho-Akt activity in dermal fibroblasts. Taken together, our data suggest that MIF released by activated T cells and dermal fibroblasts decreases the apoptosis of dermal fibroblasts through activation of ERK, Akt and Bcl-2 signalling pathways, which might be associated with excessive fibrosis in SSc.
Keywords: apoptosis, dermal fibroblasts, macrophage migration inhibitory factor, systemic sclerosis, T cells
Introduction
Systemic sclerosis (SSc) is a connective tissue disease characterized by progressive fibrosis of the skin and internal organs. The pathogenesis of SSc includes microvascular injury, inflammatory cell infiltration and the excessive proliferation of fibroblasts. Abnormal apoptosis of fibroblasts is considered to contribute to progressive fibrosis by increased accumulation of fibroblasts that produce am excessive amount of extracellular matrix [1–5]. It has been appreciated that SSc fibroblasts exhibited altered phenotype with increased production of extracellular matrix, and were shown to be resistant to apoptosis [3–5]. In particular, chronic exposure of fibroblasts to several cytokines and growth factors has been shown to affect fibroblast apoptosis in SSc [5–7].
Macrophage migration inhibitory factor (MIF) was identified originally as a soluble factor in culture medium of activated T lymphocytes that inhibited random migration of macrophage [8,9]. MIF is produced by several types of cells, such as T lymphocytes [8,9], neutrophils [10,11], macrophages [12], endothelial cells [13] and fibroblasts [14]. MIF has been implicated in various immune-mediated diseases, including rheumatoid arthritis [15–17], inflammatory bowel disease [18], multiple sclerosis [19], systemic lupus erythematosus [20], psoriasis [21] and scleroderma [22,23]. Previous studies have demonstrated that MIF enhanced the production of proinflammatory cytokines, such as tumour necrosis factor (TNF), interferon-γ, interleukin (IL)-1, IL-2, IL-6, IL-8, nitric oxide and prostaglandin E2[24]. In addition, MIF was known to play an important role in regulating cellular proliferation [25,26]. MIF also abrogates p53-dependent apoptosis of macrophages and promotes Ras-mediated transformation of fibroblasts [26–29].
Selvi et al. demonstrated that MIF was expressed in the dermal tissue of SSc, and that serum MIF levels were increased in patients with SSc compared with healthy controls [22]. However, the role of MIF in the pathogenesis of SSc remains poorly understood. In this study, we demonstrated that serum MIF levels were significantly higher in patients with SSc than healthy controls. When treated with sodium nitroprusside (SNP), SSc fibroblasts showed a lower degree of apoptosis compared with normal fibroblasts. Treatment of dermal fibroblast with MIF inhibited SNP-induced apoptosis dose-dependently of dermal fibroblasts. Such an inhibitory effect of MIF was abrogated completely by adding extracellular regulated kinase (ERK) or protein kinase B (Akt) inhibitors. In parallel, the expression of phospho-Akt (pAkt), phospho-Erk (pERK) and Bcl-2 in dermal fibroblasts were increased by the treatment with MIF. Collectively, our data suggest that up-regulated MIF in SSc may contribute to excessive skin fibrosis by inhibiting apoptosis of dermal fibroblasts.
Materials and methods
Patients
Forty-seven patients (two male and 45 female), all of whom fulfilled the criteria of the American Rheumatism Association for SSc [30], and 56 healthy controls matched for age and sex were included in the study. All the subjects gave the informed consents before the study. All samples were obtained according to the guidelines approved by the Ethics Committee of the Catholic University of Korea.
Clinical and laboratory profiles
Demographic characteristics (age, sex, disease duration) and various clinical parameters were recorded at the time of sampling. The clinical variables consisted of age, sex, disease duration, type of SSc (diffuse or limited), modified Rodnan score, presence or absence of oesophageal involvement on endoscopy and oesophageal manometry, interstitial lung disease on chest radiography and/or high-resolution computerized tomography (HRCT), diffusion capacity (DLCO; diffusion of carbon monoxide) on the pulmonary function test, Raynaud's phenomenon, arthritis, sicca syndrome, digital ulcer, pulmonary artery hypertension determined by transthoracic echocardiography and antibodies to Scl-70 or centromere using enzyme-linked immunosorbent assay (ELISA) kits (MBL, Nagoya, Japan). Interstitial lung disease was defined as bibasilar fibrosis on chest radiography and/or presence of alveolitis on HRCT. The clinical and laboratory characteristics of SSc patients are summarized in Table 1.
Table 1.
Clinical and laboratory profiles of 47 patients with systemic sclerosis.*
| Age (years) | 44·0 ± 1·7 |
| Sex (M/F) | 2/45 |
| Disease duration (months) | 62·6 ± 6·1 |
| Type of SSc (diffuse/limited) | 20/27 |
| Raynaud's phenomenon | 45/47 (95·7%) |
| Modified Rodnan score | 11·5 ± 1·5 |
| Oesophageal involvement | 23/47 (48·9%) |
| Interstitial lung disease | 27/47 (57·4%) |
| Pulmonary artery hypertension | 3/47 (6·4%) |
| Arthritis | 11/47 (23·4%) |
| Digital ulcer | 20/47 (42·6%) |
| Antibodies to Scl70 | 26/47 (55·3%) |
| Antibodies to centromere | 4/47 (8·5%) |
Data are expressed as percentage (parenthesis) or mean ± standard error of the mean.
Measurement of MIF and transforming growth factor-β
Concentrations of MIF were determined in the sera and culture supernatants using a commercial ELISA kit (R&D Systems, Minneapolis, MN, USA), as described below: 0·4 μg/ml of antibodies to human MIF was added to a 96-well plate (Nunc Inc., Roskilde, Denmark) and incubated overnight at 4°C. After blocking with phosphate buffered saline (PBS) containing 1% bovine serum albumin and 0·05% Tween 20 for 2 h at room temperature, the samples (100 μl) were added to a 96-well plate and incubated at room temperature for 2 h. After washing three times with PBS containing Tween 20, biotinylated polycolonal antibodies to MIF (200 ng/ml) were added and reactions were allowed to proceed for 2 h at room temperature. After washing, 2000-fold diluted streptavidin–horseradish–peroxidase (R&D Systems) was added, and the reaction was again allowed to proceed for 20 min at room temperature. After washing, 100 μl of tetramethylbenzidine substrate solution (R&D Systems) was added to induce a colour reaction, and 50 μl of 2 N H2SO4 (R&D Systems) was used to stop the reaction. An automated microplate reader (Vmax; Molecular Devices, Palto Alto, CA, USA) was used to measure the optical density (OD) at 450 nm. Recombinant MIF diluted in culture medium were used as a calibration standard, ranging from 10 to 2000 pg/ml. A standard curve was drawn by plotting the OD versus the log of the recombinant MIF concentration. The concentrations of transforming growth factor (TGF)-β were also determined in the sera by commercial ELISA kit (R&D Systems).
Isolation and culture of peripheral blood mononuclear cells and dermal fibroblasts
Blood samples obtained from three healthy controls were collected into heparinized tubes, and peripheral blood mononuclear cells (PBMCs) were prepared by Ficoll-Hypaque (Amersham Bioscience, Uppsala, Sweden) density gradient centrifugation. Cells (5 × 105) were dispensed into 24 multi-well plates (Nunc) in 200 μl of RPMI-1640 medium (Life Technologies, Rockville, MD, USA) containing 0·5% fetal calf serum (FCS; Gibco brl, Grand Island, NY, USA), and the culture plates were incubated for 24 h in the presence of anti-CD3 antibody (Pharmingen, San Diego, CA, USA; 1 and 5 μg/ml) and anti-CD28 antibody (Pharmingen; 1 and 5 μg/ml) or concanavalin A (ConA; 0·5, 5, and 50 μg/ml).
Dermal fibroblasts were obtained from the affected skin of three SSc patients with diffuse type at the time of initial diagnosis, and also from three healthy controls during plastic surgery. Fibroblasts were grown from explants in Dulbecco's modified Eagle's medium (DMEM) at 37°C in 5% CO2. The cells were then centrifuged at 500 g, resuspended in DMEM supplemented with 10% FCS, 2 mM glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml), and plated in 25-cm2 flasks. The culture medium was replaced by every 3 days. When cells approached confluence, they were detached with trypsin, passed after dilution 1:3 with fresh medium, and recultured until use. Cells were housed in a 37°C humidifier incubator with 5% CO2. Fibroblasts (passages 4 or 5) were seeded in 24-well plates at 3 × 104 cells per well in DMEM supplemented with 10% FCS. After 48 h of incubation, cell-free media were collected and stored at −20°C until assay. All cultures were set up in duplicate.
Apoptosis assay
Dermal fibroblasts were seeded in 24-well plates at 3 × 104 cells per well in DMEM supplemented with 0·5% FCS. Apoptosis was induced by the administration of SNP (1 mM) for 24 h. APOPercentage Apoptosis Assay (Biocolor Ltd, Newtownabbey, UK) was used to quantify apoptosis, according to the manufacturer's instructions. This assay uses a dye that stains cells as they undergo the membrane ‘flip-flop’ event when phosphatidylserine is translocated to the outer leaflet. This event is considered to be diagnostic of apoptosis but not necrosis. Digital images of APOPercentage dye-labelled cells, which appears bright pink against the white background, were used for highly sensitive apoptotic cell quantification. Using Adobe Photoshop, the level of apoptosis was measured and expressed as a pixel number. In some experiments, the degree of apoptosis was also determined by ELISA for the level of cellular DNA fragmentation (Roche Applied Science, Indianapolis, IN, USA).
Western blot analysis of Bcl-2, pAkt and pERK expression
Dermal fibroblasts (3 × 105 cells) were cultured in DMEM supplemented with 0·5% FCS and treated with 100 ng/ml of MIF for various times. The treated cells were washed twice in PBS, dissolved in sample buffer and boiled. Next, equal amounts of cellular proteins were fractionated on 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane. After immunoblot analysis with anti-pERK1/2, anti-pAkt (Cell Signalling Laboratories, Beverly, MA, USA) and anti-Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), the membranes were reprobed for total ERK1/2, total Akt and β-actin (Santa Cruz Biotechnology) for standardization.
Statistical analysis
Data are expressed as the mean ± standard error of the mean. Comparisons of the numerical data between groups were performed by Student's t-test or Mann–Whitney U-test. Correlations between the two variables were performed using the Pearson's rank correlation coefficient. P-values less than 0·05 were considered statistically significant.
Results
Elevated serum levels of MIF in patients with SSc
Demographic characteristics of the 47 patients with SSc are shown in Table 1. In addition, current medications were as follows, in order: bucillamine (26 of 47), colchicine (17 of 47), hydroxychloroquine (13 of 47), azathioprine (12 of 47), D-penicillamine (two of 47), methotrexate (one of 47) and cyclosporin (one of 47). As shown in Fig. 1a, serum levels of MIF were significantly higher in 47 patients with SSc than in 56 healthy controls (343 ± 55 pg/ml versus 185 ± 24 pg/ml, P < 0·01). However, serum MIF levels were not different between diffuse type and limited type of SSc subsets (data not shown). Moreover, there were no correlations between the serum levels of MIF and clinical variables, such as age, disease duration, modified Rodnan score, steroid dosage at the time of sampling and the presence of internal organ involvement (data not shown). Interestingly, serum levels of MIF were correlated positively with those of TGF-β in patients with SSc (Fig. 1b), but not in healthy controls (data not shown).
Fig. 1.
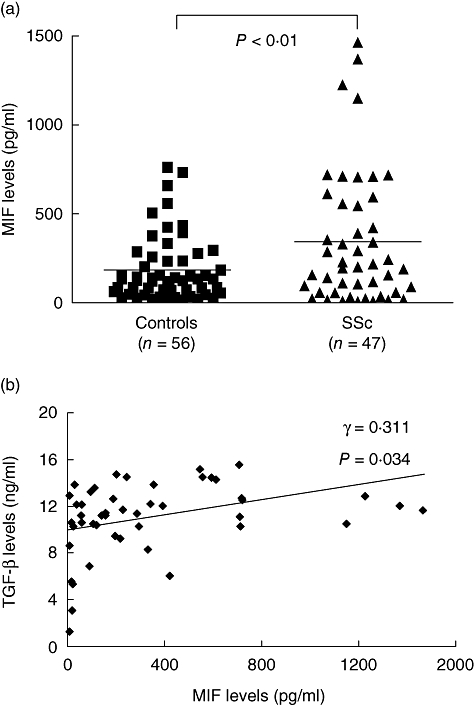
Serum concentrations of macrophage migration inhibitory factor (MIF) in patients with systemic sclerosis and healthy controls. (a) Concentrations of MIF in sera of patients with systemic sclerosis (SSc, n = 47) and healthy controls (n = 56). Data are expressed as mean ± standard error of the mean. (b) Correlation between concentrations of MIF and transforming growth factor-β in sera of patients with SSc, which were determined by enzyme-linked immunosorbent assay. TGF, transforming growth factor.
Production of MIF by peripheral blood T cells and dermal fibroblasts
Activated T lymphocytes, which are detected frequently at an early stage of SSc [31], are one of the major sources for MIF production. To determine the potential sources of circulating MIF, we examined the production of MIF in freshly isolated PBMCs from three healthy subjects by ELISA. As can be seen in Fig. 2a, stimulation of PBMCs with either ConA (5 and 50 μg/ml) or anti-CD3 (1 and 5 μg/ml) plus anti-CD28 (1 and 5 μg/ml) increased MIF production significantly. The MIF production levels by PBMCs stimulated with anti-CD3 plus anti-CD28 were higher than those with ConA (3904 ± 939 pg/ml versus 1854 ± 707 pg/ml). In addition, as reported previously [14], MIF productions from fibroblasts of SSc patients were significantly higher than those of healthy controls (39 ± 5·9 pg/ml versus 18 ± 3·1 pg/ml, P < 0·01) (Fig. 2b).
Fig. 2.
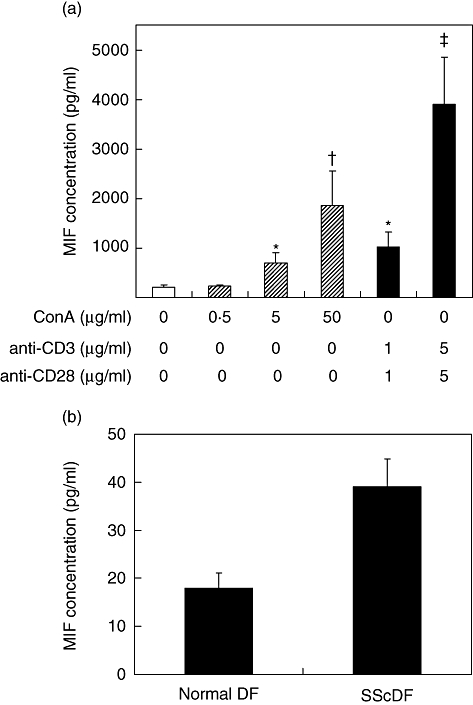
Production of macrophage migration inhibitory factor (MIF) from peripheral blood mononuclear cells and dermal fibroblasts. (a) MIF production by activated T cells. Peripheral blood mononuclear cells (5 × 105 cells) obtained from three healthy controls were cultured for 24 h with medium alone or in the presence of anti-CD3 (1 and 5 μg/ml) plus anti-CD28 (1 and 5 μg/ml), or concanavalin A (ConA) (0·5, 5, 50 μg/ml). The concentrations of MIF in the culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA). *P < 0·05; †P < 0·01; ‡P < 0·001 versus the cells stimulated with medium alone. (b) MIF production by cultured dermal fibroblasts (DF). DF (3 × 104 cells) were isolated from three healthy controls and three systemic sclerosis (SSc) patients, and then cultured for 48 h with DMEM containing 10% FCS. MIF production was determined in the culture supernatants by ELISA. Data are expressed as the mean ± standard error of the mean of three independent experiments using different cells.
Migration inhibitory factor inhibits SNP-induced apoptosis of dermal fibroblasts
To determine the role of MIF in the survival of dermal fibroblasts, we added the recombinant human MIF to culture medium of dermal fibroblasts in the presence of SNP, an apoptotic inducer. The degree of apoptosis was assessed by APOPercentage Apoptosis Assay as described in Materials and methods. As shown in Fig. 3, dermal fibroblasts underwent apoptosis by treating SNP for 24 h (Fig. 3). Apoptosis of dermal fibroblasts was also induced by limiting FCS concentration (0·5% FCS) in culture medium. The degree of apoptosis induced by SNP or serum deprivation was significantly lower in SSc fibroblasts than normal fibroblasts (Fig. 3). Of note, exogenous MIF, ranging from 1 to 100 ng/ml, blocked SNP-induced apoptosis of fibroblasts completely in a dose-dependent manner (Fig. 4). In contrast, when SSc fibroblasts were co-treated with SNP plus neutralizing anti-MIF antibody, which blocks endogenous MIF action, the degree of apoptosis was increased further compared with the cells treated with SNP alone (Fig. 5). The degree of apoptosis was verified by ELISA for the level of cellular DNA fragmentation (data not shown). These data suggest that MIF released endogenously exerts an anti-apoptotic role, and both endogenously released and exogenously added MIF therefore work together for the protection of fibroblasts from apoptotic death in our culture condition.
Fig. 3.
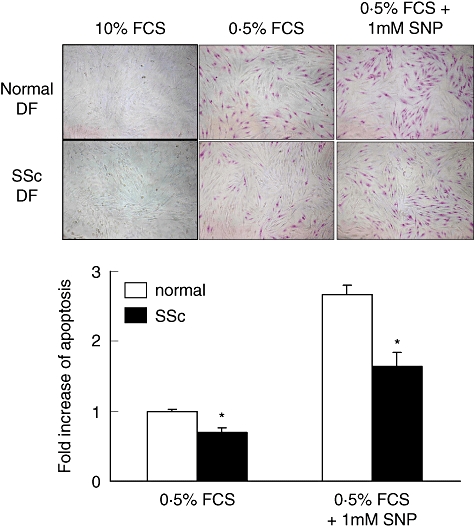
Decreased apoptosis of systemic sclerosis (SSc) fibroblasts compared with normal fibroblasts. Dermal fibroblasts (3 × 104 cells) of three normal controls and three SSc patients were cultured for 24 h with 0·5% fetal calf serum (FCS)/Dulbecco's modified Eagle's medium (DMEM) in the presence of 1 mM of sodium nitroprusside (SNP). APOPercentage Assay, a colorimetric assay, was used to assess the degree of apoptosis as described in Materials and methods. The upper panel shows the digital images of dermal fibroblasts undergoing apoptosis. Cells that appear bright pink represent apoptotic cells. In the lower panel, the level of apoptosis is measured by a pixel number using Adobe Photoshop. The data are presented as the fold increase relative to the degree of apoptosis in normal fibroblasts in DMEM supplemented with 0·5% FCS. *, P < 0·05 compared with normal dermal fibroblasts. DF, dermal fibroblasts.
Fig. 4.
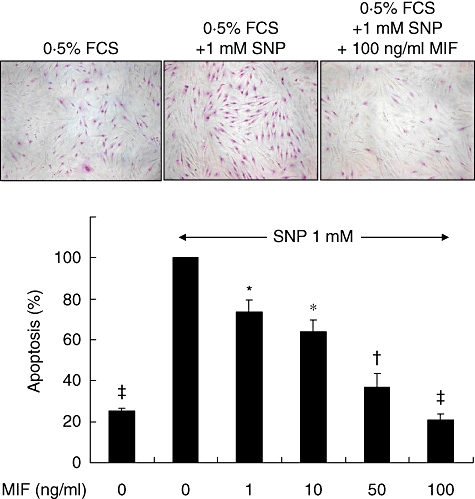
Macrophage migration inhibitory factor (MIF) inhibits sodium nitroprusside (SNP)-induced apoptosis of dermal fibroblasts in a dose-dependent manner. Normal dermal fibroblasts (3 × 104 cells) were cultured for 24 h with 0·5% fetal calf serum/Dulbecco's modified Eagle's medium in the presence of 1 mM SNP. The extent of apoptosis was assessed by APOPercentage Assay. The data are presented as the percentage of apoptosis relative to the apoptosis of untreated dermal fibroblasts (second column). *P < 0·05; †P < 0·01; ‡P < 0·001 versus the SNP-induced apoptosis without MIF. FCS, fetal calf serum.
Fig. 5.
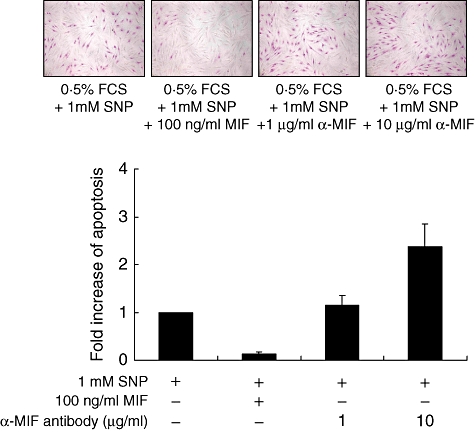
Effect of neutralizing anti-macrophage migration inhibitory factor (MIF) antibodies on the sodium nitroprusside (SNP)-induced apoptosis of dermal fibroblasts. Normal dermal fibroblasts (3 × 104 cells) were pretreated with neutralizing anti-MIF antibodies (1 and 10 μg/ml) and cultured for 24 h with 0·5% fetal calf serum/Dulbecco's modified Eagle's medium in the presence of SNP (1 mM). Apoptosis was assessed by APOPercentage Assay. The data are presented as the fold increase relative to the percentage of apoptotic cells in dermal fibroblasts treated with SNP only (first column). FCS, fetal calf serum.
Migration inhibitory factor up-regulates Bcl-2, pERK and pAkt expression in dermal fibroblasts
It is well established that Bcl-2 family members are critical to the regulation of cell survival via the modulation of mitochondrial integrity [32]. A recent study has demonstrated that MIF delays apoptosis of neutrophils by inhibiting the mitochondria-dependent death pathway [10]. Moreover, Bcl-2 seems to contribute to the resistance of apoptosis in SSc fibroblasts as Bcl-2 anti-sense oligonucleotide rendered SSc fibroblasts susceptible to apoptosis [4]. Thus, we investigated whether MIF is able to modulate Bcl-2 expression in dermal fibroblasts. As expected, the expressions of Bcl-2 in dermal fibroblasts were up-regulated after treatment with MIF (Fig. 6a). Because the activation of Akt and ERK maintains mitochondrial integrity via the up-regulation of Bcl-2 expression [33–36], we analysed the expression of pAkt and pERK in dermal fibroblasts stimulated with MIF. As shown in Fig. 6b, exogenous MIF (100 ng/ml) triggered phosphorylation of ERK1/2 and Akt in 10 min (Fig. 6b), but had little influence on the level of phospho-p38 mitogen activated protein kinase (MAPK) (data not shown). Moreover, the protective effect of MIF on the apoptosis of dermal fibroblasts was abrogated completely by treating cells with either ERK inhibitor PD98059 or Akt inhibitor LY294002 (Fig. 6c), suggesting involvement of the pAkt and pERK pathways in MIF-induced fibroblast survival.
Fig. 6.
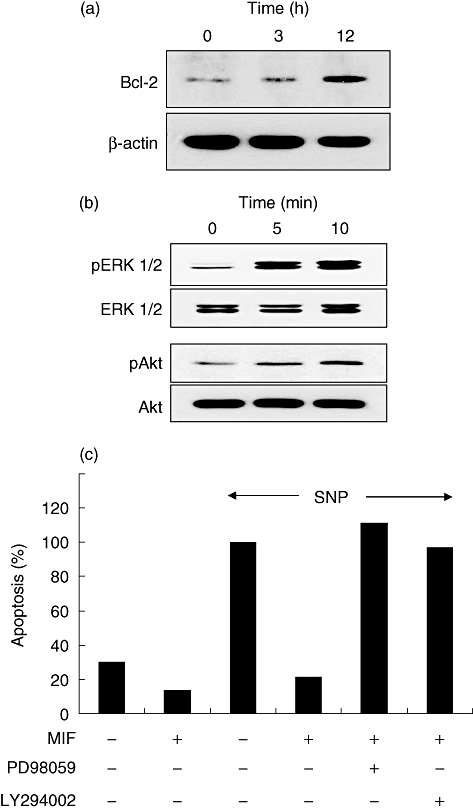
Up-regulation of Bcl-2, phospho-extracellular regulated kinase (pERK) or phospho-protein kinase B (Akt) inhibitors by macrophage migration inhibitory factor (MIF). (a, b) Effects of MIF on expression of Bcl-2, and on phosphorylation of ERK mitogen activated protein kinase and Akt kinase in dermal fibroblasts. Normal dermal fibroblasts treated with MIF (100 ng/ml) for the indicated times were subjected to Western blot using anti-Bcl-2 antibodies (a), as well as anti-pERK1/2 and anti-pAkt antibodies (b). The expression levels of β-actin, total ERK and total Akt were used to verify equal protein loading. (c) Effects of ERK or Akt inhibitor on the MIF-induced fibroblast survival. Normal dermal fibroblasts were preincubated for 30 min in the presence of ERK inhibitor PD98059 (20 μM) or Akt inhibitor LY294002 (20 μM), and then cultured with MIF (100 ng/ml) plus sodium nitroprusside (SNP) (1 mM) for 24 h. Apoptosis was assessed by APOPercentage Assay. The data are presented as a relative value to the percentage of apoptotic cells in dermal fibroblasts treated with SNP only (third column). A representative result from three similar experiments is shown.
Discussion
We demonstrate here that serum levels of MIF were elevated in patients with SSc than in healthy controls, which is consistent with the previous report [22]. We also found a positive correlation of MIF level with TGF-β concentration in the sera of SSc patients, suggesting the potential profibrogenic role of MIF in SSc pathogenesis. There are some reports regarding the relationship of MIF with TGF-β[37,38]. TGF-β has been shown to stimulate MIF mRNA expression in some types of cells [37], and the reverse seems to be true. For example, Leung et al. reported that MIF increased synthesis of TGF-β1 in cultured mesangial cells [38]. Moreover, they found that in mouse kidney with experimental IgA nephropathy, anti-MIF treatment reduced TGF-β1 expression significantly, indicating that MIF may contribute to the regulation of TGF-β1 expression. Although we did not find the positive effect of TGF-β on the production of MIF in the dermal fibroblasts (data not shown), further studies on relationship of two cytokines in SSc patients are required to clarify this issue.
In the early stage of SSc, perivascular mononuclear cells consist mainly of activated T cells [39,40]. Activated T cells stimulate fibroblast via cell-to-cell contact like effector cells, leading to the progression of fibrosis [41]. A previous study also detected the positive staining of MIF in dermal fibroblasts and infiltrating mononuclear cells using immunohistochemical method [22]. In the present study, MIF production in freshly isolated PBMCs was increased markedly by stimulation with anti-CD3 plus CD28 (Fig. 2a). Dermal fibroblasts of healthy subjects and SSc also produced MIF, the levels of which were greater in SSc fibroblasts than in normal fibroblasts (Fig. 2b). These data suggest that elevated serum MIF levels in SSc patients may come from activated T cells in the affected skin or peripheral blood, as well as dermal fibroblasts, although we did not compare directly the MIF levels in PBMCs between SSc and healthy controls. Based on our findings, it is intriguing to speculate that fibroblast-derived MIF could trigger secondarily T cell activation of blood-derived leucocytes infiltrated in dermis. Simultaneously, activated T cell-derived MIF, in turn, might augment the further activation of fibroblasts by secreting cytokines involved in the tissue remodelling of SSc, such as TNF-α, IL-6 and possibly MIF in an autocrine/paracrine fashion [23,42].
Migration inhibitory factor has been considered to be involved in the control of cell proliferation in several types of tumours and inflammatory disease such as rheumatoid arthritis [25,26]. In our study, MIF almost completely inhibited the SNP-induced apoptosis of dermal fibroblasts exogenously in a dose-dependent manner (Fig. 4). Moreover, neutralizing anti-MIF antibody, which blocks endogenous MIF, enhanced the SNP-induced apoptosis (Fig. 5), suggesting that endogenous MIF also confers apoptotic resistance to SSc fibroblasts. Considering that MIF production was higher in SSc fibroblasts than normal fibroblasts, it can be postulated that apoptotic resistance of SSc fibroblasts (Fig. 3) may be caused, at least in part, by overproduced MIF. Some reports showing that MIF secreted endogenously as well as added exogenously stimulates fibroblast proliferation support this notion [25,43]. In this regard, MIF blockade would be a desirable pro-apoptotic strategy to control excessive fibroproliferation in SSc.
The ERK1/2 pathway is crucial to fibroblast proliferation [44]. Lacey et al. demonstrated that MIF induction of fibroblast-like synoviocyte proliferation is dependent on ERK MAPK [44]. Similarly, higher levels of activated ERK have been detected in dermal and lung fibroblasts of SSc patients compared with control subjects [45]. On the other hand, the phosphoinositol-3-kinase/Akt pathway (PI3K/Akt) activation was also shown to promote growth and survival of cells [34–36]. Jun et al. observed that a higher proportion of skin fibroblasts derived from SSc was pAkt-positive compared with those with normal subjects or other inflammatory disease [46]. In this study, MIF treatment resulted in an increase in the expression of Bcl-2 in dermal fibroblasts, and also elevated levels of pAkt and pERK, both of which are known to be located upstream of the Bcl-2 signalling pathway. In addition, the anti-apoptotic effect of MIF was cancelled by the Akt or ERK inhibitor, suggesting that both signals mediate the MIF-induced increase in fibroblast survival in SSc. These observations indicate that MIF may function in concert with pAkt and pERK to modulate the expression of Bcl-2, thereby protecting SSc fibroblasts from apoptosis.
In conclusion, we showed that serum MIF levels were significantly higher in patients with SSc than in healthy controls. In addition, both the endogenously and exogenously secreted MIF significantly protected SNP-induced apoptosis of dermal fibroblasts via up-regulation of Bcl-2 as well as activation of the ERK1/2 MAPK and PI3K/Akt pathways. These data indicate that MIF contribute to the skin sclerosis observed in scleroderma by inhibiting the apoptosis of dermal fibroblasts. Therefore, MIF antagonism might be useful as a therapeutic strategy in the regulation of excessive fibrosis in SSc.
Acknowledgments
This work was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (0405-DB01-0104-0006) and SRC/ERC programme MOST/KOSEF (R11-2002-098-05001-0).
References
- 1.Jelaska A, Arakawa M, Broketa G, Korn JH. Heterogeneity of collagen synthesis in normal and systemic sclerosis skin fibroblasts. Increased proportion of high collagen-producing cells in systemic sclerosis fibroblasts. Arthritis Rheum. 1996;39:1338–46. doi: 10.1002/art.1780390811. [DOI] [PubMed] [Google Scholar]
- 2.Botstein GR, Sherer GK, LeRoy EC. Fibroblast selection in scleroderma: an alternative model of fibrosis. Arthritis Rheum. 1982;25:189–95. doi: 10.1002/art.1780250212. [DOI] [PubMed] [Google Scholar]
- 3.LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest. 1974;54:880–9. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santiago B, Galindo M, Rivero M, Pablos JL. Decreased susceptibility to Fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis Rheum. 2001;44:1667–76. doi: 10.1002/1529-0131(200107)44:7<1667::AID-ART291>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 5.Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000;43:2230–9. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Tamm I, Kikuchi T, Zychlinsky A. Acidic and basic fibroblast growth factors are survival factors with distinctive activity in quiescent BALB/c 3T3 murine fibroblasts. Proc Natl Acad Sci USA. 1991;88:3372–6. doi: 10.1073/pnas.88.8.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J. 1994;13:3286–95. doi: 10.1002/j.1460-2075.1994.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–2. doi: 10.1126/science.153.3731.80. [DOI] [PubMed] [Google Scholar]
- 9.David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell–antigen interaction. Proc Natl Acad Sci USA. 1966;56:72–7. doi: 10.1073/pnas.56.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumann R, Casaulta C, Simon D, Conus S, Yousefi S, Simon HU. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J. 2003;17:2221–30. doi: 10.1096/fj.03-0110com. [DOI] [PubMed] [Google Scholar]
- 11.Riedemann NC, Guo RF, Gao H, et al. Regulatory role of C5a on macrophage migration inhibitory factor release from neutrophils. J Immunol. 2004;173:1355–9. doi: 10.4049/jimmunol.173.2.1355. [DOI] [PubMed] [Google Scholar]
- 12.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishihira J, Koyama Y, Mizue Y. Identification of macrophage migration inhibitory factor (MIF) in human vascular endothelial cells and its induction by lipopolysaccharide. Cytokine. 1998;10:199–205. doi: 10.1006/cyto.1997.0276. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi T, Nishihira J, Koyama Y, Sasaki S, Yamamoto Y. Decreased prostaglandin E2 production by inflammatory cytokine and lower expression of EP2 receptor result in increased collagen synthesis in keloid fibroblasts. J Invest Dermatol. 2006;126:990–7. doi: 10.1038/sj.jid.5700227. [DOI] [PubMed] [Google Scholar]
- 15.Leech M, Metz C, Santos L, et al. Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum. 1998;41:910–7. doi: 10.1002/1529-0131(199805)41:5<910::AID-ART19>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Morand EF, Bucala R, Leech M. Macrophage migration inhibitory factor: an emerging therapeutic target in rheumatoid arthritis. Arthritis Rheum. 2003;48:291–9. doi: 10.1002/art.10728. [DOI] [PubMed] [Google Scholar]
- 17.Leech M, Metz C, Hall P, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42:1601–8. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 18.de Jong YP, Abadia-Molina AC, Satoskar AR, et al. Development of chronic colitis is dependent on the cytokine MIF. Nat Immunol. 2001;2:1061–6. doi: 10.1038/ni720. [DOI] [PubMed] [Google Scholar]
- 19.Niino M, Kikuchi S, Fukazawa T, Yabe I, Tashiro K. Estrogen receptor gene polymorphism in Japanese patients with multiple sclerosis. J Neurol Sci. 2000;179:70–5. doi: 10.1016/s0022-510x(00)00381-6. [DOI] [PubMed] [Google Scholar]
- 20.Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J Rheumatol. 2004;31:268–73. [PubMed] [Google Scholar]
- 21.Donn RP, Plant D, Jury F, et al. Macrophage migration inhibitory factor gene polymorphism is associated with psoriasis. J Invest Dermatol. 2004;123:484–7. doi: 10.1111/j.0022-202X.2004.23314.x. [DOI] [PubMed] [Google Scholar]
- 22.Selvi E, Tripodi SA, Catenaccio M, et al. Expression of macrophage migration inhibitory factor in diffuse systemic sclerosis. Ann Rheum Dis. 2003;62:460–4. doi: 10.1136/ard.62.5.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu SP, Leng L, Feng Z, et al. Macrophage migration inhibitory factor promoter polymorphisms and the clinical expression of scleroderma. Arthritis Rheum. 2006;54:3661–9. doi: 10.1002/art.22179. [DOI] [PubMed] [Google Scholar]
- 24.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–6. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 26.Nishihira J, Ishibashi T, Fukushima T, Sun B, Sato Y, Todo S. Macrophage migration inhibitory factor (MIF). Its potential role in tumor growth and tumor-associated angiogenesis. Ann NY Acad Sci. 2003;995:171–82. doi: 10.1111/j.1749-6632.2003.tb03220.x. [DOI] [PubMed] [Google Scholar]
- 27.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–82. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fingerle-Rowson G, Petrenko O, Metz CN, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petrenko O, Fingerle-Rowson G, Peng T, Mitchell RA, Metz CN. Macrophage migration inhibitory factor deficiency is associated with altered cell growth and reduced susceptibility to Ras-mediated transformation. J Biol Chem. 2003;278:11078–85. doi: 10.1074/jbc.M211985200. [DOI] [PubMed] [Google Scholar]
- 30.American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 31.Sakkas LI, Xu B, Artlett CM, Lu S, Jimenez SA, Platsoucas CD. Oligoclonal T cell expansion in the skin of patients with systemic sclerosis. J Immunol. 2002;168:3649–59. doi: 10.4049/jimmunol.168.7.3649. [DOI] [PubMed] [Google Scholar]
- 32.Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–9. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Pugazhenthi S, Nesterova A, Sable C, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–6. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 34.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J Exp Med. 2005;202:1363–74. doi: 10.1084/jem.20051283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chadha KS, Khoury T, Yu J, et al. Activated Akt and Erk expression and survival after surgery in pancreatic carcinoma. Ann Surg Oncol. 2006;13:933–9. doi: 10.1245/ASO.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi N, Nishihira J, Sato Y, et al. Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. Mol Med. 1998;4:707–14. [PMC free article] [PubMed] [Google Scholar]
- 38.Leung JC, Chan LY, Tsang AW, et al. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol Dial Transplant. 2004;19:1976–85. doi: 10.1093/ndt/gfh323. [DOI] [PubMed] [Google Scholar]
- 39.Gruschwitz MS, Vieth G. Up-regulation of class II major histocompatibility complex and intercellular adhesion molecule 1 expression on scleroderma fibroblasts and endothelial cells by interferon-gamma and tumor necrosis factor alpha in the early disease stage. Arthritis Rheum. 1997;40:540–50. doi: 10.1002/art.1780400321. [DOI] [PubMed] [Google Scholar]
- 40.Gruschwitz M, von den Driesch P, Kellner I, Hornstein OP, Sterry W. Expression of adhesion proteins involved in cell–cell and cell–matrix interactions in the skin of patients with progressive systemic sclerosis. J Am Acad Dermatol. 1992;27:169–77. doi: 10.1016/0190-9622(92)70165-c. [DOI] [PubMed] [Google Scholar]
- 41.Sato S. Abnormalities of adhesion molecules and chemokines in scleroderma. Curr Opin Rheumatol. 1999;11:503–7. [PubMed] [Google Scholar]
- 42.Feghali CA, Bost KL, Boulware DW, Levy LS. Mechanism of pathogenesis in scleroderma. Overproduction of interleukin 6 by fibroblast cultured from affected skin sites of patients with scleroderma. J Rheumatol. 1992;19:1207–11. [PubMed] [Google Scholar]
- 43.Leech M, Lacey D, Xue JR, et al. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003;48:1881–9. doi: 10.1002/art.11165. [DOI] [PubMed] [Google Scholar]
- 44.Lacey D, Sampey A, Mitchell R, et al. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–9. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- 45.Tourkina E, Gooz P, Pannu J, et al. Opposing effects of protein kinase Calpha and protein kinase Cepsilon on collagen expression by human lung fibroblasts are mediated via MEK/ERK and caveolin-1 signaling. J Biol Chem. 2005;280:13879–87. doi: 10.1074/jbc.M412551200. [DOI] [PubMed] [Google Scholar]
- 46.Jun JB, Kuechle M, Min J, et al. Scleroderma fibroblasts demonstrate enhanced activation of Akt (protein kinase B) in situ. J Invest Dermatol. 2005;124:298–303. doi: 10.1111/j.0022-202X.2004.23559.x. [DOI] [PubMed] [Google Scholar]