

Abstract
Regulatory T cells (Tregs) may inhibit immunity against cancer. Induction and expansion of Tregs in the immunosuppressive microenvironment created by a growing tumour appear to be one of the mechanisms by which it can evade host defence. We studied the impact of CD25+ Tregs in a B cell lymphoma model in which Rag2–/– mice received adoptive transfer of wild-type spleen cells with or without CD25+ cells, and concurrently subcutaneous inoculation of the B cell lymphoma cell line A20. We also examined the effect of engaging the glucocorticoid-induced tumour necrosis factor receptor (GITR) − an approach reported previously to abrogate the suppressive effects of Tregs. Mice that received spleen cells depleted of CD25+ Tregs showed significantly slower tumour growth and increased survival compared with mice that received unsorted spleen cells. The Treg-depleted group also had significantly more CD8+ T cells infiltrating the tumours and higher levels of serum immunoglobulin G subclasses. The anti-GITR treatment had no significant effect on tumour growth, survival or immunoglobulin production. In the CD25-depleted group four of 10 mice developed clinical signs of autoimmunity, in contrast to none in the non-depleted group. Forkhead box P3+ T cells were found in tumour-draining lymph nodes in mice in the CD25-depleted group, suggesting an in vivo induction or expansion of rare transferred donor Tregs. Thus, our study showed that removal of CD25+ Tregs enhanced anti-tumour immunity against local growth of a B cell lymphoma and that induction or expansion of Tregs could be one mechanism by which the growing tumour evades immune surveillance.
Keywords: anti-tumour immunity, GITR, lymphoma, regulatory T cells
Introduction
Host resistance against cancer includes both innate and adaptive components of the immune system. In humans, however, anti-tumour immunity is often inefficient and short-lived [1]. Developing cancers generate a growth-promoting and immunosuppressive microenvironment mediated by apoptotic cells and production of regulatory factors including interleukin (IL)-10, transforming growth factor (TGF)-β, vascular endothelial growth factor and prostaglandin E2, enzymes such as indoleamine 2,3-dioxygenase and intrinsic down-regulation of major histocompatibility complex molecules on tumour cells [2–4].
Recent studies, both in experimental animals and humans, have documented a role for regulatory T cells (Tregs) in the suppression of anti-tumour immunity [5,6]. Tregs are generally divided into two major subsets, the best-studied being CD4+ CD25+ forkhead box P3+ (FoxP3+) naturally occurring Tregs[7]. The transcription factor FoxP3 is particularly vital to the function of these cells and is considered specific for Tregs in mice [8]. Regrettably, FoxP3 cannot be used for sorting of viable cells for experimental purposes because of its nuclear localization. Naturally occurring Tregs develop in the thymus during ontogeny and have been implicated primarily in inhibiting autoimmune reactions through direct cell contact [9]. The other major category of Tregs is the inducible or adaptive subset, characterized by production of immunosuppressive cytokines, particularly IL-10 and TGF-β[10]. This subset can develop in the periphery from naive or memory T cells stimulated under tolerogenic conditions [11]. Tregs expressing FoxP3 have been shown to be induced from CD4+ CD25− T cells in the periphery through ligation of the T cell receptor in the presence of TGF-β[12].
Cancer patients reportedly have increased numbers of Tregs in peripheral blood and malignant effusions and these cells accumulate in tumours [13–17]. Several human studies have reported that numerous tumour-associated Tregs may predict poor clinical outcome [15,18]. Removal of CD4+ CD25+ Tregs has been shown to enhance anti-tumour immunity in various mouse models [19,20]. Recently, Levitsky's group demonstrated in a systemic mouse model of the A20 B cell lymphoma that these malignant cells induced both expansion of naturally occurring CD25+ Tregs and de novo generation of inducible Tregs[21]. Although multiple immune evasion mechanisms have been described recently for this lymphoma cell line, induction of Tregs was shown to dominate [22].
Administration of an agonistic monoclonal antibody (mAb) to glucocorticoid-induced tumour necrosis factor receptor (GITR) in experimental fibrosarcoma [23] and melanoma [24] was found to elicit immune responses that eradicated established tumours. GITR is expressed constitutively on naturally occurring Tregs, but it is also up-regulated and may function as a co-stimulatory molecule on effector T cells [25]. The mechanisms responsible for the effects of engaging this molecule are debated [26].
Here, we investigated the effect of adoptive transfer of wild-type (WT) Balb/c spleen cells with or without predepletion of CD4+ CD25+ Tregs on local tumour growth of A20 B cell lymphoma cells and survival of immunodeficient Rag2−/− recipients. We also examined the potential effect of the anti-GITR mAb DTA-1 on anti-tumour immunity.
Materials and methods
Mice and tumour cell lines
Wild-type Balb/c and Rag2−/− mice on a Balb/c background were bred locally and maintained under pathogen-free conditions. A20 mouse B cell lymphoma cells were kindly donated by Dr Bjarne Bogen, Institute of Immunology, University of Oslo. All experiments were performed in accordance with institutional and international guidelines for laboratory animal research.
Adoptive cell transfer
Splenic mononuclear cells from WT Balb/c mice (n = 35) were obtained by mincing spleens and lysing the red blood cells. CD25+ cells were depleted by means of magnetic antibody cell sorting beads (Miltenyi Biotec, Bergisch Gladbach, Germany). After washing three times in phosphate-buffered saline (PBS), cells were pooled and transferred adoptively to Balb/c recipients deficient for Rag2. One group of mice (n = 10) received 3 × 107 unsorted WT spleen cells, whereas one group (n = 10) received 3 × 107 WT spleen cells depleted of the CD25+ cell population. One group (n = 5) of Rag2−/− mice served as controls and received no adoptive transfer or other treatment. Two groups received unsorted WT spleen cells and subsequently subcutaneous (s.c.) administration of 1 mg of affinity-purified rat anti-GITR mAb (clone DTA-1) (n = 10) or 1 mg of rat immunoglobulin G (IgG) isotype control (n = 5) as intraperitoneal (i.p.) injections on days 2, 8 and 14 after lymphoma cell injection. The anti-GITR mAb was a generous gift from Dr S. Sakaguchi, Kyoto, Japan.
Injection of tumour cells and tumour growth measurement
On day 1 after adoptive transfer of 3 × 107 unsorted or CD25-depleted WT spleen cells, all mice received a s.c. injection of 1·25 × 105 A20 cells in the neck region. This cell number had been determined as sufficient by titration in a pilot study. Mice were examined for malignant growth by measuring tumour size every 3–4 days, and killed when the tumour had reached a diameter of approximately 20 mm (because of rapid growth, some tumours were of larger size).
It is well established that immunocompromised mice receiving lymphoid cells depleted of CD25+ T cells may develop autoimmunity. Mice that developed symptoms of systemic disease, i.e. appeared ill even in the absence of a tumour, were also killed. Symptoms of general illness, including wasting and colitis (rectal bleeding, rectal prolapse and macroscopically inflamed mucosa) were considered signs of autoimmunity.
Flow-cytometric intracellular and cell surface analyses
Immunophenotyping of spleen cells was performed by surface staining with the following rat anti-mouse mAbs: anti-CD3 peridinin chlorophyll (clone 145–2C11), anti-CD4 fluorescein isothiocyanate (clone RM4-5) and anti-CD25 phycoerythrin (clone PC61) (all purchased from BD Pharmingen, San Jose, CA, USA). Intracellular staining for FoxP3 was performed with the FoxP3 Staining Set from Biosciences (Nordic Biosite, Täby, Sweden), including an allophycocyanin-conjugated anti-mouse/rat FoxP3 mAb (clone FJK-16 s). Fixation, permeabilization and staining were carried out according to the manufacturer's recommendations. Analysis of stained cells was performed with a FACSCalibur flowcytometer (BD Pharmingen).
Enzyme-linked immunosorbent assay
For determination of serum Ig concentrations, 96-well flat-bottomed plates (Costar, Corning, NY, USA) were coated overnight with Fc-specific anti-mouse IgG mAb (Sigma, Saint Louis, MO, USA). Plates were blocked for 1 h at room temperature with 0·5% (w/v) gelatin in PBS, washed in PBS containing 0·05% Tween 20 (PBS-Tween) and incubated with dilutions of mouse serum samples and standard mouse IgG (Sigma) for 2 h at room temperature. The wells were then incubated for 3 h with biotinylated rat anti-mouse IgG, IgG1 or IgG2, and the binding of these mAbs was revealed by peroxidase-conjugated streptavidin (R&D Systems, Minneapolis, MN, USA) with the appropriate substrate (tetramethylbenzidene peroxidase substrate; KPL, Gaithersburg, MD, USA).
Immunohistochemistry
Tissue specimens of tumours and draining lymph nodes were embedded in octreotide, snap-frozen in liquid nitrogen and stored at −70°C until sectioning. Cryosections were cut at 4 μm and acetone-fixed for 10 min at room temperature. For detection of CD8+ T cells and FoxP3+ T cells, tissue sections were incubated for 1 h with the following mAbs of mouse specificities: anti-CD3 (Armenian hamster IgG, clone 145–2C11; Nordic Biosite AB), anti-CD8 (rat IgG2a, clone 53–6·7; Pharmingen, San Diego, CA, USA) and anti-FoxP3 (rat IgG2a, clone FJK-16 s; Nordic Biosite AB). After a brief wash in PBS, sections were incubated for 1 h with combinations of the following secondary mAbs: Cy3-conjugated goat anti-Armenian hamster IgG; Cy3-conjugated donkey anti-rat IgG (H + l) (both from Jackson ImmunoResearch, Baltimore, PA, USA); and Alexa Fluor 488-conjugated goat anti-rat IgG (Molecular Probes, Eugene, OR, USA). Appropriate isotype- and concentration-matched control reagents ensured immunostaining specificity.
For enumeration of CD8+ T cells, slides were examined blindly by the same investigator (I. H.). CD3+ CD8+ cells were counted in an average of 68 fields (range 36–93) from each sample at × 200 magnification.
Statistics
spss 12·0.1 for Windows (Chicago, IL, USA) and GraphPad Prism 4 (San Diego, CA, USA) were used for the statistical analyses. Differences and correlations between groups were analysed by the Mann–Whitney two-tailed test and Spearman's rank correlation coefficient respectively. Log-rank test with Kaplan–Meier curves was used for survival analysis.
Results
Depletion of CD25+ cells
Among the mouse WT spleen cells used for adoptive transfer, 6–10% of CD4+ T cells co-expressed the Treg markers CD25 and FoxP3 (Fig. 1a). After depletion of CD25+ cells by magnetic beads, less than 0·05% of CD4+ T cells were found to be double-positive for these markers (Fig. 1b), while approximately 1% were found to be FoxP3+ among the remaining CD4+ CD25– T cells. Although some of the latter may have regulatory activity [27], these results show that the vast majority of Tregs were depleted by this method.
Fig. 1.
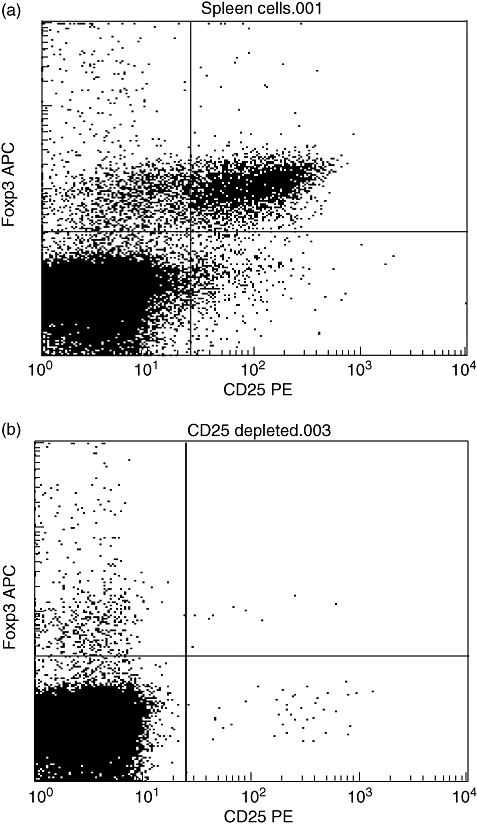
Flow-cytometric analysis of CD25 and FoxP3 expression in unsorted wild-type Balb/c spleen cells before (a) and after CD25-depletion (b). Plots are gated on CD3+ CD4+ cells and show a near complete removal of CD25+ FoxP3+ cells by the magnetic CD25 depletion procedure. Results are representative of at least five independent experiments. FoxP3, forkhead box P3; PE, phycoerythrin; APC, allophycocyanin.
Tumour growth and survival
Rag2−/− mice were injected with 1·25 × 105 A20 cells s.c. in the neck region 1 day after being transferred adoptively with 3 × 107 unsorted or CD25-depleted WT spleen cells. One group (n = 5) served as controls and received only tumour cells but no adoptive transfer of WT spleen cells. Solid tumours began to appear between day 14 and day 19 after inoculation. The mice were killed when the tumour was at least 20 mm in diameter as measured with a caliper.
The mice that had received unsorted spleen cells did not differ from the controls with regard to tumour growth (Fig. 2a). All mice in the control group were killed because of large tumour size within day 37, and all mice in the group that had received unsorted spleen cells were killed by day 48 (Fig. 3). Conversely, the group that had received CD25-depleted spleen cells showed significantly smaller tumour sizes on days 19–31 compared with the group that had received unsorted spleen cells (Fig. 2a), although great variability was noted within the group (Fig. 2b). After day 31, statistical comparisons of tumour sizes could not be made because of the small numbers of remaining live mice.
Fig. 2.
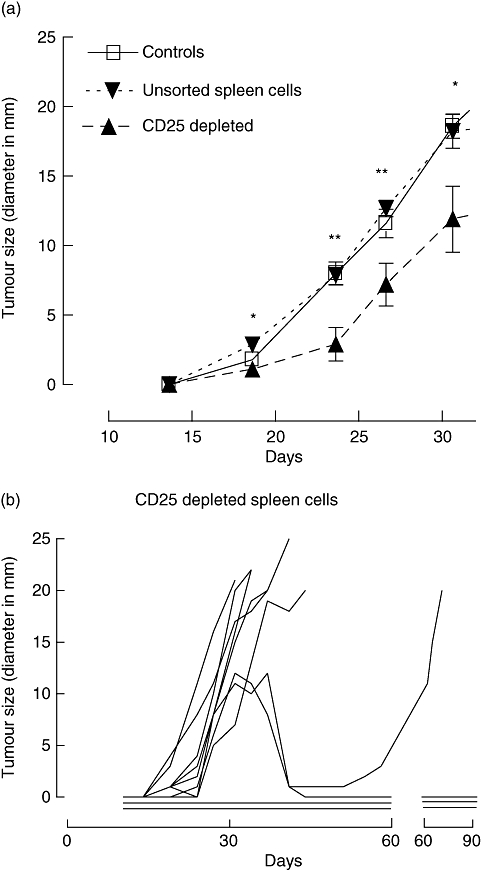
(a) Growth of B cell lymphoma tumours in control Rag2–/– mice that did not receive adoptive spleen-cell transfer (n = 5), and the two groups that received either unsorted wild-type spleen cells (n = 10) or CD25-depleted cells (n = 10) (*P < 0·05, **P < 0·01, Mann–Whitney test, two-tailed). Statistical comparisons are between the groups that received unsorted or CD25-depleted cells. Error bars indicate standard error of the mean. (b) Tumour growth shown as tumour size (diameter measured in mm) at various time-points in individual mice of the group that received CD25-depleted spleen cells. Each line represents one animal. Horizontal lines indicate no measurable tumour.
Fig. 3.
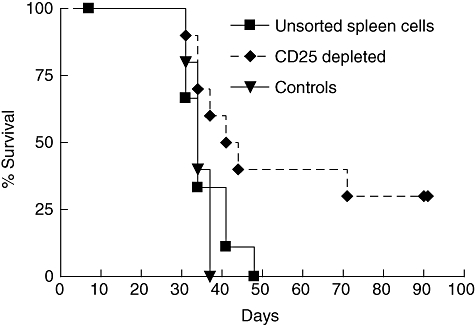
Survival in the same three groups of mice described in Fig. 2a. Kaplan–Meier plot shows significantly increased survival in mice that received CD25-depleted cells compared with mice that received unsorted wild-type spleen cells and controls (P = 0·036, log-rank test).
In the group that had received CD25-depleted WT spleen cells (n = 10), two mice never developed tumours and in two others the tumours regressed between day 31 and day 41 (Fig. 2b). In one of the latter the tumour recurred from day 55, whereas the other mouse remained tumour-free. Altogether, three mice in this group showed no signs of tumour growth when killed after 90 days. Notably, the mice that had received CD25-depleted cells showed significantly increased survival compared with those that had received unsorted cells; 71 days after tumour cell inoculation, four mice in the former group remained alive (log-rank test, P = 0·036, Fig. 3).
It is well known that adoptive transfer of WT spleen cells depleted of Tregs to immunodeficient mice leads to the development of autoimmune disease [28]. In line with this, all the four long-term survivors (i.e. those that lived for ≥ 71 days; Figs 2b and 3) in the CD25-depleted group developed symptoms of systemic disease, including wasting (one mouse) and colitis with rectal prolapse (three mice). Mice that had received unsorted spleen cells showed no clinical signs of autoimmune disease.
Administration of anti-GITR mAb
In vivo administration of agonistic mAbs to GITR has been shown to eradicate several different tumour types [23,24]. However, in this experimental mouse model, we observed no effect on tumour growth or overall animal survival by i.p. injection of 1 mg anti-GITR mAb compared with isotype-matched control mAb on days 2, 8 and 14 after adoptive lymphoma cell transfer (data not shown).
Measurement of humoral immunity
Rag2−/− mice lack productive rearrangement of Ig variable regions and T cell receptors; as a consequence, they show negligible serum Ig levels compared with WT mice. Adoptive transfer of spleen cells resulted in substantial Ig levels in all mice, as revealed in serum samples collected on the day they were killed. IgG1 and IgG2a were significantly higher (P = 0·046 and P = 0·036 respectively) in the mice that had received CD25-depleted cells compared with those that had received unsorted cells (Fig. 4a and b), and a similar trend was observed for total IgG (P = 0·055, Fig. 4c). However, there was no significant correlation between Ig levels and tumour size or survival in the group that received CD25-depleted cells (data not shown). The Ig production also did not appear to relate directly to development of autoimmunity, as mice that developed either wasting or colitis with rectal prolapse did not differ in Ig levels from those without such symptoms (P = 0·41, data not shown). No effect of anti-GITR treatment on Ig production was revealed (data not shown).
Fig. 4.
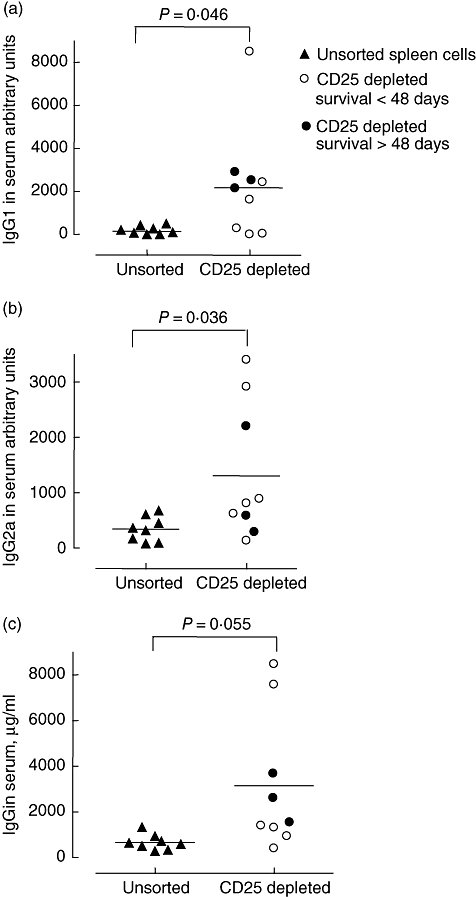
Serum levels of IgG1, IgG2a and total IgG in Rag2–/– mice that received unsorted wild-type spleen cells or CD25-depleted cells and showed a survival > 48 days or < 48 days. Serum was collected on the day the mice were killed and measurements were performed by enzyme-linked immunosorbent assay. Significance levels indicated for Mann–Whitney two-tailed test. Ig, immunoglobulin.
Immunophenotyping in situ
Traditionally, cytotoxic CD8+ T cells have been considered the main effectors of the adaptive immune system contributing to anti-tumour immunity [29], although the role of CD4+ T helper cells is becoming increasingly appreciated [30]. We wanted to examine the density of CD8+ T cells in tumour specimens from mice in the groups that had received unsorted or CD25-depleted WT spleen cells. In the latter group, intratumour CD8+ T cells were increased significantly compared with the former (P = 0·038, Fig. 5).
Fig. 5.
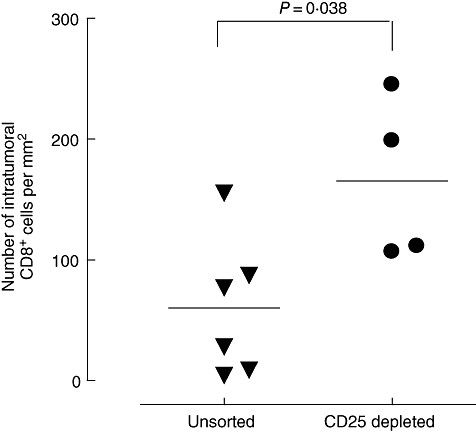
Number of tumour-infiltrating CD8+ T cells was increased significantly in Rag2–/– mice that had received CD25-depleted cells compared with unsorted wild-type spleen cells. Data show the number of CD8+ cells mm2 of tumour tissue (P = 0·038, Mann–Whitney two-tailed test).
FoxP3+ CD3+ cells (Fig. 6) were identified in tumour-draining lymph nodes from all the four examined mice in the group that had received unsorted spleen cells, and in three of the five mice that had received CD25-depleted cells. Unfortunately, because of very small size, lymph nodes were not obtained from the long-term survivors in the CD25-depleted group, which were without tumours. Because all the lymphoma cell recipients were Rag2−/− mice, they had no T cells of self-origin; and as shown in Fig. 1, the fraction of transferred WT splenic CD25+ FoxP3+ T cells after CD25 depletion was always less than 0·05%. Therefore, the FoxP3+ T cells found in mice that had received CD25-depleted cells were most probably a result of Treg-differentiation from adoptively transferred CD25− T cells.
Fig. 6.
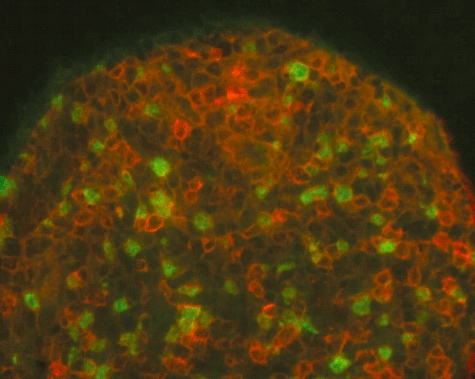
Immunohistochemistry of tumour-draining lymph node in Rag2–/– mice that had received CD25-depleted wild-type spleen cells. Paired immunofluorescence staining shows the presence of CD3+ (red) cells concurrently expressing nuclear FoxP3 (green); ×400 magnification.
Discussion
Here we show that depletion of CD25+ putative Tregs from adoptively transferred WT spleen cells increased survival and inhibited local tumour growth after s.c. injection of A20 lymphoma B cells in Rag2−/− mice compared with mice that received unsorted spleen cells. The former mice also revealed signs of autoimmune disease and elevated IgG levels, whereas no signs of autoimmunity and only low IgG levels were observed in the latter group. Together these findings suggested strongly that depletion of CD25+ T cells affords enhanced immune activation, resulting in improved anti-tumour immunity, sometimes accompanied by autoimmunity.
Growing malignant tumours, including the A20 B cell lymphoma [22], are known to generate an immunosuppressive environment, and induction of Tregs is considered to be one mechanism of immune evasion. In three of five examined mice that had received CD25-depleted spleen cells, lymph nodes draining the site of lymphoma cell injection were found to contain FoxP3+ T cells. This suggested that transferred CD25− T cells could differentiate to putative Tregs in the recipient mice or that a minor contaminating population of Tregs could expand after transfer. Such possibilities might explain, at least in part, that some mice in this group were unable to mount significant anti-tumour responses. Thus, decision-making in the immune system may be based on a delicate balance, with only a few T cells having the potential to tip the outcome in favour of either productive immunity or tolerance. Similar findings have been demonstrated by Levitsky's group in a systemic model of the A20 B cell lymphoma [21]. Here we show that Tregs also have an effect on local tumour growth of these malignant cells.
Upon adoptive T cell transfer to a lymphopenic environment, such as in Rag2−/− recipients, acute homeostatic proliferation takes place, which has been associated with immune activation, development of autoimmunity and enhanced anti-tumour immunity [31,32]. We observed no significant difference in tumour growth between the group that received unsorted WT spleen cells and the control Rag2−/− group without adoptive transfer. This showed that the mere presence of B and T cells, although activated through homeostatic proliferation in our experimental model, was insufficient to influence tumour growth.
Interestingly, we detected elevated numbers of CD8+ T cells accumulating in tumours in mice that had received CD25-depleted spleen cells. Because three of the long-term survivors in the CD25-depleted group had no local tumour growth at the time they were killed, it was impossible to correlate the number of intratumour CD8+ T cells with survival. Nevertheless, our findings suggested that cytotoxic CD8+ T cells represent one mechanism controlling tumour growth in the absence of CD4+ CD25+ Tregs.
Engagement of the Treg-associated GITR-molecule did not affect tumour growth or host survival in this mouse model, although the employed mAb has been shown to enhance tumour elimination in other cancer models, such as chemically induced sarcoma and melanomas [23,24]. GITR+ Tregs have been found within lymphomas, suggesting that this molecule might also play a role in such malignancies, but the mechanism behind the GITR-mediated anti-tumour effect is debated [26]. This effect may become apparent at different time-points of the carcinogenic process and depend on the type of cancer.
Serum Ig levels were increased significantly in mice that had received CD25-depleted WT spleen cells, implying enhanced humoral immunity after removal of CD4+ CD25+ Tregs. However, the possible contribution of antibodies to cancer resistance remains uncertain, being less well described than cell-mediated mechanisms. Nevertheless, the observed clinical effect of therapeutic mAbs, such as rituximab (anti-CD20) [33], suggests that humoral immunity could play a role in host resistance against malignant B cell lymphomas.
In conclusion, we showed that depletion of CD25+ putative Tregs results in local growth inhibition of a malignant B cell lymphoma without the support of tumour vaccination. Decreased tumour growth and increased survival were associated with immune stimulation with increased IgG levels and autoimmune disease. However, as shown by others [21], we found that FoxP3+ cells were induced or expanded in tumour-bearing mice, implying that development of Tregs in the recipient may enhance tumour tolerance instead of protective immunity, even in the face of therapeutic cancer vaccines [34]. This underscores the need to develop new adjuvants for cancer immunotherapy which reduce Treg-inducing activity to stimulate preferentially the effector arm of the adaptive immune system [35].
References
- 1.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Gajewski TF, Meng Y, Blank C, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 3.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–36. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad M, Rees RC, Ali SA. Escape from immunotherapy: possible mechanisms that influence tumor regression/progression. Cancer Immunol Immunother. 2004;53:844–54. doi: 10.1007/s00262-004-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–11. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 6.Wang RF. Immune suppression by tumor-specific CD4+ regulatory T-cells in cancer. Semin Cancer Biol. 2006;16:73–9. doi: 10.1016/j.semcancer.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 10.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–7. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 11.Akbar AN, Vukmanovic-Stejic M, Taams LS, Macallan DC. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat Rev Immunol. 2007;7:231–7. doi: 10.1038/nri2037. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-{beta} induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–12. [PubMed] [Google Scholar]
- 14.Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood. 2006;107:3639–46. doi: 10.1182/blood-2005-08-3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 16.Chen YQ, Shi HZ, Qin XJ, et al. CD4+CD25+ regulatory T lymphocytes in malignant pleural effusion. Am J Respir Crit Care Med. 2005;172:1434–9. doi: 10.1164/rccm.200504-588OC. [DOI] [PubMed] [Google Scholar]
- 17.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–8. [PubMed] [Google Scholar]
- 18.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–18. [PubMed] [Google Scholar]
- 20.Tanaka H, Tanaka J, Kjaergaard J, Shu S. Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother. 2002;25:207–17. doi: 10.1097/00002371-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Zhou G, Levitsky HI. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol. 2007;178:2155–62. doi: 10.4049/jimmunol.178.4.2155. [DOI] [PubMed] [Google Scholar]
- 22.Elpek KG, Lacelle C, Singh NP, Yolcu ES, Shirwan H. CD4+CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J Immunol. 2007;178:6840–8. doi: 10.4049/jimmunol.178.11.6840. [DOI] [PubMed] [Google Scholar]
- 23.Ko K, Yamazaki S, Nakamura K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–91. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen AD, Diab A, Perales MA, et al. Agonist anti-GITR antibody enhances vaccine-Induced CD8+ T-cell responses and tumor immunity. Cancer Res. 2006;66:4904–12. doi: 10.1158/0008-5472.CAN-05-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nocentini G, Riccardi C. GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur J Immunol. 2005;35:1016–22. doi: 10.1002/eji.200425818. [DOI] [PubMed] [Google Scholar]
- 26.Shevach EM, Stephens GL. The GITR–GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol. 2006;6:613–18. doi: 10.1038/nri1867. [DOI] [PubMed] [Google Scholar]
- 27.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 29.Emens LA. Roadmap to a better therapeutic tumor vaccine. Int Rev Immunol. 2006;25:415–43. doi: 10.1080/08830180600992423. [DOI] [PubMed] [Google Scholar]
- 30.Gerloni M, Zanetti M. CD4 T cells in tumor immunity. Springer Semin Immunopathol. 2005;27:37–48. doi: 10.1007/s00281-004-0193-z. [DOI] [PubMed] [Google Scholar]
- 31.Dummer W, Niethammer AG, Baccala R, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–92. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baccala R, Theofilopoulos AN. The new paradigm of T-cell homeostatic proliferation-induced autoimmunity. Trends Immunol. 2005;26:5–8. doi: 10.1016/j.it.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359–68. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 34.Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood. 2006;107:628–36. doi: 10.1182/blood-2005-07-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baecher-Allan C. Anderson DE. Immune regulation in tumor-bearing hosts. Curr Opin Immunol. 2006;18:214–19. doi: 10.1016/j.coi.2006.01.010. [DOI] [PubMed] [Google Scholar]