

Abstract
Thiol-containing drugs such as WR1065, the free thiol form of amifostine, have been shown to induce a delayed radioprotective effect in both malignant and non-malignant cells. In mammalian cells exposed to a dose as low as 40 μM WR1065, the redox-sensitive nuclear transcription factor κB (NFκB) is activated, leading to an elevation in the expression of the antioxidant gene manganese superoxide dismutase (SOD2) and a concomitant increase in active SOD2 enzyme levels that peaks 24 to 32 h later. Exposure of cells to ionizing radiation during the period of elevated SOD2 enzymatic activity results in an enhanced radiation resistance. This is seen as an increase in surviving fraction as determined by standard colony formation assays. To determine whether this delayed radioprotection can be maintained over a prolonged period in cells of either malignant or non-malignant origin, both human microvascular endothelial cells (HMEC) and SA-NH mouse sarcoma cells were grown to confluence and exposed to 40 μM WR1065 using three administration protocols: (1) daily drug exposure for 10 days followed each day by irradiation with 2 Gy; (2) drug exposure once every 48 h followed by irradiation with 2 Gy 48 h later for 14 days; and (3) drug exposure every 72 h followed by irradiation with 2 Gy 72 h later for 12 days. As a function of each experimental condition, cell numbers and associated SOD2 enzymatic activities were measured at the time of each irradiation. None of the treatment conditions were toxic to either HMEC or SA-NH cells. SOD2 activity was elevated 5.3- and 1.8-fold over background on average for HMEC exposed to 40 μM WR1065 every 24 or 48 h, respectively. Likewise, SOD2 activity was elevated in SA-NH mouse sarcoma cells 7.8- and 4.9-fold after daily exposure to WR1065 or exposure to WR1065 once every 48 h, respectively. Both HMEC and SA-NH cells exhibited enhanced radiation resistance that correlated with the increase in SOD2 activity. The average respective increases in cell survival were 1.33 ± 0.01 (SEM), 1.23 ± 0.01 and 1.04 ± 0.01 for HMEC exposed to WR1065 every 24, 48 and 72 h, respectively, and 1.27 ± 0.01, 1.18 ± 0.02 and 1.02 ± 0.02 for SA-NH cells exposed to WR1065 every 24, 48 and 72 h, respectively. Both the elevation in WR1065-induced SOD2 enzymatic activity and the corresponding increase in radiation resistance were completely inhibited in HMEC and SA-NH cells transfected with human or mouse SOD2 siRNA oligomers and irradiated 24 h later. These data demonstrate that a delayed radioprotective effect can be induced and maintained over a prolonged period in both non-malignant and malignant cells exposed to thiol-containing drugs such as WR1065. For non-malignant cells this represents a novel paradigm for radiation protection. The ability of WR1065 to induce a persistent elevated radiation resistance in malignant cells, however, suggests a new potential concern regarding the issue of tumor protection in patients exposed to thiol-containing drugs.
INTRODUCTION
Mitochondria have long been recognized as playing an important role in cellular responses to ionizing radiation. Processes leading to apoptosis are known to be induced by ionizing radiation, including the release of cytochrome c and the activation of caspase 3 that leads to PARP cleavage and subsequent DNA strand breaks (1–3). The mitochondria as centers of respiration are naturally subject to the damaging events of reactive oxygen species (ROS). In particular, the mitochondria are susceptible to damage from the highly reactive superoxide, which is released as a consequence of Complex I and Complex III activities in normal respiration, as well as by ROS generated by ionizing radiation and other stress-inducing agents. Once formed, superoxide anion can participate in a number of pathways, including the induction of damage (4) and survival pathways (5), as well as growth-related signal transduction processes (6). At the biochemical level, superoxide can inactivate iron-sulfur cluster-containing enzymes, resulting in the liberation of iron into cells, which can then facilitate Fenton chemistry and generate highly reactive hydroxyl radicals as well as initiate lipid peroxidation of polyunsaturated fatty acids. Superoxide can also react with carbonyl compounds and halogenated carbons to create toxic peroxy radicals and interact with nitric oxide (NO) to form the highly damaging and reactive unstable anion and oxidant peroxynitrite ONOO− (7, 8).
Manganese superoxide dismutase (SOD2) is a mitochondrial matrix protein that serves as the primary mitochondrial defense against superoxide formation. Its primary function is to facilitate the dismutation of two molecules of superoxide anion into water and hydrogen peroxide. In the presence of glutathione peroxidase or catalase, hydrogen peroxide is then converted into water and oxygen. SOD2 is nuclear-encoded and has been shown to be highly protective not only against superoxide produced by normal respiratory processes but also against exogenously induced ROS production by agents such as ionizing radiation that can lead to cell lethality (1, 2, 9, 10). Because SOD2 is encoded by a nuclear gene, a signaling pathway(s) must exist to activate the nuclear transcription of SOD2 in response to a buildup of superoxide within the mitochondria, resulting in the production and translocation of SOD2 back to the mitochondria to facilitate the detoxification of the ROS. The SOD2 gene contains binding motifs for a number of transcription factors that include activator proteins 1 (AP1) and 2 (AP2), specificity protein 1 (Sp1), and adenosine 3′,5′-cyclic monophosphate-regulator element-binding factor (CREB), which are all involved in its constitutive expression (11, 12). Two families of transcription factors have been identified, however, that regulate the inducible rather than the constitutive expression of SOD2. These are nuclear transcription factor κB (NFκB) and the Forkhead Box O3a transcription factor (FOXO3a, FKHRL1) (13–15). The protein kinase B (PKB/c-Akt)-regulated FOXO3a has been shown to regulate the activation of SOD2 activity in quiescent cells in response to a buildup of the oxidant hydrogen peroxide (15, 16). Presumably, oxidative stress induced during the initiation of the processes that lead to apoptosis can lead to an up-regulation of FOXO3a at the transcriptional level facilitating the activation of SOD2 gene expression and the buildup of active SOD2 enzyme (15, 17). A similar signaling mechanism has been identified for the induction of SOD2 activity after the buildup of ROS in proliferating cells through NFκB activation (4). It has been demonstrated that an elevation in mitochondrial ROS can activate a signal relay pathway in which serine/threonine protein kinase D (PKD) promotes NFκB activation and its translocation to the nucleus followed by increased SOD2 transcription (5). Thus mitochondrial-associated oxidative stress, whether induced by endogenous metabolically related or exogenously administered insults, can initiate a signaling process that leads to the stimulation or induction of the antioxidant gene SOD2 whose protein will facilitate the detoxification of mitochondrial ROS and lead to enhanced cell survival.
The activation of nuclear localized genes coding antioxidant proteins in cells as a result of mitochondrial-associated oxidative stresses is an example of an adaptive survival response. Presumably, if such antioxidant enzyme levels could be elevated in cells prior to exposure to oxidative stress-inducing agents such as ionizing radiation, cellular survival responses would also be enhanced due to their increased ability to minimize ROS damage. SOD2 is an effective antioxidant enzyme that can, when present at elevated levels at the time of irradiation, exhibit radioprotective properties (18–20). Using SOD2 transgene constructs and adenovirus vectors, SOD2 therapy has been demonstrated to be effective in enhancing radiation resistance under in vivo conditions (19, 20). Furthermore, it has been clearly demonstrated that mitochondrial localization of the SOD2 transgene product is a requirement for protection of cells against radiation-induced cell killing, an indication of both the importance of the mitochondria as a target for cellular responses to radiation exposure and the effectiveness of SOD2 as a radioprotector (9). SOD2 and SOD mimetics have now become the focus of research into the development of novel radioprotective agents (21).
We earlier identified a novel radiation protection paradigm known as the thiol-induced SOD2-mediated “delayed radioprotective effect” (22–25). This radioprotection paradigm is based on earlier observations that the thiol reducing agents N-acetylcysteine, dithiothreitol and 2-mercaptoethanol had the ability to activate NFκB and subsequently elevate SOD2 gene expression (26). Additional reports followed indicating that the thiol-containing drugs oltipraz, captopril, mesna and WR1065, the active free thiol form of amifostine, could also elevate SOD2 gene expression through their ability to activate NFκB in exposed malignant and non-malignant cells of human and mouse origin (27, 28). After exposure of cells to these thiols, activation of NFκB occurred within 30 min and was followed by elevated SOD2 gene expression and a 10- to 20-fold buildup of active SOD2 enzyme that peaked about 24 to 30 h later (22–28). Cells irradiated at times of maximum SOD2 buildup after thiol exposure exhibited a 20 to 40% increase in resistance to ionizing radiation: hence the term delayed radioprotective effect (22–25). This effect could be inhibited by treating cells with the NFκB inhibitors BAY11-7082 or Helenalin, by stably transfecting them with a mutant IκBα gene under the control of a CMV promoter to prevent NFκB activation after thiol exposure (23, 24), or by transfecting cells with SOD2 siRNA to prevent thiol-induced elevation of SOD2 enzyme levels and activity (25). The proposed mechanism of action underlying the delayed radioprotective effect is that reducing agents having an active thiol moiety can reduce cysteine residues on the p50 and p65 subunits of NFκB that results in activation and enhanced binding of this transcription factor to the NFκB binding site(s) of SOD2 (29). This in turn induces SOD2 gene expression, resulting in the buildup and translocation of active SOD2 enzyme to the mitochondria. The time course of this process ranges from 24 to 30 h. Cells exposed to ionizing radiation at this time exhibit an enhanced radiation resistance as seen in a 20 to 40% increase in surviving fraction as determined by standard colony-forming assay methods (22–25).
The purpose of the present study was to determine whether the delayed radioprotective effect is only transitory or whether it can be maintained over a prolonged period as a function of multiple exposures of cells to low doses of a thiol-containing drug. Furthermore, it has been suggested that the intracellular redox system of tumor compared to normal cells is compromised, rendering malignant cells more susceptible to redox imbalance and stress due to increased SOD2 activity (30). Presumably elevated SOD2 activity might enhance the production of hydrogen peroxide as a product of superoxide dismutation, which in tumor compared to normal cells could build up to toxic levels because of insufficient catalase or glutathione peroxidase activities. To address these issues, two mammalian cell systems were used. Both human microvascular endothelial cells (HMEC) as a representative of a non-malignant cell type and SA-NH cells derived from a murine sarcoma were exposed to 40 μM of WR1065 every 24, 48 or 72 h for up to 14 days and assessed at these times with regard to their clonogenic survival after exposure to 2 Gy of ionizing radiation as a function of the elevation in their respective SOD2 enzyme activities compared to matched control cell populations.
METHODS AND MATERIALS
Cells and Culture Conditions
Human microvascular endothelial cells (HMEC) were obtained from the Biologic Products Branch, Centers for Disease Control, Atlanta, GA, and were maintained in endothelial basal medium MCDB131 (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 15% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 10 ng/ml epidermal growth factor (Invitrogen Life Technologies), 1 μg/ml hydrocortisone (Sigma, St. Louis, MO), penicillin and streptomycin (Invitrogen Life Technologies). New cell stocks were used every 3 months. SA-NH cells derived from an SA-NH murine sarcoma tumor and adapted for in vitro growth in culture medium were obtained from the laboratory of Dr. Luka Milas, Department of Experimental Radiation Oncology, M.D. Anderson Cancer Center, Houston, TX. SA-NH cells were maintained in modified McCoy’s 5A medium (Invitrogen Life Technologies) supplemented with 10% FBS (Atlanta Biologicals), penicillin and streptomycin (Invitrogen Life Technologies). Both cell lines were subcultured weekly using 0.25% trypsin and 1 mM EDTA (Invitrogen Life Technologies) and were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C. To be consistent with earlier studies regarding the growth conditions and the induction of an acute delayed radioprotective effect, cells were grown to confluence as fed cultures in all experiments (22–25) and refed every 3 days. This approach also had the advantage of minimizing the stress and the chance of contamination of cell cultures that might result from multiple trypsinizations and reseedings that would be required to maintain a 10–14-day continuous drug exposure if cells were also required to be constantly in an exponential phase of growth.
Drug Treatment Conditions
WR1065 (2-[{aminopropyl}amino]ethanethiol), the active thiol metabolite of amifostine, was supplied by the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment, National Cancer Institute. Immediately prior to use, the drug was dissolved in phosphate-buffered saline (PBS) at a concentration of 1 M and was then passed through a 0.2-μm syringe filter for sterilization. Cells were exposed to WR1065 at a final concentration of 40 μM.
Irradiation Conditions and Survival Assay
HMEC and SA-NH cells were irradiated at room temperature using a Philips X-ray generator operating at 250 kVp and 15 mA at a dose rate of 1.65 Gy/min. Three different experimental protocols were followed to evaluate the effects of administering multiple doses of WR1065 at a concentration of 40 μM on the radiation response of HMEC and SA-NH cells. Each dose of WR1065 was administered to fed confluent cultures either once every 24 h with exposure of cells to 2 Gy 24 h later for a total of 10 days, once every 48 h with exposure to 2 Gy 48 h later for a total of 14 days, or once every 72 h with exposure to 2 Gy 72 h later for a total of 12 days. The radiation responses of cells from each of the drug treatment protocols were compared to matched controls irradiated in the absence of WR1065 treatment. After irradiation, regardless of the drug treatment protocol followed, cells were trypsinized, counted and plated at appropriate numbers to give rise to about 100 colonies per dish, five dishes per experimental point. At 21 or 10 days later for HMEC and SA-NH cells, respectively, plates were stained with 20% crystal violet and colonies were counted. Each experiment was repeated three times. Pairwise comparisons of surviving fractions, determined from 15 plates of cells at each experimental point, at 2 Gy between selected experimental conditions were performed using a Student’s two-tailed t test.
Cell Toxicity Assay
The effects of the various drug treatment protocols on cell numbers as a function of duration of treatment were determined. Total cell numbers for each culture plate were obtained each day for cells exposed to 40 μM WR1065 every 24, 48 or 72 h and were compared to matched controls not exposed to the drug. Fed confluent cell cultures growing in 60-mm dishes were trypsinized and suspended in 5 ml growth medium, and total cell numbers determined by counting with a hemocytometer. Potential drug-induced toxic effects on cells were determined from the ratio of cell counts for drug-treated cells relative to untreated cells. From the resulting data, ratios of cell counts as a function of time of treatment were plotted, best fit regression lines drawn, and slope and correlation coefficients calculated.
SOD2 siRNA Transfection
HMEC and SA-NH cells grown to confluence in 100-mm dishes were transfected with 100 nM of the appropriate SOD2 siRNA. Small interfering RNA sequences for human and mouse were (5′ AAT GCT ACA ATA GAG CAG CTT 3′) and (5′ AAG GAA CAA CAG GCC TTA TTC 3′), respectively. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. Briefly, siRNA oligomer (100 nM final concentration) was diluted in 1.5 ml serum-free medium. Thirty microliters of Lipofectamine 2000 reagent was diluted in 1.5 ml of serum-free medium, mixed gently, and then incubated for 5 min at room temperature. The siRNA oligomer was then combined with the diluted Lipofectamine 2000, gently mixed, and incubated for 20 min at room temperature to allow complex formation. During this incubation the growth medium was aspirated from the dishes and the cells were washed with PBS at 37°C to remove traces of serum. The siRNA-Lipofectamine 2000 complexes were added to the dishes and gently swirled to ensure a uniform distribution. The cells were incubated for 24 h with the transfection complexes under their normal growth conditions of 37°C and 95% air/5% CO2. The cells were then washed with PBS at 37°C and fresh complete growth medium was added. Parallel Western blot experiments were performed using sham-, scrambled sequence SOD2 oligomer-, and SOD2 siRNA oligomer-transfected cells to demonstrate the specificity and functionality of the SOD2 siRNA. No difference in SOD2 levels was observed for sham-transfected or scrambled SOD2 siRNA-transfected cells, whereas a knockdown of SOD2 levels was observed in the SOD2 siRNA-transfected cells (data not shown). Surviving fractions were determined for sham-transfected cells and cells transfected with SOD2 siRNA treated with WR1065 and irradiated as described above.
SOD2 Enzyme Activity Assay
SOD2 activity was measured in both HMEC and SA-NH cells using the Superoxide Dismutase Assay Kit from Trevigen (Gaithersburg, MD) following the manufacturer’s instructions. Briefly, cells grown to confluence were trypsinized and counted. Five million cells were lysed with 500 μl of lysis solution. The resulting suspension was centrifuged at 14,000g for 5 min at 4°C and then transferred to a clean 1.5-ml tube. Activity was determined at room temperature using a colorimetric assay based on the ability of SOD to form H2O2 from superoxide radicals generated by an exogenous reaction involving xanthine and xanthine oxidase which converts nitroblue tetrazolium (NBT) to NBT-diformazan. NBT-diformazan absorbs light at 550 nm. The extent of reduction in the appearance of NBT-diformazan is a measure of SOD activity. Activity was measured using 400 μl of cell suspension in the presence of 5 mM sodium cyanide (NaCN) (Sigma) to inhibit copper/zinc SOD (SOD1) activity. Absorbance changes were recorded for 5 min, and the percentage inhibition was calculated using an SOD standard curve generated with known concentrations of purified SOD supplied with the kit.
RESULTS
Radiation Response after Multiple Treatments
The effects of multiple WR1065 treatments on the induction and maintenance of the delayed radioprotective effect was demonstrated using both non-malignant HMEC and murine tumor-derived SA-NH cells. A concentration of 40 μM WR1065 was chosen for the multiple treatments because this dose is sufficient to activate NFκB and induce SOD2 gene expression and the delayed radioprotective effect while still being too low to induce a direct or “immediate” radioprotective effect on cells (22–24). The effects of multiple treatments with WR1065 on the response of HMEC to 2-Gy doses of ionizing radiation are presented in Fig. 1. WR1065 administration once every 24 or 48 h significantly enhanced the resistance of HMEC to the cytotoxic effects of ionizing radiation at each time for the duration of each treatment, a demonstration of a persistent thiol-induced delayed radioprotective effect (see Fig. 1a and b). By taking a ratio of the resulting surviving fractions of WR1065-exposed and corresponding untreated cells at each time, protection factors along with P values calculated using a two-tailed t test were determined. Cumulative protection factors representing the average of all individual protection factors for each treatment protocol were also determined. Average protection factors of 1.33 (P < 0.001) and 1.23 (P = 0.001) were determined for the 24- and 48-h WR1065 treatments, respectively. Cells exposed at 72-h intervals to WR1065, however, exhibited no such increase in radiation resistance (protection factors of 1.04, P = 0.567; see Fig. 1c).
FIG. 1.
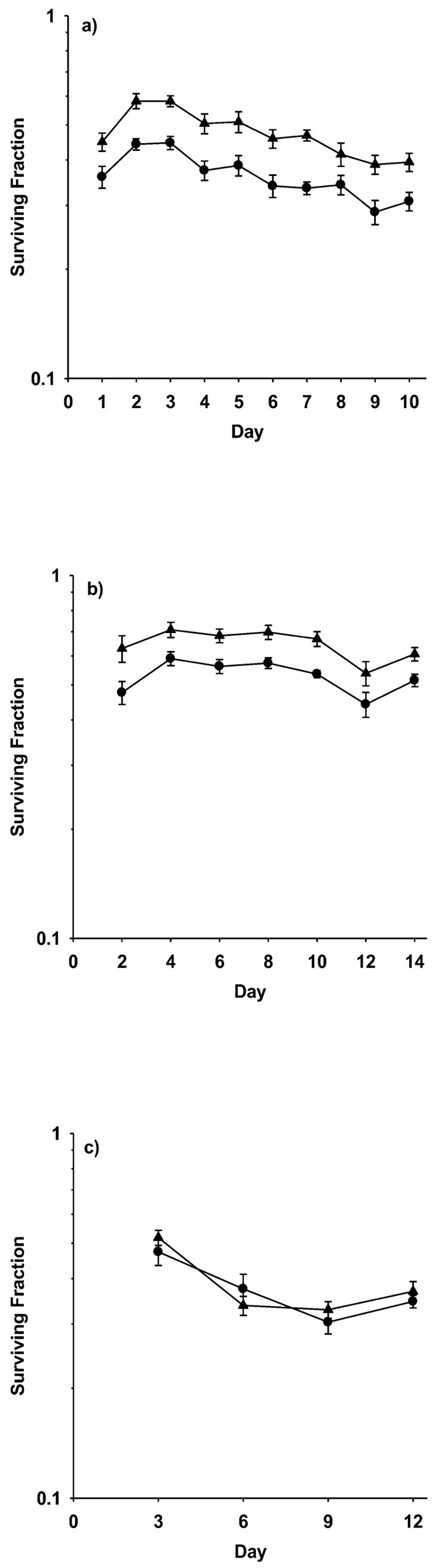
Survival of HMEC grown and maintained as fed confluent cultures exposed to 2 Gy of 250 kVp X rays as a function of exposure to 40 μM WR1065 under three different drug administration protocols: (panel a) WR1065 administered at 24-h intervals with irradiation 24 h later, for a total time of 10 days; (panel b) WR1065 administered at 48-h intervals with irradiation 48 h later, for a total time of 14 days; and (panel c) WR1065 administered at 72-h intervals with irradiation 72 h later, for a total time of 12 days. Survival of WR1065-treated cells (▲) for each interval is compared to corresponding matched controls irradiated in the absence of WR1065 treatment (●). Each bar represents the mean ± SEM of colony counts from 15 plates obtained from three experiments.
SOD2 activities were also monitored in HMEC as a function of WR1065 administration. Individual changes in SOD2 activity as a function of each WR1065 treatment protocol were determined and an average change in SOD2 activity relative to untreated controls was calculated. Consistent with the increases in radiation resistance observed, administration of WR1065 every 24 and 48 h resulted in average increases in SOD2 activities of 5.27- (P = 0.002) and 1.85-fold (P < 0.001), respectively. These data are summarized in Table 1.
TABLE 1.
Magnitude of Delayed Radioprotection and Associated SOD2 Activity in HMEC after WR1065 Exposure
| Treatment | Mean surviving fraction ± SEMa | Protection factor ± SEM | P valueb | Mean SOD2 activity ± SEMc | Relative increase | P valued |
|---|---|---|---|---|---|---|
| 2 Gy | 0.36 ± 0.01 | 1.33 ± 0.01 | <0.001 | 0.048 ± 0.02 | 5.27 | 0.002 |
| 40 μM WR1065, 24 h – 2 Gy | 0.48 ± 0.01 | 0.163 ± 0.02 | ||||
| 2 Gy | 0.53 ± 0.01 | 1.23 ± 0.01 | <0.001 | 0.127 ± 0.01 | 1.85 | <0.001 |
| 40 μM WR1065, 48 h – 2 Gy | 0.65 ± 0.02 | 0.232 ± 0.01 | ||||
| 2 Gy | 0.37 ± 0.02 | 1.04 ± 0.01 | 0.567 | 0.142 ± 0.03 | 0.99 | 0.972 |
| 40 μM WR1065, 72 h – 2 Gy | 0.39 ± 0.02 | 0.140 ± 0.03 |
Average from three separate experiments for 24- and 48-h exposures, and two separate experiments for the 72-h exposure.
Comparison between cells irradiated with 2 Gy and cells treated with WR1065 and then irradiated 24 to 72 h later. P values were calculated using a two-tailed t test.
The average activity in U/106 cells was calculated from individual measurements over the entire treatment period.
Comparison between WR1065-treated cells and untreated control cells. P values were calculated using a two-tailed t test.
The data obtained for SA-NH cells are similar to those reported for HMEC. As shown in Fig. 2, both the daily and 48-h exposures of SA-NH cells to 40 μM WR1065 resulted in both a persistent elevation in resistance to radiation-induced cell killing and increased SOD2 enzymatic activity. Cumulative protection factors of 1.27 (P < 0.001) and 1.18 (P < 0.001) were obtained for cells exposed to WR1065 every 24 and 48 h, respectively. These elevated protection factors correlated with corresponding elevated average SOD2 activities, e.g., 7.75- and 4.88-fold increases after daily and 48-h exposures, respectively (see Table 2). Exposure of cells to WR1065 at 72-h intervals followed 72 h later with exposure to 2 Gy radiation failed to elicit an increase in cellular radiation resistance.
FIG. 2.
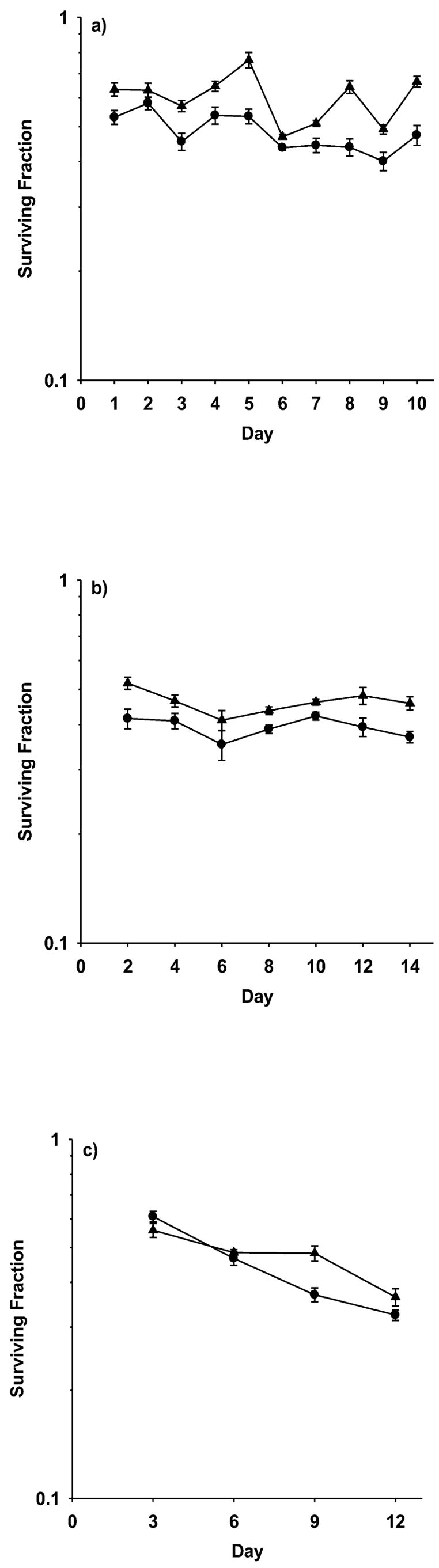
Survival of SA-NH murine tumor cells grown and maintained as fed confluent cultures exposed to 2 Gy of 250 kVp X rays as a function of exposure to 40 μM WR1065 under three different drug administration protocols: (panel a) WR1065 administered at 24-h intervals with irradiation 24 h later, for a total time of 10 days; (panel b) WR1065 administered at 48-h intervals with irradiation 48 h later, for a total time of 14 days; and (panel c) WR1065 administered at 72-h intervals with irradiation 72 h later, for a total time of 12 days. Survival of WR1065-treated cells (▲) for each interval is compared to corresponding matched controls irradiated in the absence of WR1065 treatment (●). Each bar represents the mean ± SEM of colony counts from 15 plates obtained from three experiments.
TABLE 2.
Magnitude of Delayed Radioprotection and Associated SOD2 Activity in SA-NH Cells after WR1065 Exposure
| Treatment | Mean surviving fraction ± SEMa | Protection factor ± SEM | P valueb | Mean SOD2 activity ± SEMc | Relative activity | P valued |
|---|---|---|---|---|---|---|
| 2 Gy | 0.48 ± 0.06 | 1.27 ± 0.01 | <0.001 | 0.013 ± 0.002 | 7.75 | 0.002 |
| 40 ±M WR1065, 24 h – 2 Gy | 0.61 ± 0.09 | 0.087 ± 0.03 | ||||
| 2 Gy | 0.39 ± 0.08 | 1.18 ± 0.02 | <0.001 | 0.015 ± 0.005 | 4.88 | 0.008 |
| 40 ±M WR1065, 48 h – 2 Gy | 0.46 ± 0.08 | 0.052 ± 0.02 | ||||
| 2 Gy | 0.47 ± 0.12 | 1.02 ± 0.02 | 0.152 | 0.029 ± 0.01 | 0.93 | 0.937 |
| 40 ±M WR1065, 78 h – 2 Gy | 0.48 ± 0.08 | 0.027 ± 0.01 |
Average from three separate experiments for 24- and 48-h exposures, and two separate experiments for the 72-h exposure.
Comparison between cells irradiated with 2 Gy and cells treated with WR1065 and then irradiated 24 h to 72 h later. P values were calculated using a two-tailed t test.
The average activity in U/106 cells was calculated from individual measurements over the entire treatment period.
Comparison between WR1065-treated cells and untreated control cells. P values were calculated using a two-tailed t test.
To assess whether prolonged WR1065 dosing of HMEC and SA-NH cells and the subsequent persistent elevation of SOD2 activity resulted in a general toxicity or reduction of cell numbers, cell counts were obtained over time as a function of the treatment protocol and were compared to their corresponding untreated controls. No significant toxic effect of WR1065 treatment at a concentration of 40 μM was observed for either HMEC (see Fig. 3a) or SA-NH cells (see Fig. 3b) regardless of the treatment protocol followed. There was a slight negative slope for each of the curves describing the change in cell numbers as a function of time ranging from 0.001 to −0.02, but no correlation between the magnitude of this change and the frequency of WR1065 treatments could be discerned. Rather, these results suggest that the slight changes in cell number observed after each of the treatment protocols was more a function of the time that cells were maintained under the general growth conditions required to maintain them as fed confluent cultures.
FIG. 3.
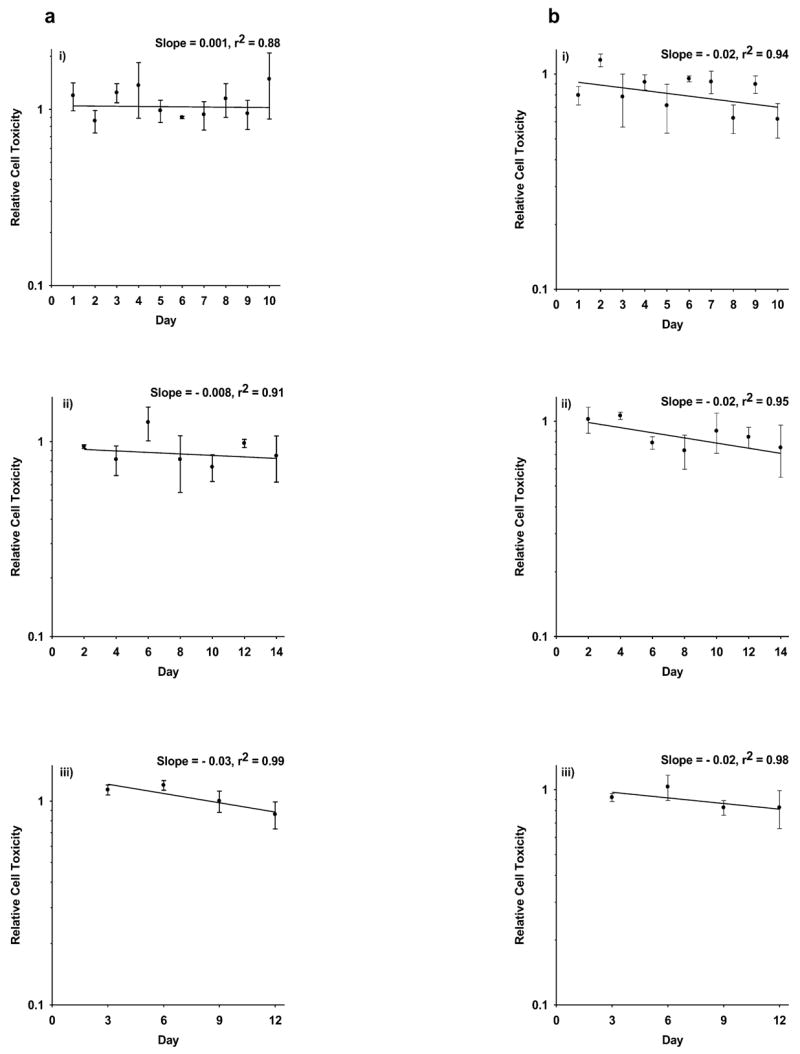
Relative cell toxicity measured as the ratio of the total number of HMEC (panel a) and SA-NH cells (panel b) in WR1065-treated culture plates compared to corresponding HMEC and SA-NH cells, respectively, not treated with WR1065 as a function of the drug administration protocol followed. Cells were exposed to 40 μM WR1065 (i) every 24 h for up to 10 days, (ii) every 48 h for up to 14 days, and (iii) every 72 h for up to 12 days. Data points from 15 plates in three separate experiments were acquired for each experimental protocol and plotted, and regression lines with corresponding slopes and correlation coefficients were calculated. Each error bar represents mean ± SEM of relative cell numbers obtained from 15 plates in three separate experiments.
Exposure of HMEC and SA-NH cells to WR1065 under the three treatment protocols resulted in both increased resistance to ionizing radiation, e.g., the delayed radioprotective effect, and a corresponding increase in SOD2 enzymatic activity with the increase in SOD2 activity directly correlating with the corresponding increase in the magnitude of radiation resistance. To better evaluate the relationship between these two phenomena, both HMEC and SA-NH cells were transfected with SOD2 siRNA and were then compared to corresponding sham-transfected controls with respect to changes in radiation sensitivity and SOD2 activity after exposure to 40 μM WR1065 24 h earlier. After WR1065 exposure, sham-transfected HMEC exhibited both an enhanced radiation resistance, e.g., protection factor of 1.34, and a 5.9-fold increase in SOD2 activity from 0.048 units (U)/106 cells to 0.282 U/106 cells (see Fig. 5a). In contrast, SOD2 siRNA-transfected HMEC exposed to WR1065 exhibited neither an increase in radiation resistance, e.g., protection factor = 0.94, nor a change in SOD2 activity, e.g., 0.046 U/106 cells. The effects of SOD2 si-RNA transfection on SA-NH were more dramatic than observed for HMEC. While WR1065 treatment of sham-transfected SA-NH cells resulted in an elevation in radiation resistance, e.g., protection factor = 1.35, and in a 6.5-fold increase in SOD2 activity from 0.018 U/106 cells to 0.117 U/106 cells, SOD2 siRNA-transfected cells exhibited not only an enhanced radiation sensitivity, e.g., protection factor = 0.75, but also a reduction in SOD2 activity below the sham-transfected control cells, e.g., 0.012 U/106 cells. These data are presented in Fig. 5b for comparison.
FIG. 5.

Survival of HMEC (panel a) and SA-NH cells (panel b) transfected with SOD2 siRNA and exposed to 2 Gy of 250 kVp X rays 24 h after treatment with 40 μM WR1065, sham-transfected cells treated with 40 μM WR1065 and irradiated 24 h later, and cells irradiated in the absence of thiol treatment. Each bar represents the mean ± SEM of three separate experiments. Panel a: Compared to the radiation-only group, HMEC protection factors (PF) of 0.94, P = 0.94, and 1.34, P < 0.001, were calculated for the SOD2 siRNA + 40 μM WR1065 and the 40 μM WR1065 treatment conditions, respectively. HMEC not exposed to WR1065, exposed to 40 μM and assayed 24 h later, and transfected with SOD2 siRNA, exposed to 40 μM WR1065 and assayed 24 h later exhibited SOD2 activities of 0.048, 0.282 and 0.046 U/106 cells, respectively. Panel b: Compared to the radiation-only group, SA-NH cell protection factors of 0.75, P < 0.001, and 1.35, P < 0.001, were calculated for the SOD2 siRNA + 40 μM WR1065 and the 40 μM WR1065 treatment conditions, respectively. SA-NH cells not exposed to WR1065, exposed to 40 μM WR1065 and assayed 24 h later, and transfected with SOD2 siRNA, exposed to 40 μM WR1065 and assayed 24 h later exhibited SOD2 activities of 0.018, 0.117 and 0.012 U/106 cells, respectively. P values were calculated using a two-tailed t test.
DISCUSSION
The purpose of the present study was to determine whether a thiol-induced SOD2-mediated enhancement of radioresistance in cells could be maintained for prolonged periods after multiple administrations of WR1065 in both malignant SA-NH cells and non-malignant HMEC. A dose of 40 μM was identified for multiple administrations because it has been demonstrated to be too low to protect cells directly but high enough to induce an activation of NFκB and to subsequently enhance SOD2 gene expression (22–25, 31). The kinetics of SOD2 buildup in cells after their exposure to thiols was fairly consistent in all of the cell lines evaluated thus far, i.e., rising 8 to 10 h after exposure, reaching a maximum at between 20 and 28 h, and then falling back to pre-thiol exposure background levels by 30 to 40 h (22–25). For this reason three drug administration protocols were chosen for evaluation. Since maximal levels of SOD2 protein and associated activity occurred at about 24 h after thiol administration in all of the three cell systems tested to date, a 24-h treatment schedule was chosen with the expectation that if this effect could be maintained at a persistent level, then thiol administered at 24-h intervals would result in a prolonged maximal radiation resistance of cells, consistent with the magnitude of enhanced radioresistance characteristic of the delayed radioprotective effect observed 24 h after administration of a single dose of thiol. Likewise, since SOD2 levels returned to background levels in cells by 40 h after thiol administration, a schedule of thiol administration every 72 h followed by irradiation 72 h later should result in no change in the radiation resistance of cells compared to their corresponding untreated controls. Finally, an intermediate schedule of drug administration every 48 h was followed to determine whether the magnitude of the delayed radio-protective effect could be sustained at a detectable and significant level.
As shown in Figs. 1 and 2 and Tables 1 and 2, the most effective drug treatment protocol for elevating SOD2 enzymatic activity and enhancing radiation resistance in both HMEC and SA-NH cells is the administration of WR1065 every 24 h. The average protection factors determined for HMEC after daily exposure to thiol was 1.33 (see Table 1) compared to 1.34 observed for HMEC after only a single thiol exposure and then irradiated 24 h later (see Fig. 5). The average increase in SOD2 activity over the 10 day period was 5.3-fold (see Table 1) compared to a single-dose enhancement of 5.9-fold (see Fig. 5a). These data demonstrate that multiple daily treatments over a 10-day period with a low dose of thiol will result in the maintenance of the delayed radioprotective effect at the maximal level that can be demonstrated after a single-dose administration. While the magnitude and persistence of the effect were similar for both HMEC and SA-NH cells (see Fig. 2, Table 2), the inherent endogenous SOD2 activity measured was greater in non-malignant HMEC, 0.048 ± 0.02 U/106 cells, than in mouse tumor SA-NH cells, 0.013 ± 0.002 U/106 cells, consistent with reports that cancer cells compared to normal cells are generally low in the enzymatic activities of both SOD1 and SOD2 (30, 32, 33). SA-NH cells also appeared to require a relatively greater elevation in SOD2 activity than did HMEC to achieve comparable levels of increased radiation protection (see Fig. 4).
FIG. 4.
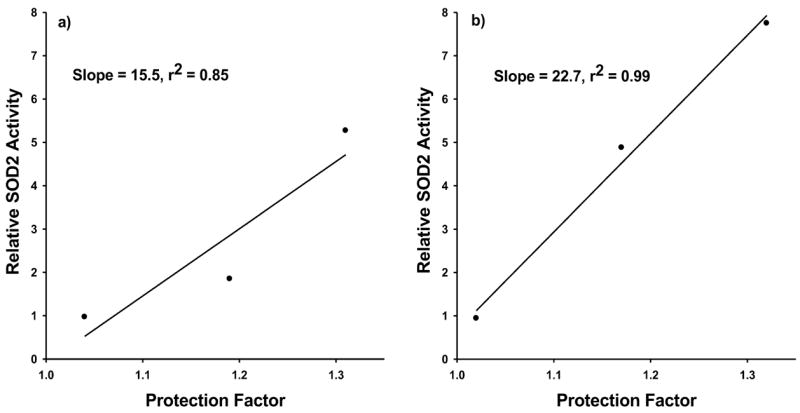
Relationship between the relative change in SOD2 activity and the relative change in radiation resistance as measured by enhanced survival in the form of protection factors for HMEC (panel a) and SA-NH cells (panel b) as a function of multiple treatments with WR1065. Data are from three separate experiments.
Maintenance of an elevated level of SOD2 enzymatic activity for a prolonged period might lead to toxicity, especially in tumor cells due to their relatively unstable redox state compared to normal cells and the potential of over-production of H2O2, the by-product of the dismutation process of superoxide anion. As shown in Fig. 3a and b, no evidence of significant cell loss due to toxicity induced by WR1065 exposure either directly or as a result of elevated SOD2 activity was observed regardless of the frequency or the number of WR1065 doses administered under the three different treatment protocols followed for either HMEC or SA-NH cells.
To verify the role of SOD2 in the delayed radioprotective effect, both HMEC and SA-NH cells were transfected with SOD2 siRNA and then exposed to WR1065 and irradiated 24 h later. HMEC transfected with SOD2 siRNA and exposed to WR1065 failed to show either an enhanced resistance to ionizing radiation (see Fig. 5a) or an increase in SOD2 activity over non-thiol-treated cells. The effect of SOD2 siRNA transfection on radiation response and SOD2 activity was more dramatic in mouse tumor SA-NH cells exposed to WR1065. Not only did this treatment reduce SOD2 levels in thiol exposed compared to sham-transfected control cells, but it also significantly sensitized the cells to ionizing radiation (see Fig. 5b). These data demonstrate the important role of SOD2 in the cellular response to radiation exposure. Furthermore, because SOD2 is localized within the mitochondria and this localization is required for demonstrating protection against radiation-induced cellular damage (9), these findings support the importance of mitochondria as intracellular targets in the overall radiation response paradigm.
The delayed radioprotective effect can be considered a form of an adaptive response described originally as a phenomenon by which exposure of certain types of cells to a relatively low dose of radiation induces an enhanced resistance to the deleterious effects of a second but much larger dose of ionizing radiation (34, 35). This phenomenon appears to require de novo protein synthesis since exposure of these cells to the protein synthesis inhibitor cyclohexamide after irradiation with the initial low radiation dose will inhibit the adaptive response to the second higher dose (36). Recently it was demonstrated that an initial exposure of mouse skin JB6P+ epithelial cells to 10 cGy resulted in an enhanced resistance to a second higher dose of 2 Gy (10). This low-dose-induced adaptive response was demonstrated to be due to the activation of NFκB and elevation of a group of NFκB-regulated genes including p65, SOD2, phosphorylated extracellular signal-related kinase, cyclin B1 and 14-3-3ζ. Treatment of these cells with the NFκB inhibitor IMD-0354 diminished the adaptive resistance effect, while transfection of cells with siRNA against mouse SOD2 was shown to inhibit the development of the adaptive response (10).
In contrast to the low-dose radiation-induced adaptive response, which is induced by oxidative damage and is transient, the thiol-induced adaptive response is induced through a reductive process, resulting in an enhanced survival response that can be maintained over a prolonged period. The consequence, however, of the thiol-induced adaptive response is the accumulation of elevated levels of active SOD2 in both malignant and non-malignant cells that translates to an enhanced level of radiation resistance. Enhanced radiation resistance induced in this manner can be maintained for a prolonged period, giving rise to two important but disparate implications for radiation protection. Radiation workers or individuals at risk for radiation exposure may be able to exploit this phenomenon to enhance their inherent radiation resistance through the addition of thiol-containing drugs such as NAC for use as a supplement to their daily diet. Conversely, patients undergoing radiation and/or chemotherapy treatments for their cancers should use caution and should refrain not only from using thiol-containing dietary supplements but also should consider the risk–benefit implications of using thiol-containing medications such as the ACE inhibitor captopril, which can be prescribed for the control of hypertension, or the medically approved cytoprotectors amifostine and mesna.
Acknowledgments
This work was supported in part by NIH/NCI R01 Grant CA99005 (DJG) and DOE Grant DE-FG02-05ER64086 (DJG).
References
- 1.Zamzami N, Susin SA, Marchetti P, Hirsh T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou L, Yuan R, Serggio L. Molecular mechanisms of irradiation-induced apoptosis. Front Biosci. 2003;8:d9–19. doi: 10.2741/927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belka C, Jendrossek V, Pruschy M, Vink S, Verheij M, Budach W. Apoptosis-modulating agents in combination with radiotherapy-current status and outlook. Int J Radiat Oncol Biol Phys. 2004;58:542–554. doi: 10.1016/j.ijrobp.2003.09.067. [DOI] [PubMed] [Google Scholar]
- 4.Storz P. Reactive oxygen species-mediated mitochondria-to-nucleus signaling: A key to aging and radical-caused diseases. Sci STKE. 2006;332:re3. doi: 10.1126/stke.3322006re3. [DOI] [PubMed] [Google Scholar]
- 5.Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloomfield G, Pears C. Superoxide signaling required for multicellular development of Dictyostelium. J Cell Sci. 2003;116:3387–3397. doi: 10.1242/jcs.00649. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am J Physiol Cell Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 9.Epperly MW, Gretton JE, Sikora CA, Jefferson M, Bernarding M, Nie S, Greenberger JS. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res. 2003;160:568–578. doi: 10.1667/rr3081. [DOI] [PubMed] [Google Scholar]
- 10.Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. Nuclear factor-κB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 2007;67:3220–3228. doi: 10.1158/0008-5472.CAN-06-2728. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Porntadavity S, St Clair DK. Transcriptional regulation of human manganese superoxide dismutase gene: The role of specificity protein 1 (SP1) and activating protein-2 (AP2) Biochem J. 2002;362:401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porntadavity S, Xu Y, Kiningham K, Rangnekar VM, Prachayasitikul V, St Clair DK. TPA-activated transcription of the human MnSOD gene: Role of transcription factors SP1 and Egr1. DNA Cell Biol. 2001;20:473–481. doi: 10.1089/104454901316976109. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Kiningham K, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St Clair DK. An intronic NFκB element is essential for induction of human manganese superoxide dismutase gene by tumor necrosis factor-β and interleukin 1β. DNA Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- 14.Guozheng G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, Ogi J, Khaletskiy A, Li Z, Weydert C, Li JJ. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol. 2003;23:2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kops GJPL, Dansen TB, Polderman PE, Saarloos I, Wirtz KWA, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BMT. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 16.Essers MAG, Weijzen S, de Vries-Smits AMM, Saarloos I, de Ruiter ND, Bos JL, Burgering BMT. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu JW, Chandra D, Rudd MD, Butler AP, Pallotta V, Brown D, Coffer PJ, Tang DG. Induction of prosurvival molecules by apoptotic stimuli: Involvement of FOXO3a and ROS. Oncogene. 2005;24:2020–2031. doi: 10.1038/sj.onc.1208385. [DOI] [PubMed] [Google Scholar]
- 18.Hirose K, Longo DL, Oppenheim JJ, Matsushima K. Overexpression of mitochondrial manganese superoxide dismutase promotes survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anti-cancer drugs, and ionizing radiation. FASEB J. 1993;7:361–368. doi: 10.1096/fasebj.7.2.8440412. [DOI] [PubMed] [Google Scholar]
- 19.Epperly MW, Bray JA, Kraeger S, Zwacka R, Engelhardt J, Travis E, Greenberger JS. Prevention of late effects of irradiation lung damage by manganese superoxide dismutase gene therapy. Gene Ther. 1998;5:196–208. doi: 10.1038/sj.gt.3300580. [DOI] [PubMed] [Google Scholar]
- 20.Epperly MW, Bray JA, Krager S, Berry LA, Gooding W, Engelhardt JF, Zwacka R, Travis EL, Greenberger JS. Intratracheal injection of adenovirus containing the human MnSOD transgene protects athymic nude mice from irradiation-induced organizing alveolitis. Int J Radiat Oncol Phys. 1999;43:169–181. doi: 10.1016/s0360-3016(98)00355-1. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Park JW. A manganese porphyrin complex is a novel radiation protector. Free Radic Biol Med. 2004;37:272–283. doi: 10.1016/j.freeradbiomed.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Murley JS, Kataoka Y, Weydert CJ, Oberley LW, Grdina DJ. Delayed cytoprotection after enhancement of SOD2 (Mn-SOD) gene expression in SA-NH mouse sarcoma cells exposed to WR1065, the active metabolite of amifostine. Radiat Res. 2002;158:101–109. doi: 10.1667/0033-7587(2002)158[0101:dcaeos]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 23.Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFκB-mediated induction of SOD2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162:536–546. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 24.Murley JS, Kataoka Y, Weydert CJ, Oberley LW, Grdina DJ. Delayed radioprotection by nuclear transcription factor κB-mediated induction of manganese superoxide dismutase in human microvascular endothelial cells after exposure to the free radical scavenger WR1065. Free Radic Biol Med. 2006;40:1004–1016. doi: 10.1016/j.freeradbiomed.2005.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murley JS, Kataoka Y, Baker LL, Diamond AL, Morgan WF, Grdina DJ. Manganese superoxide dismutase (SOD2)-mediated delayed radioprotection induced by the free thiol form of amifostine and tumor necrosis factor α. Radiat Res. 2007;167:465–474. doi: 10.1667/RR0758.1. [DOI] [PubMed] [Google Scholar]
- 26.Das KC, Lewis-Molock Y, White CW. Activation of NF-κB and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am J Physiol. 1995;269:L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- 27.Antras-Ferry J, Maheo K, Chevanne M, Dubos MP, Morel F, Guillouzo A, Cillard P, Cillard J. Oltipraz stimulates the transcription of the manganese superoxide dismutase gene in rat hepatocytes. Carcinogenesis. 1997;18:2113–2117. doi: 10.1093/carcin/18.11.2113. [DOI] [PubMed] [Google Scholar]
- 28.Murley JS, Kataoka Y, Hallahan DE, Roberts JC, Grdina DJ. Activation of NFκB and MnSOD gene expression by free radical scavengers in human microvascular endothelial cells. Free Radic Biol Med. 2001;30:1426–1439. doi: 10.1016/s0891-5849(01)00554-8. [DOI] [PubMed] [Google Scholar]
- 29.Matthews JR, Wakasugi N, Virelizier J, Yodoi Y, Hay RT. Thioredoxin regulates the binding activity of NF-κB by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oberley LW. Anticancer therapy by overexpression of superoxide dismutase. Antioxid Redox Signal. 2001;3:461–472. doi: 10.1089/15230860152409095. [DOI] [PubMed] [Google Scholar]
- 31.Grdina DJ, Shigematsu N, Dale P, Newton GL, Aguilera JA, Fahey RC. Thiol and disulfide metabolites of the radiation protector and potential chemopreventive agent WR-2721 are linked to both its anti-cytotoxic and anti-mutagenic mechanisms of action. Carcinogenesis. 1995;16:767–774. doi: 10.1093/carcin/16.4.767. [DOI] [PubMed] [Google Scholar]
- 32.Venkataraman S, Jiang X, Weydert C, Zhang Y, Zhang HJ, Goswami PC, Ritchie JM, Oberley LW, Buettner GR. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene. 2005;24:77–89. doi: 10.1038/sj.onc.1208145. [DOI] [PubMed] [Google Scholar]
- 33.Ough M, Lewis A, Zhang Y, Hinkhouse MM, Ritchie JM, Oberley LW, Cullen JJ. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic Res. 2004;38:1223–1233. doi: 10.1080/10715760400017376. [DOI] [PubMed] [Google Scholar]
- 34.Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science. 1984;223:594–597. doi: 10.1126/science.6695170. [DOI] [PubMed] [Google Scholar]
- 35.Shadley JD. Chromosomal adaptive response in human lymphocytes. Radiat Res. 1994;138(Suppl):S9–S12. [PubMed] [Google Scholar]
- 36.Youngblom JH, Wiencke JK, Wolff S. Inhibition of the adaptive response of human lymphocytes to very low doses of ionizing radiation by the protein synthesis inhibitor cycloheximide. Mutat Res. 1989;227:257–261. doi: 10.1016/0165-7992(89)90107-3. [DOI] [PubMed] [Google Scholar]