

Abstract
Macrophage migration inhibitory factor (MIF) is a pleiotropic pro-inflammatory cytokine with many cellular targets in rheumatoid arthritis (RA). MIF has been reported to activate cells via mitogen-activated protein kinase and serine/threonine kinase (AKT or protein kinase B)-dependent signal transduction pathways. Its contribution to T cell activation and signalling in RA is not known. Using MIF −/− mice and a T cell-mediated model of RA, antigen-induced arthritis, we investigated the role of MIF in T cell activation and signalling. Arthritis severity was significantly reduced in MIF −/− mice compared with wildtype mice. This reduction was associated with decreased T cell activation parameters including footpad delayed type hypersensitivity, antigen-induced splenocyte proliferation and cytokine production. Splenocyte proliferation required extracellular signal-regulated kinase (ERK)1/2 phosphorylation, and decreased T cell activation in MIF −/− mice was associated with decreased phosphorylation of ERK1/2 but not AKT. Collectively, these data suggest that MIF promotes antigen-specific immune responses via regulation of ERK phosphorylation in T cells.
Keywords: arthritis, ERK, MIF, T cell
Macrophage migration inhibitory factor (MIF) is a broad-spectrum pro-inflammatory cytokine implicated in many inflammatory diseases, including rheumatoid arthritis (RA) (reviewed in [1]). Increased expression of MIF in RA correlates with disease activity [1,2] and there is increasing evidence that polymorphisms in the MIF gene are associated with inflammatory arthritis [3,4]. Functions of MIF relevant to RA include activation of cell proliferation, regulation of macrophage and fibroblast inflammatory mediators including cytosolic phospholipase A2, cyclooxygenase-2, interleukin (IL)-1, tumour necrosis factor (TNF), IL-8 and IL-6, and inhibition of apoptosis [5–10].
T cells are prominent in RA synovium and small but physiological levels of T cell products have been detected at the site of injury [11]. Many animal models that are clearly T cell dependent are routinely used in the investigation of RA pathogenesis [12,13]. MIF is expressed in resting and activated T cells [14,15] and MIF mRNA and protein are abundantly expressed in mouse delayed type hypersensitivity (DTH) lesions [15]. Treatment with rMIF can exacerbate DTH reactions [15] while anti-MIF antibody treatment is inhibitory [16]. Studies using MIF antagonism [14–16] and MIF deficient mice [17] have shown a requirement for MIF in aspects of T cell activation including, proliferation, cytokine and chemokine production.
Recently, the cellular source of an important T cell-derived proinflammatory cytokine, IL-17 has been identified in the mouse and termed T helper 17 (Th17) [18]. RA is the best studied clinical condition regarding IL-17 (reviewed in [19]. In addition to interferon (IFN)-γ, IL-17 has been detected in RA and both are proposed to mediate immune responses, fibroblast activation and bone destruction [11]. MIF has also been shown to be required in the homing of T cells to inflammatory sites [20–22] acting as a noncognate ligand of CXC chemokine receptors and inducing T cell transmigration via CXCR4 [22].
Delayed type hypersensitivity reactions are contributed to both by T cell and macrophage functions and while T cell-derived MIF is not detected in DTH lesions [15], T cells can release MIF upon mitogenic or antigenic stimulation [14]. Macrophages have been shown to be responsible for much of MIF produced in vivo and several studies have reported the signalling pathways utilized by macrophage MIF in exerting its various activities [23–26]. In contrast, the signalling mechanism of T cell derived-MIF is comparatably understudied.
The role of MIF in disease models mediated by T cell-initiated adaptive immune responses, including experimental autoimmune encephalomyelitis [27], and glomerulonephritis [28], suggest a potentially pivotal role of MIF in the activation and regulation of T cell function in RA. To test this hypothesis we investigated the contribution of MIF to T cell activation and signalling in a T cell-mediated model of RA, antigen-induced arthritis (AIA). As MIF has been reported to activate cells via extracellular signal-regulated kinase (ERK) mitogen-activated protein (MAP) kinase [5,10] and serine/threonine kinase (AKT or protein kinase B)-dependent signal transduction pathways [29,30], we also investigated antigen-induced T cell utilization of these pathways. We report that MIF promotes antigen-specific immune responses and the activation of ERK MAP kinase in T cells.
Materials and methods
Animals
Migration inhibitory factor deficient mice were generated as previously described [31]. MIF−/− and wildtype (WT) littermates (B6/129) [31] were used. Mice were bred and housed under specific pathogen-free conditions in the animal facility of Monash Medical Centre until used for experiments, during which they were maintained in conventional housing. All animal experiments were performed in accordance with the regulations of Monash University Animal Ethics Committee.
Induction and assessment of arthritis
Antigen-induced arthritis was induced and assessed in mice (10–14 weeks of age) as described previously [12]. Briefly, mice were immunized on day 0 with 200 μg of methylated bovine serum albumin [mBSA; Sigma Chemical Co., Castle Hill, Australia) emulsified in 0·2 ml of Freund's complete adjuvant (CFA; Sigma) injected subcutaneously in the flank skin. At day 7, mice were given 100 μg mBSA in 0·1 ml CFA by intradermal injection at the base of the tail. Arthritis was induced by intra-articular injection of 30 μg mBSA in 10 μl sterile saline into one knee. The contralateral knee received 10 μl of saline to serve as control.
At day 28 after first immunization, knee joint thickness of both mBSA and saline-injected knees was measured across the patella using micro callipers (Mitutoyo, Kawasaki-shi, Japan) and results expressed in mm. Joints were then fixed in formalin, decalcified in 15% EDTA-Tris buffer (BDH Chemicals, Sydney, Australia), and 6 μm-thick sagittal knee sections stained and counter stained with safranin-O and fast green/iron haematoxylin (ICN Biomedicals, Ohio, USA) respectively. Histological sections were scored 0–3 for four parameters [12]: synovitis was defined as hypercellularity of the synovium including pannus formation, joint space exudate was identified as leucocytes, discretely or in aggregates, in the joint space, cartilage degradation was defined as loss of safranin-O staining of articular cartilage (0 = full stained cartilage, 3 = totally unstained cartilage), and bone damage was graded on the extent and depth of subchondral bone damage.
Induction and assessment of cutaneous DTH
Mice were challenged on day 27 following first immunization by a single intradermal injection of 50 μg mBSA/20 μl saline in the right footpad, with 20 μl saline injected in the left footpad serving as control [12]. Mice were killed 24 h later and footpad swelling quantified using micro callipers. DTH measurements were performed by an observer blinded to mouse genotype. Results were expressed as the difference in footpad swelling between mBSA and saline-injected footpads, and expressed as change in footpad thickness (mm).
T cell proliferation and cytokine production
T cell proliferation was assessed by measuring the cellular DNA incorporation of [3H]-thymidine as previously described [12]. Briefly, spleens were removed aseptically at day 28 after the first immunization. Single-cell suspensions were prepared by gently teasing tissue apart using a needle and syringe. Erythrocytes were lysed by incubation in Boyle's solution (0·17 M Tris/0·16 M ammonium chloride) for 1 min at 37°C. Cell suspensions were washed in RPMI (JRH Biosciences, Victoria Australia) containing 5% fetal calf serum (FCS; JRH) and a single cell suspension prepared in RPMI/5%FCS/0·05% 2-mercaptoethanol (2ME; Sigma). Cells (1 × 105/200 μl) were cultured in triplicate in the presence or absence of mBSA (10, 100 μg/ml) for 48 h (37°C, 5% CO2). For ERK1/2 inhibition experiments cells were pretreated with 50 μM PD98059 [32] or DMSO control 30 min prior to addition of mBSA. Cells were pulsed with [3H]-thymidine (GE Healthcare, New South Wales, Australia) in the final 18 h then harvested. Incorporation of thymidine into DNA was measured with a Wallac 1409 liquid scintillation counter (Pharmacia, Turku, Finland). Results were expressed as counts per minute (cpm) and proliferation index (mBSA-treated cpm/baseline cpm).
Cytokine production was measured by enzyme-linked immunosorbent assay (ELISA) of supernatant from splenocytes cultured as described [12]. Briefly, splenocytes (2 × 106/500 μl) were cultured in duplicate for 48 h (RPMI/5%FCS/0·05% 2ME, 37°C, 5% CO2) in the presence or absence of mBSA (10 μg/ml). Antibodies used were rat antimouse IFN-γ (R46A2; Pharmingen, San Diego, CA) and biotinylated XMG1·2 (Pharmingen) for IFN-γ. The sensitivity of the assays was 12 pg/ml.
Cell lysate preparation and Western blot analysis
Non-enriched splenocytes (10 × 106) were cultured for 6 h (RPMI/5% FCS/0·05% 2ME, 37°C, 5% CO2) in the presence or absence of mBSA (10 μg/ml). For phosphorylated ERK (P-ERK) detection, cells were lysed in lysis buffer containing 20 mM HEPES pH 7·7, 2·5 mM MgCl2, 0·1 mM EDTA, 20 mM β-glycerophosphate, 100 mM NaCl, Triton X-100, 1 M DL-dithiothreitol, 0·1 nM sodium orthovanadate, 20 μg/ml leupeptin, 100 μg/ml phenylmethylsulphonyl fluoride, and 20 μg/ml aprotinin (all reagents from Sigma) as previously described [10]. For phosphorylated-AKT (P-AKT) detection, cells were washed with cold phosphate buffered saline (PBS) then lysed with 2x sodium dodecyte sulphate sample buffer. The protein samples were boiled for 10 min then stored at −20°C.
Immunoblotting was performed using antibodies directed against P-ERK and P-AKT (Ser473) and β-actin according to the manufacturer's protocol (Cell Signalling Laboratories, Beverly, MA). Briefly, equal amounts of cellular proteins were fractionated on 10% SDS-polyacrylamide electrophoresis gels and transferred to Hybond-C extra nitrocellulose membranes (Millipore, Bedford, MA). Blots were blocked with blocking buffer then incubated sequentially with appropriate primary and fluorescence-conjugated secondary antibodies. Blots were scanned using Odyssey (Li-Cor Biotechnology, Lincoln, NE). Densitometry ratios were normalized to β-actin content and results expressed as relative density.
Flow cytometric analysis
The following antibodies were used for two colour labelling for flow cytometry: anti-CD4-FITC, anti-CD4-PE, anti-CD54-PE, anti-CD25-FITC, anti-CD69-PE and antiphospho-ERK1/2-Alexa Fluor 488 (BD Pharmingen, San Diego, CA). For detection of CD4 positive lymphocytes expressing CD54, CD25 and CD69, splenocytes were cultured in the presence and absence of mBSA (10 μg/ml) for 48 h (RPMI/5% FCS/0·05% 2ME, 37°C, 5% CO2) then washed and incubated with relevant monoclonal antibodies for 30 min at 4°C.
For detection of P-ERK, splenocytes were cultured in the presence or absence of mBSA (10 ug/ml) for 15 min (RPMI/0·05% 2ME without serum, 37°C, 5% CO2) then washed in 2% BSA/PBS (wash buffer). Cells were fixed in 65 μl of 10% formaldehyde (Polysciences Inc, Warrington, PA) for 10 min at room temperature (RT) followed by addition of 1·0 ml Triton X-100 (Sigma) diluted in PBS to obtain a 0·1% final concentration. After 30 min incubation at RT cells were washed in cold wash buffer then incubated in 1 ml 50% methanol in PBS for 15 min at 4°C. Cells were washed then incubated simultaneously with antiphospho-ERK1/2-Alexa Fluor 488 and anti-CD4-PE. Cells were analysed on a MoFlo flow cytometer (Dako-Cytomation, Fort Collins, CO).
Antibody response
Serum levels of antimBSA immunoglobulin G (IgG) were determined by ELISA as previously described [12]. Briefly, polyvinyl microtiter plates were coated with mBSA (100 μg/ml) for 24 h at 4°C. The plates were blocked with 2% casein (Sigma) in PBS with 0·05% Tween-20 (PBS-T, Science Supply Australia, Seven Hill, NSW, Australia) at RT for 1 h. After washing, 100 μl of diluted (1/100) serum samples were incubated for 24 h at 4°C. Plates were then washed, and sequentially incubated with biotinylated rabbit antimouse-IgG (1:2000, DAKO) or biotinylated rabbit anti-isotype-specific (IgG1, IgG2a; DAKO) antibodies for 2 h then streptavidin-horseradish peroxidase conjugate (1:2000) for 30 min at RT. The enzyme reaction was detected using the TMB peroxidase-substrate (Sigma) with hydrogen peroxide (Sigma) and optical density at 450 nm determined.
Statistical analysis
Data were analysed using the Mann–Whitney test for comparisons of group means of histological scores, or Student's t-test for comparisons of continuous variables. Results are expressed as the mean ± standard error of the mean. For each test, values of P < 0·05 were regarded as statistically significant.
Results
Decreased arthritis severity in MIF −/− mice
Methylated BSA (mBSA)-challenged WT mice knee joints exhibited significantly increased thickness in comparison with contralateral saline-injected knees (P < 0·001) (Fig. 1a). In contrast, mBSA injection did not result in increased knee thickness in MIF −/− littermates, and MIF −/− littermates had significantly lower knee joint thickness in comparison with WT (P < 0·0001) (Fig. 1a).
Fig. 1.
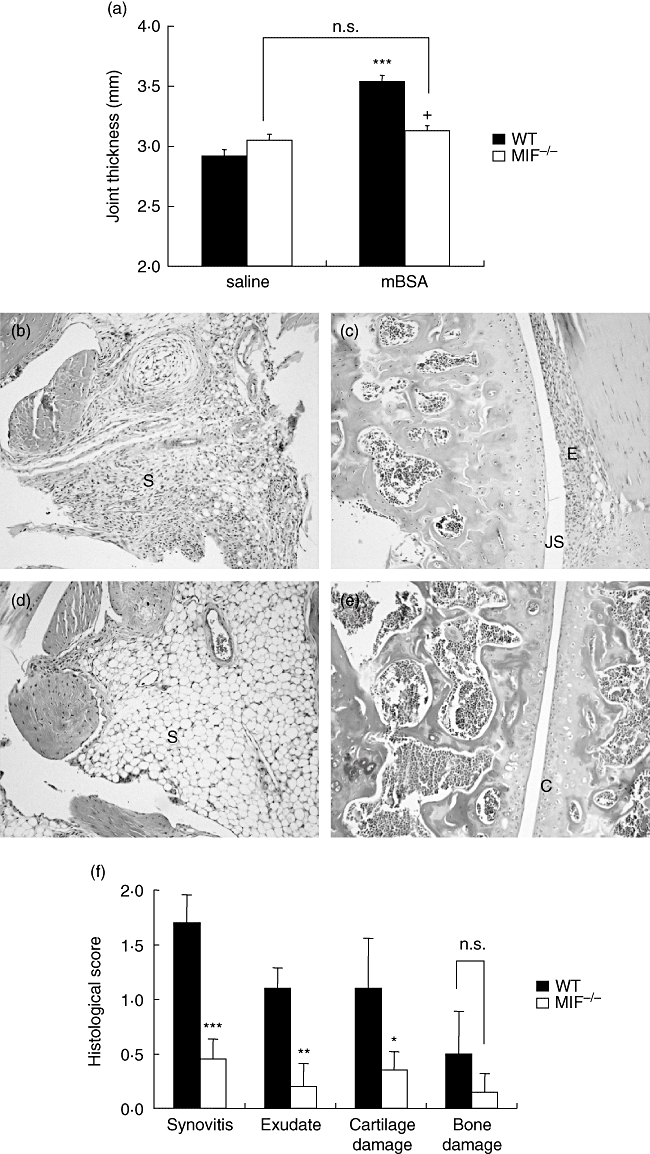
Arthritis induction in migration inhibitory factor (MIF)−/−and wildtype (WT) mice. Mice received intra-articular injections of methylated bovine serum albumin (mBSA) (30 μg; test knee) or saline (control knee) on day 21 after first immunization. On day 28, arthritis severity was measured by thickness of mBSA- and saline-injected knee joints (a) WT, but not MIF −/− mice, exhibited significantly increased joint thickness in response to mBSA (***P < 0·001). MIF −/− mice joint thickness after mBSA injection was significantly lower than WT (+P < 0·0001). Histological scores (f) of Safranin-O-stained joint sections on a scale of 0–3 for each individual feature, synovitis, joint space exudate, cartilage degradation and bone damage (as described in Materials and Methods). WT mice (b and c) exhibited more severe arthritis in comparison with MIF −/− mice (d and e) as evidenced by significantly higher scores for all but one histopathological feature (f) (*P < 0·05, **P < 0·01, ***P < 0·001 for MIF −/−versus WT mice). S = synovium, JS = joint space, E = exudate, C = articular cartilage. Magnification ×200. n.s., not significant.
Histological examination of synovial sections of WT mice showed significant synovitis and exudate in the synovial space, as well as marked cartilage degradation (Fig. 1b and c). In contrast, MIF −/− joint sections had little or no synovitis (P < 0·001), joint exudate (P < 0·01) or cartilage degradation (P < 0·05 cf WT) (Fig. 1d and e). Bone damage was minimal in both groups; significant bone damage is not usually seen at this timepoint of AIA [12] (Fig. 1f). Saline-injected knee joint histology was comparable in both groups and revealed no evidence of arthritis (data not shown).
Impaired T cell activation in MIF −/− mice
Indices of T cell activation showed significant reductions in MIF −/− mice. In comparison with WT littermates, footpad DTH was significantly decreased in MIF −/− mice (P < 0·0001) (Fig. 2a). As DTH reactions are contributed to both by T cell-dependent adaptive immune responses, we sought to investigate antigen-specific T cell function. Basal splenocyte proliferation was significantly lower in MIF −/− cells (P < 0·01) compared with WT (Fig. 2b). Antigen stimulation was associated with increased WT splenocyte proliferation, and this was significantly higher than MIF −/− splenocyte proliferation (Fig. 2b). Correcting for the lower basal proliferation in MIF −/− splenocytes by calculating mBSA-induced proliferation as a proliferation index, demonstrated that both WT and MIF −/− splenocytes exhibit comparable responses to antigen at 10 μg/ml mBSA dose. However, MIF −/− splenocyte proliferation index was significantly lower at 100 μg/ml mBSA (P < 0·05) (Fig. 2c). Basal and antigen-induced IFN-γ production was significantly lower in MIF −/− cells (P < 0·05) (Fig. 2d).
Fig. 2.
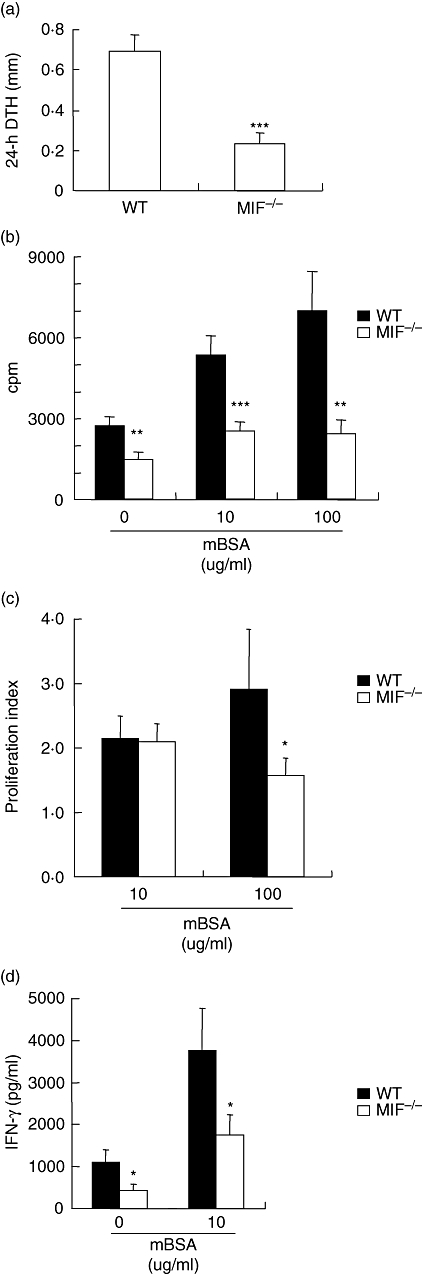
T cell activation in migration inhibitory factor (MIF) −/− and wildtype (WT) mice. Cutaneous delayed type typersensitivity was induced in sensitized mice by intradermal challenge with methylated bovine serum albumin (mBSA) in the footpad and footpad swelling measured 24 h later. Results are expressed as the difference between mBSA and saline-injected footpad thickness measured using micro callipers (a). Splenocyte proliferation was measured by [3H]-thymidine incorporation after 48 h in the presence or absence of mBSA (10, 100 μg/ml). Results are expressed as cpm (b) and proliferation index (see methods) (c). In a separate experiment, IFN-γ (d) from splenocyte culture supernatants was measured by enzyme-linked immunosorbent assay. MIF −/− mice had significantly decreased evidence of T cell activation in all parameters measured. All results are expressed as mean ± standard error of the mean of at least 7 mice in each group (*P < 0·05, **P < 0·01, ***P < 0·001). cpm, counts per minute; IFN, interferon.
The expression of T cell surface molecules on CD4+ cells was assessed by flow cytometry. Firstly, there was no difference in the basal number of CD4+ cells in the spleen between WT and MIF −/− mice (data not shown). Basal CD54 (Fig. 3a), CD25 (Fig. 3b), and CD69 (Fig. 3c) expressions were similar in MIF −/− and WT splenocytes. Treatment with mBSA did not increase (Fig. 3a–c) expression of surface molecules CD54, CD25 and CD69 on either WT or MIF −/− CD4+ cells.
Fig. 3.
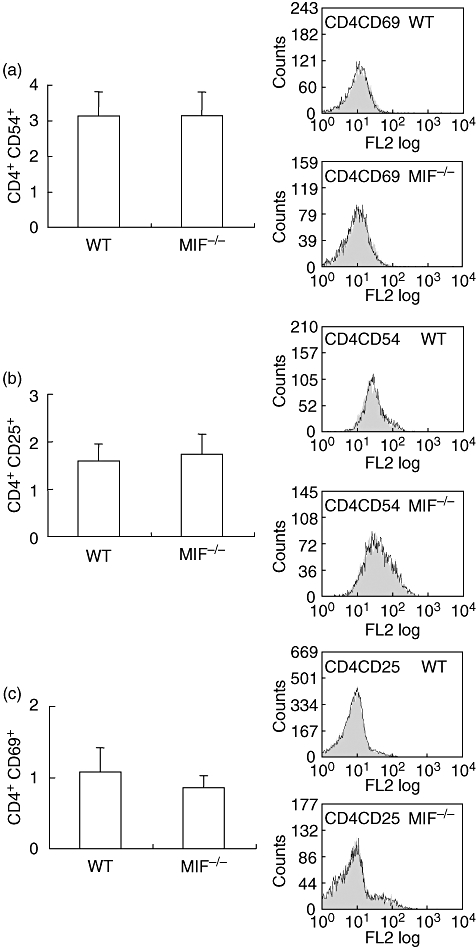
Flow cytometric analysis of T cell surface antigens. Splenocytes were labelled with antibodies to CD54, CD25 and CD69. Results represent percent positive for indicated surface antigens gated on CD4-positive lymphocyte population. Representative histograms show percent positive stain of untreated (clear) and methylated bovine serum albumin (mBSA) (grey)-treated cells. Migration inhibitory factor (MIF) −/− and wildtype (WT) CD4-positive cells had comparable basal expression of CD54 (a) CD25 (b) and CD69 (c). In both groups, treatment with mBSA did not affect all three surface molecules on CD4-positive cells.
Reduced antibody response to mBSA in MIF −/− mice
Sera were collected from mice at day 28 after first immunization and mBSA-specific serum antibody levels measured by ELISA. MIF −/− mice exhibited significantly reduced antigen-specific total IgG (Fig. 4a), IgG1 (Fig. 4b) and IgG2a (Fig. 4c) compared with WT (Fig. 4) (all P < 0·0001).
Fig. 4.
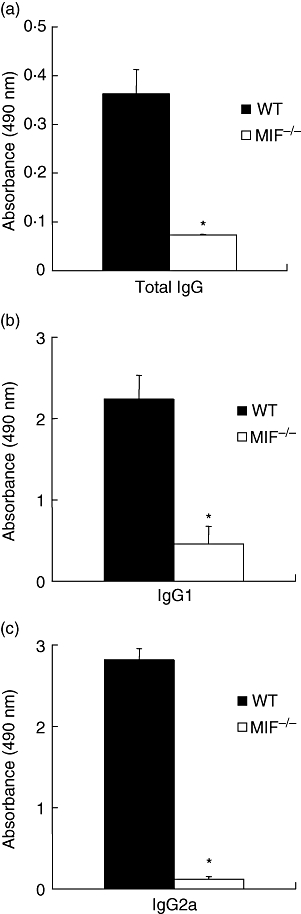
Methylated bovin serum albumin-specific serum antibody titres. Serum levels of total antigen-specific immunoglobulin G (IgG), IgG1 and IgG2a were determined by enzyme-linked immunosorbent assay and results expressed as absorbance at 450 nm. Migration inhibitory factor (MIF) −/− serum levels of total IgG (a), and isotype IgG1 (b) and IgG2a (c) were all significantly lower compared with wildtype (WT) levels (*P < 0·0001).
Utilization of signalling pathways in antigen-stimulated T cells
Two signalling pathways activated during T cell activation are known to be influenced by MIF. Basal ERK activation was comparable in lysates from MIF −/− and WT littermate splenocytes. Antigen stimulation increased ERK phosphorylation in WT cells but this was not observed in MIF −/− cells (Fig. 5a). Flow cytometric analysis of phospho-ERK expression by CD4+ splenocytes demonstrate significantly increased phospho ERK in WT cells stimulated with antigen (P < 0·01). This effect was absent in MIF −/− cells (Fig. 5b). The dependence of T cell proliferation on the ERK pathway was studied by use of the ERK1/2 inhibitor PD98059 in a 3H-thymidine proliferation assay. Inhibition of ERK1/2 resulted in significantly decreased antigen-stimulated proliferation (Fig. 5c) compared with DMSO-treated antigen-stimulated control. Measurement of AKT-phosphorylation showed comparable basal and antigen-stimulated AKT phosphorylation in WT and MIF −/− cells. No increase in AKT-phosphorylation was induced by antigen treatment of WT or MIF −/− cells.
Fig. 5.
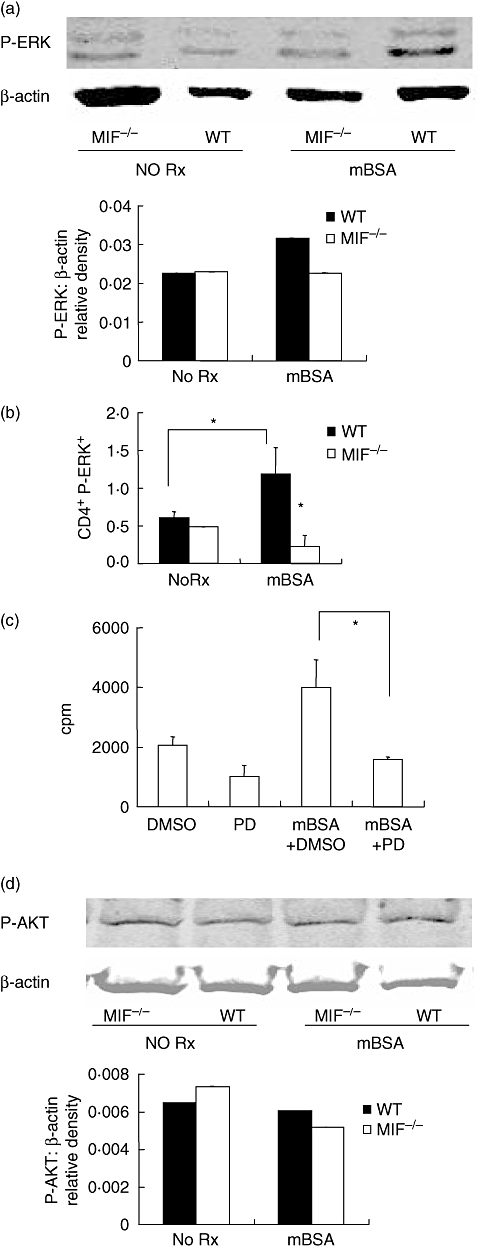
Analysis of phosphorylated-extracellular signal-regulated kinase (P-ERK) and serine/threonine kinase (AKT). Splenocytes were cultured for 6 h with and without methylated bovine serum albumin (mBSA). Phospho-ERK and phospho-AKT were measured by Western blotting and the membranes reprobed for β-actin as loading control. Blots are representative of three independent experiments. Migration inhibitory factor (MIF) −/− cells had decreased P-ERK compared with wildtype (WT) mice following mBSA stimulation (a). To identify the lymphocyte population expressing P-ERK, splenocytes were simultaneously labelled with CD4-phycoerythrin and P-ERK-fluorescein isothiocyanate for flow cytometric analysis. Results are expressed as percent positive for P-ERK in gated population and are data from a representative of nine experiments (b). Stimulation with mBSA resulted in significantly increased P-ERK in WT CD4+. MIF −/− cells did not exhibit an increase in P-ERK in response to mBSA. Inhibition of ERK1/2 by 30 min pretreatment with PD98059 inhibited antigen-stimulated cell proliferation (c) (*P < 0·05). DMSO control pretreatment had no inhibitory effect. Western blotting for P-AKT shows comparable basal and antigen-stimulated P-AKT in both WT and MIF −/− cells (d). Antigen-stimulation did not increase P-AKT in either group (d).
Discussion
We show here that arthritis severity, cutaneous DTH responses, and splenocyte IFN-γ production and proliferation were significantly reduced in the absence of MIF. These observations are in keeping with the reported modulation of T cell function by anti-MIF antibody in vitro[14,33] and as a result of MIF deficiency [17]. Although not required for disease induction in AIA, antigen-specific antibodies are present in diseased animals and are regulated by the Th1 immune response [34]. Levels of antigen-specific total IgG, IgG1 and IgG2a were reduced in MIF −/− mice, suggesting an effective reduction of the immune response in the absence of MIF. These observations are in keeping with a previous study showing reduction of antigen-specific T cell proliferation and antibody production by MIF antagonism [14].
Activation by MIF of intracellular signal pathways including those utilizing ERK MAP kinase and PI3 kinase has been reported in various cell types [5,10,23,24,26,29]. ERK MAP kinase and PI3 kinase pathways mediate T cell activation particularly in response to IL-2 stimulation [35], but the signalling pathways influenced by MIF in the regulation of T cell activation have not been previously established. In the current study, basal ERK phosphorylation was similar in WT and MIF −/− splenocytes. In response to antigen stimulation however, WT splenocytes exhibited increased ERK phosphorylation, but this was not observed in MIF −/− splenocytes. This was confirmed by flow cytometric analysis of antigen-stimulated T cells, which demonstrated increased ERK phosphorylation in WT but not MIF −/− CD4+ T cells. Impairment of antigen-induced ERK phosphorylation is a potential mechanism for the observed down-regulation of T cell activation in MIF −/− mice.
The importance of the PI3 kinase pathway to joint inflammation has been reported [36] and the PI3 kinase/AKT pathway has shown to be responsive to MIF in other systems [25,29]. In the current study, however, P-AKT, a downstream effector kinase in the PI3 kinase pathway, was not induced after antigen stimulation, and was not different in WT compared with MIF −/− mice. Utilization of signalling pathways is not uniform in all cell types, and the current findings do not suggest MIF effects on the PI3 kinase pathway are central to its effect on T cell activation.
The mechanism of action of MIF modulation of the Th1 immune response is not completely defined. MIF has been shown to modulate expression of key molecules, including MHC class II, ICAM-1, B7-1, B7-2, CD40 and CD40L, in adaptive immune responses in the mouse [21]. Besides interaction with antigen-presenting cells (APCs), successful immune responsiveness requiring T cell activation also requires integrin activation [37,38]. In the current study however, there was no difference observed in both basal and antigen stimulated CD54 (ICAM-1) and CD25 expression of CD4+ T cell from WT and MIF −/− mice suggesting a lack of effect of MIF on these molecules, at least in this setting. CD69 is an early activation marker present on T cells and appears unlikely to be relevant to MIF–mediated T cell activation as comparable expression of CD69 were observed in MIF −/− and WT mice.
An additional role for MIF in T cell homing to inflammatory sites has been reported [20–22]. A recent study by Bernhagen and others went further to suggest MIF as a noncognate ligand of CXC chemokine receptors, in that MIF was shown to induce T cell transmigration in a chemotactic process that was transduced through CXCR4 [22].
Elevated MIF levels have been reported in RA serum, synovial fluid and synovial tissues, with the latter correlating with disease activity [2,7]. Cellular sources of MIF in RA include macrophages, fibroblast-like synoviocytes (FLS), endothelial cells and to a lesser extent T cells [7]. T cells have a central role in the orchestration of the immune pathways that initiate inflammation and joint destruction in RA [39]. Despite this, little is known about the effects of MIF on T cell function in RA.
T cells are prominent in cellular infiltrates of RA synovium [40]. Small but physiologically relevant amounts of T cell cytokines such as IFN-γ and IL-17 are expressed in RA and are proposed to mediate immune responses, fibroblast activation and bone destruction [11]. IL-17 is an important T cell-derived cytokine (reviewed in [19]). Its cellular source in the mouse has only recently been identified and termed Th17 [18]. IL-17 interacts synergistically with TNF and to a lesser extent, IL-1 and this may be a possible mechanism by which T cell can directly influence inflammatory responses [11]. To date there is no known study on MIF and Th17 interaction. A recent study in experimental diabetes show MIF −/− mice were less susceptible to disease induction and this was associated with lower lymphocyte proliferation, adhesion and production of IL-23 in the spleen [41]. As IL-23 can enhance IL-17 production [42,43] it is possible that MIF can regulate IL-17 via its effects on IL-23.
Animal models of RA including collagen-induced arthritis, adjuvant arthritis and AIA are all initiated by an adaptive immune response in which T cells play a central role [12,13,44]. For example, IL-12 is pivotal in the differentiation of naïve T cells into IFN-γ-producing Th1 cells [45] and in AIA [12]. A cofactor role for MIF in the promotion of T cell priming has been suggested [14], but studies on the intracellular signalling mechanism of MIF in T cells MIF are absent. In a study by Kitaichi and others, anti-MIF treatment was found to decrease antigen-specific responses of IFN-γ- and IL-4-producing T cells, related to blockade of signalling via T cell receptor and not via IL-2 receptor [33]. We have reported impaired MAP kinase activation in response to IL-1 and TNF in MIF-deficient fibroblasts [46]. In the current study, we demonstrate that diminished arthritis severity and T cell activation in MIF −/− mice is associated with impaired antigen-induced ERK MAP kinase phosphorylation. This suggests the ERK pathway is utilized by MIF in exerting its positive effects on T cell activation.
Acknowledgments
We are grateful to Jin Xue, Georgia Milenkovski, and Pamela Hall for technical assistance, Paul Hutchinson and James Ngui for flow cytometric analysis.
References
- 1.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 2.Morand EF, Leech M, Weedon H, Metz C, Bucala R, Smith MD. Macrophage migration inhibitory factor in rheumatoid arthritis: clinical correlations. Rheumatology (Oxford) 2002;41:558–62. doi: 10.1093/rheumatology/41.5.558. [DOI] [PubMed] [Google Scholar]
- 3.Radstake TR, Sweep FC, Welsing P, et al. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52:3020–9. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 4.Baugh JA, Chitnis S, Donnelly SC, et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3:170–6. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 5.Lacey D, Sampey A, Mitchell R, et al. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–9. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- 6.Leech M, Lacey D, Xue JR, et al. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003;48:1881–9. doi: 10.1002/art.11165. [DOI] [PubMed] [Google Scholar]
- 7.Leech M, Metz C, Hall P, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42:1601–8. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 8.Onodera S, Nishihira J, Koyama Y, et al. Macrophage migration inhibitory factor up-regulates the expression of interleukin-8 messenger RNA in synovial fibroblasts of rheumatoid arthritis patients: common transcriptional regulatory mechanism between interleukin-8 and interleukin-1beta. Arthritis Rheum. 2004;50:1437–47. doi: 10.1002/art.20190. [DOI] [PubMed] [Google Scholar]
- 9.Sampey AV, Hall PH, Mitchell RA, Metz CN, Morand EF. Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum. 2001;44:1273–80. doi: 10.1002/1529-0131(200106)44:6<1273::AID-ART219>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Santos LL, Lacey D, Yang Y, Leech M, Morand EF. Activation of synovial cell p38 MAP kinase by macrophage migration inhibitory factor. J Rheumatol. 2004;31:1038–43. [PubMed] [Google Scholar]
- 11.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–14. [PubMed] [Google Scholar]
- 12.Santos LL, Milenkovski GP, Hall PH, et al. IL-18 is redundant in T-cell responses and in joint inflammation in antigen-induced arthritis. Immunol Cell Biol. 2006;84:166–73. doi: 10.1111/j.1440-1711.2005.01406.x. [DOI] [PubMed] [Google Scholar]
- 13.Williams RO, Mason LJ, Feldmann M, Maini RN. Synergy between anti-CD4 and anti-tumor necrosis factor in the amelioration of established collagen-induced arthritis. Proc Natl Acad Sci USA. 1994;91:2762–6. doi: 10.1073/pnas.91.7.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bacher M, Metz CN, Calandra T, et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA. 1996;93:7849–54. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernhagen J, Bacher M, Calandra T, et al. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med. 1996;183:277–82. doi: 10.1084/jem.183.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos L, Hall P, Metz C, Bucala R, Morand EF. Role of macrophage migration inhibitory factor (MIF) in murine antigen–induced arthritis: interaction with glucocorticoids. Clin Exp Immunol. 2001;123:309–14. doi: 10.1046/j.1365-2249.2001.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Huang X, Wolters PJ, et al. Cutting edge: deficiency of macrophage migration inhibitory factor impairs murine airway allergic responses. J Immunol. 2006;177:5779–84. doi: 10.4049/jimmunol.177.9.5779. [DOI] [PubMed] [Google Scholar]
- 18.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–56. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 19.Miossec P. Interleukin-17 in fashion, at last: ten years after its description, its cellular source has been identified. Arthritis Rheum. 2007;56:2111–15. doi: 10.1002/art.22733. [DOI] [PubMed] [Google Scholar]
- 20.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J Immunol. 2003;170:1274–82. doi: 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- 21.Stavitsky AB, Xianli J. In vitro and in vivo regulation by macrophage migration inhibitory factor (MIF) of expression of MHC-II, costimulatory, adhesion, receptor, and cytokine molecules. Cell Immunol. 2002;217:95–104. doi: 10.1016/s0008-8749(02)00516-6. [DOI] [PubMed] [Google Scholar]
- 22.Bernhagen J, Krohn R, Lue H, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 23.Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414:920–4. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell RA, Liao H, Chesney J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA. 2002;99:345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amin MA, Haas CS, Zhu K, et al. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood. 2006;107:2252–61. doi: 10.1182/blood-2005-05-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu X, Lin SG, Huang XR, et al. Macrophage migration inhibitory factor induces MMP-9 expression in macrophages via the MEK-ERK MAP kinase pathway. J Interferon Cytokine Res. 2007;27:103–9. doi: 10.1089/jir.2006.0054. [DOI] [PubMed] [Google Scholar]
- 27.Powell ND, Papenfuss TL, McClain MA, et al. Cutting edge: macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5611–14. doi: 10.4049/jimmunol.175.9.5611. [DOI] [PubMed] [Google Scholar]
- 28.Yang N, Nikolic-Paterson DJ, Ng YY, et al. Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): potential role of MIF in regulating glucocorticoid production. Mol Med. 1998;4:413–24. [PMC free article] [PubMed] [Google Scholar]
- 29.Amin MA, Volpert OV, Woods JM, Kumar P, Harlow LA, Koch AE. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res. 2003;93:321–9. doi: 10.1161/01.RES.0000087641.56024.DA. [DOI] [PubMed] [Google Scholar]
- 30.Lue H, Thiele M, Franz J, et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26:5046–59. doi: 10.1038/sj.onc.1210318. [DOI] [PubMed] [Google Scholar]
- 31.Fingerle-Rowson G, Petrenko O, Metz CN, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 33.Kitaichi N, Ogasawara K, Iwabuchi K, et al. Different influence of macrophage migration inhibitory factor (MIF) in signal transduction pathway of various T cell subsets. Immunobiology. 2000;201:356–67. doi: 10.1016/s0171-2985(00)80090-x. [DOI] [PubMed] [Google Scholar]
- 34.Miossec P, van den Berg W. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum. 1997;40:2105–15. doi: 10.1002/art.1780401203. [DOI] [PubMed] [Google Scholar]
- 35.Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest. 2004;33:109–42. doi: 10.1081/imm-120030732. [DOI] [PubMed] [Google Scholar]
- 36.Camps M, Ruckle T, Ji H, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–43. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 37.Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:6327–33. doi: 10.4049/jimmunol.175.10.6327. [DOI] [PubMed] [Google Scholar]
- 38.Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr Opin Immunol. 2005;17:251–8. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Tran CN, Lundy SK. Fox DA. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology. 2005;12:183–9. doi: 10.1016/j.pathophys.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burger D, Dayer JM. The role of human T-lymphocyte-monocyte contact in inflammation and tissue destruction. Arthritis Res. 2002;4(Suppl. 3):S169–76. doi: 10.1186/ar558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stosic-Grujicic S, Stojanovic I, Maksimovic-Ivanic D, et al. Macrophage migration inhibitory factor (MIF) is necessary for progression of autoimmune diabetes mellitus. J Cell Physiol. 2007 doi: 10.1002/jcp.21346. on line. [DOI] [PubMed] [Google Scholar]
- 42.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 43.Hoeve MA, Savage ND, de Boer T, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–70. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 44.Sakaguchi S, Sakaguchi N. Animal models of arthritis caused by systemic alteration of the immune system. Curr Opin Immunol. 2005;17:589–94. doi: 10.1016/j.coi.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 45.O'Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–50. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 46.Toh ML, Aeberli D, Lacey D, et al. Regulation of IL-1 and TNF receptor expression and function by endogenous MIF. J Immunol. 2006;177:4818–25. doi: 10.4049/jimmunol.177.7.4818. [DOI] [PubMed] [Google Scholar]