

Abstract
The mature bovine cathepsin C (CC) molecule is composed of four identical monomers, each proteolytically processed into three chains. Five intrachain disulfides and three nonpaired cysteine residues per monomer were identified. Beside catalytic Cys234 in the active site, free-thiol Cys331 and Cys424 were characterized. Cys424 can be classified as inaccessible buried residue. Selective modification of Cys331 results in dissociation of native CC tetramer into dimers. The 3D homology-based model of the CC catalytic core suggests that Cys331 becomes exposed as the activation peptide is removed during procathepsin C activation. The model further shows that exposed Cys331 is surrounded by a surface hydrophobic cluster, unique to CC, forming a dimer–dimer interaction interface. Substrate/inhibitor recognition of the active site in the CC dimer differs significantly from that in the native tetramer. Taken together, a mechanism is proposed that assumes that the CC tetramer formation results in a site-specific occlusion of endopeptidase-like active site cleft of each CC monomeric unit. Thus, tetramerization provides for the structural basis of the dipeptidyl peptidase activity of CC through a substrate access-limiting mechanism different from those found in homologous monomeric exopeptidases cathepsin H and B. In conclusion, the mechanism of tetramer formation as well as specific posttranslational processing segregates CC in the family of papain proteases.
Keywords: Cathepsin C, cysteine protease, 3D model, dipeptidyl peptidase I, free-thiol cysteine, oligomerization
Cathepsin C (CC) or dipeptidyl peptidase I (E.C. 3.4.14.1) is a lysosomal cysteine protease belonging to the papain family (Metrione et al. 1966; McDonald et al. 1969; Ishidoh et al. 1991; Nikawa et al. 1992). Cathepsin C is widely expressed in many tissues with high levels in lung, kidney, and placenta, as well as in the spleen and liver, that serve as the main source for its purification (Pham et al. 1997; Rao et al. 1997). Like other lysosomal cysteine proteases, CC is involved in nonspecific intracellular protein degradation (McDonald and Schwabe 1977). In addition, CC appears to play a central role in activation of zymogens of many serine proteases in immune/inflammatory cells. These leukocyte and mast cell granule-associated proteases include cathepsin G and elastase (McGuire et al.1993), mast cell chymase and tryptase (Murakami et al. 1995; Sakai et al. 1996; McEuen et al. 1998), and lymphocyte granzymes A and B (Smyth et al. 1995; Kummer et al. 1996; Wilharrm et al. 1999). CC-deficient cytotoxic T lymphocytes or natural killer cells were found unable to induce apoptosis in target cells (Pham and Ley 1999). During degranulation of mast cells the active CC is released, and its extracellular role in degradation of matrix-associated proteins was proposed (Wolters et al. 2000). Recently, mutations in the CC gene have been found responsible for autosomal recessive disorders known as Papillon-Lefévre syndrome and Haim-Munk syndrome associated with palmoplantar hyperkeratosis and premature periodontitis (Hart et al. 1999, 2000; Toomes et al. 1999).
CC is capable of sequentially removing dipeptides from the free amino terminus of various peptide and protein substrates, thus acting in the exopeptidase (specifically dipeptidyl peptidase) mode (McDonald et al. 1969; Lindley 1972; McGuire et al. 1992). The cleavage is ineffective if the fragmented bond has on either side a proline residue, or the N-terminal residue is lysine or arginine. Natural class-selective inhibitors of cysteine proteases are generally effective against CC. Among them, the proteinaceous inhibitors of cystatin family are the most potent (Nicklin and Barrett 1984; Dolenc et al. 1996), while the low molecular mass inhibitors such as E-64 are effective at higher concentration (Barrett et al. 1982; Nikawa et al. 1992). Another specific inhibitor Gly-Phe-diazomethyl ketone was designed based on a sequence of a favorite CC substrate (Green and Shaw 1981). Finally, a new cathegory of specific peptidic inhibitors containing the N-terminal cluster of arginine residues has recently been discovered (Horn et al. 2000).
The cDNA sequences encoding rat, human, murine, dog, bovine, and Schistosoma CCs have been determined (Ishidoh et al. 1991; Paris et al. 1995; McGuire et al. 1997; Hola-Jamriska et al. 1998; Wolters et al. 1998; Frye et al. 2000). Their comparison demonstrates that the enzyme primary structure is highly conserved among different species. The CC sequence comprises a signal peptide, propeptide, and catalytic core. In general, the amino acid sequence of the CC core is homologous to the sequences of other mature proteases of the papain family. Particularly, the phylogenetic analysis demonstrated the closest evolutionary relationship of CC to cathepsin B group (Berti and Storer 1995). The propeptide of CC is substantially longer than propeptides of other members of the family due to an extension at its N-terminus. This N-terminal portion, unique to CC molecule, is retained in the mature enzyme (here referred to as residual pro-part) (Nikawa et al. 1992; Dolenc et al. 1995). The C-terminal portion of propeptide is homologous with propeptides of other cysteine proteases (Hola-Jamriska et al. 1998), and, analogously, it functions as an activation peptide liberated from the proenzyme during maturation (Dahl et al. 2001).
Beside the residual pro-part, CC differs in its quaternary structure from other, monomeric members of the papain family. Although there is a variation in reported data and ambiguity in terminology, the wild-type form of mammalian CC is a tetrameric homo-oligomer of 160–200 kD (McDonald et al. 1969; Metrione et al. 1970; McGuire et al. 1992; Nikawa et al. 1992; Dolenc et al. 1995). In rat macrophages, procathepsin C (proCC) monomer with mass of 55 kD is found associated in a dimer (Muno et al. 1993) similar to that reported for human proCC (Dahl et al. 2001). The dimer is further proteolytically processed and forms a tetramer during its transport to lysosomes by a mannose-6-phosphate-dependent pathway (Muno et al. 1993). At present, the structural basis for CC assembly into tetramer as well as the impact of oligomerization on the function of the molecule and its possible physiological relevance is unknown. In our work, we characterized the covalent structure of the bovine CC, and used this information to build its spatial molecular model. We discovered structural determinants involved in CC tetramer formation, and on this basis propose a novel model of relationship among processing, quaternary structure, and exopeptidase activity of this enzyme.
Results
Proteolytic processing of chains
Bovine spleen cathepsin C (bsCC) displays a pattern of three bands with mass of 24, 21, and 8 kD on SDS-PAGE under nonreducing conditions (Fig. 1 ▶). Their N-terminal amino acid sequences determined from a Western blot correspond to a residual pro-part (starting D1), heavy (starting L207), and light (starting D371) chain, respectively, according to alignment with the cDNA sequence of bovine CC (Frye et al. 2000) (Fig. 2 ▶). These data correspond to the N-termini of chains found for rat and human CC (Nikawa et al. 1992; Dolenc et al. 1995). The pro-part mass of bovine CC is higher than the mass of the heavy chain. This result is just opposite to results found for rat and human CC (Nikawa et al. 1992; Dolenc et al. 1995). On reduction, the SDS-PAGE pattern of the bsCC chains was not significantly changed. Nevertheless, an additional minor band appeared that migrated with mass 13 kD. Its N-terminal sequence originated from the interior of the pro-part starting from G34. Taken together, these results demonstrate noncovalent association of the three main chains. The heterogeneity of residual pro-part chain encompasses the intact major form and the derived minor form cleaved proteolytically in two disulfide-connected parts (Fig. 2 ▶).
Fig. 1.

SDS-PAGE pattern of bovine spleen cathepsin C. The reduced (lane 2) and nonreduced (lane 3) sample of the purified enzyme with indicated positions of the chains. The labeling of cysteine residues with IAEDANS fluorogen (lanes 4,5): the enzyme denatured in 8 M urea to label all cysteines (lane 4); the selective labeling of buried cysteine on light chain (lane 5)—the accessible cysteines were blocked by DTNB under native conditions, and this material was labeled with IAEDANS in 8 M urea. The samples in lanes 4 and 5 were run under nonreducing conditions. The molecular mass markers are shown in lane 1.
Fig. 2.

Posttranslational processing and primary structure of bovine cathepsin C. Amino acid sequence found in mature enzyme is in bold, the regions removed during processing are in italics (according to cDNA by Frye et al. 2000). The N-termini ( ) and C-termini (
) and C-termini ( ) of residual pro-part, heavy, and light chains are indicated. A minor fragmentation is marked with a down triangle, and N-glycosylated asparagine residues with solid circle. Paired half-cystine residues in identified disulfides D1 to D5 are interconnected by full lines. In case of cysteine residues, the SH-group is displayed: catalytic Cys234 (SH*), buried Cys424, and Cys331 on the dimer–dimer interface. The portion of the molecule sequenced in this study is underlined. The numbering of the sequence is according to the proenzyme.
) of residual pro-part, heavy, and light chains are indicated. A minor fragmentation is marked with a down triangle, and N-glycosylated asparagine residues with solid circle. Paired half-cystine residues in identified disulfides D1 to D5 are interconnected by full lines. In case of cysteine residues, the SH-group is displayed: catalytic Cys234 (SH*), buried Cys424, and Cys331 on the dimer–dimer interface. The portion of the molecule sequenced in this study is underlined. The numbering of the sequence is according to the proenzyme.
The three main chains of the denatured RCM-bsCC (see Materials and Methods) were separated by gel chromatography and RP-HPLC. Their C-terminal sequences were determined by sequencing the C-terminal peptide fragments recovered from digests of pro-part, heavy, and light chain with N-Asp proteinase (D104–N128), cyanogen bromide (A347–G368), and trypsin (G420–L439), respectively. Further, the C-terminals were corroborated by carboxypeptidase Y digestion of the intact chains. The obtained sequences were compared to cDNA data (Fig. 2 ▶). The results show that the residual pro-part chain in the mature CC molecule corresponds to the N-terminal half (D1–N128) of a coded propeptide sequence, and that the C-terminal half of the full-length propeptide (T129–H206) was removed. The newly formed C-terminus of residual pro-part is nine amino acid residues longer compared to that of human CC (Dolenc et al. 1995). Another proteolytic processing site was found in the sequence originally connecting heavy and light chains. In this case, dipeptide LR370 from the C-terminus of the heavy chain was proteolytically removed (see Discussion).
Disulfide structure
A total number of 13 cysteines/halfcystine residues in bsCC monomer was established by amino acid analysis, and their status was determined by protein chemical methods. The CM-bsCC molecule (see Materials and Methods) was fragmented by trypsin or by a combined trypsin–chymotrypsin digest. Disulfide positive peptides were recovered by RP-HPLC and characterized by amino acid sequencing. This data enabled identification of pairing of five disulfides with the following connectivities: C6–C94 (D-1), C30–C112 (D-2) in the pro-part chain, and C231–C274 (D-3), C267–C307 (D-4), C297–C313 (D-5) in the heavy chain (Fig. 2 ▶). The pro-part disulfides have no counterparts in other cysteine proteases, as the N-terminal half of propeptide (residual pro-part) is a unique structural feature of the CC molecule. The core of the CC molecule (heavy and light chain), on the other hand, is homologous with other members of the papain family, which is also reflected in the disulfide distribution pattern. The disulfides D-3 and D-4 are completely conserved throughout the family, while the counterpart of disulfide D-5 can only be found in the cathepsin B molecule (Baudyš et al. 1990).
Characterization of cysteine residues
The presence of three cysteine residues per monomer was indicated by DTNB titration of bsCC under denaturing conditions (exact value 2.7). This number was corroborated on the structural level through sequence analysis of peptide fragments containing a modified CM–cysteine residue, which were recovered from peptide digests of CM-bsCC (see above). The identified residues correspond to C234 and C331 of the heavy chain, and C424 of the light chain (Fig. 2 ▶).
The chemical properties of cysteine residues were further examined. Titration of bsCC with DTNB under native conditions revealed two cysteines per monomer (value 1.8). However, only one cysteine (value 0.8) was found if bsCC, pretreated with IAA under native conditions, was titrated. It indicates (1) there is one cysteine residue fully exposed to the solvent (for IAA or DTNB modification), (2) one cysteine residue is accessible only for bulky hydrophobic reagents (DTNB), and (3) one cysteine residue becomes available for titration only after complete unfolding. Of these, C234 is evidently freely accessible, as it is completely conserved through the papain family representing catalytic residue in the active site. In support of this, we found that IAA-treated native bsCC is completely inactive. The SDS-PAGE pattern of selectively labeled chains was used to discriminate between the character of C331 and C424. BsCC, modified with DTNB under native conditions, was denatured and treated with thiol-labeling chromogen DABIA or fluorogen IAEDANS. In this material, the label was exclusively incorporated only in the light chain containing C424 (Fig. 1 ▶).
On the basis of these results, C424 can be classified as buried, while C331 behaves as partially obstructed residue. According to sequence alignment of homologous cysteine proteases (Berti and Storer 1995), C331 is unique and specific for CC molecules, while C424 has its only homolog in the cathepsin B sequence. Moreover, the cathepsin B C424 homolog displays the same properties of a buried cysteine as found for bsCC in this work (Baudyš et al. 1988).
Quaternary structure
Mass of bsCC was determined by gel chromatography on a Superdex 200 column and by ultracentrifugation, giving an analogous result of 203, respectively 209 kD (Fig. 3 ▶). Because the sum of the mass of the chains (analyzed on SDS-PAGE) is about 53 kD (monomer mass), the mature bsCC is a tetramer. The bsCC oligomerization degree was corroborated by titration of dipeptidyl peptidase activity of bsCC with chicken cystatin inhibitor, giving a value of 3.7 cystatin molecules bound per bsCC tetramer. The tetrameric structure of bsCC in the cystatin–bsCC complex was further analyzed using gel chromatography. The elution volume of a single symmetrical peak corresponded to a mass of about 250 kD (the cystatin mass is 13 kD).
Fig. 3.

Gel chromatography of bovine spleen cathepsin C (bsCC) on a Superdex 200 HR10/30 column. The column (1 × 30 cm) was equilibrated with 0.1 M Na-acetate buffer, pH 5.5, 1 mM EDTA, 0.2 M NaCl, and 40–50 μg protein samples were chromatographed. The elution peak is depicted for the bsCC tetramer (wild-type [—] and deglycosylated [- -]) and for the bsCC dimer produced by pCMB modification of the respective tetramer (wild-type [-•-] and deglycosylated [. . .]). Their mass values in kD are indicated (solid down triangle). The position of calibration standards is marked (↓) for catalase (232 kD), aldolase (158 kD), transferrin (80 kD), serum albumin (67 kD), and ovalbumin (43 kD).
Disassembly of the tetramer
The mass of bsCC modified with various thiol-labeling agents under native conditions was monitored using gel chromatography on a Superdex 200 column (Table 1). No change in mass of the tetramer (203 kD) was detected for bsCC selectively modified at the active site residue C234. For this purpose, the treatment with IAA or with a specific low mass inhibitor E-64 was used, which produced inactive bsCC material. On the contrary, modification with hydrophobic aromatic DTNB resulted in the disappearance of the 203 kD peak and formation of a 100 kD peak (Fig. 3 ▶), both having the same SDS-PAGE pattern. These results can only be interpreted as dissociation of the tetramer to dimers due to modification of the cysteine residue(s).
Table 1.
Modifications used to study oligomeric structure of bovine spleen cathepsin C a
| Modification | Mass (kD) | Oligomer |
| no | 203 | tetramer |
| IAA | 203 | tetramer |
| pCMB | 100 | dimer |
| pCMB → SH | 203 | tetramer |
| IAA → pCMB | 100 | dimer |
| deglycosylation | 150 | tetramer |
| deglycosylation → pCMB | 75 | dimer |
| sulfo-NHS acetate | 203 | tetramer |
| pCMB → sulfo-NHS acetate → SH | 100 | dimer |
a The mass of CC and its derivatives was determined on a Superdex 200 column. The modification reagent DTNB used instead of pCMB gave the same results. An alternative reagent to IAA is the E-64 inhibitor. SH indicates dialysis against 1 mM mercaptoethanol, and → marks particular steps of a given procedure.
Because the treatment with DTNB is directed against both C234 and C331 (see Characterization of cysteine residues paragraph), the two-step modification procedure was tested to discriminate between them. First, the active site C234 was blocked with IAA (or E-64), and, afterwards, the DTNB treatment of the remaining C331 was applied. This material was also eluted as the dimeric species. Taken together, this clearly indicates that modification of C331 directly results in conversion of the tetramer into the dimer.
Effect of another hydrophobic thiol-modifying reagent pCMB was studied, and the same results as found for DTNB were obtained. Further, a reversible character of the conversion was demonstrated by dialysis of the modified dimer against solution with low concentration of a thiol compound such as β-mercaptoethanol. Under such conditions, the DTNB or pCMB label on cysteine can be specifically removed (Veronese et al. 1976). The removal of a modifying group from C331 in the dimer always resulted in reconstitution of the tetrameric bsCC. The recovery yield was 100%. Moreover, the recovered tetramer was fully active (provided that the dimer used for reconstitution did not have its active site C234 previously irreversibly modified with IAA or E-64).
Oligomeric status of bsCC was also studied after enzymatic removal of four N-linked oligosaccharides attached to the molecule (Fig. 2 ▶). bsCC was fully deglycosylated by endoglycosidase F/ N-Glycosidase F under native conditions and analyzed by gel chromatography (Fig. 3 ▶). Deglycosylated bsCC was eluted as a peak corresponding to a mass of about 150 kD. Treatment of this material with pCMB, however, yielded a mass of 75 kD only on the gel filtration column. These values correspond to the mass of deglycosylated tetramer and deglycosylated dimer, respectively, as calculated from SDS-PAGE data of deglycosylated chains (see above). Further, the activity of deglycosylated tetramer was tested and found only slightly reduced (80%) compared to the wild-type CC control. We conclude that the wild-type and deglycosylated bsCC display an analogous quaternary structure as well as activity and, thus, N-linked glycans are not involved in maintaining the quaternary structure of CC.
Model of tertiary structure
A three-dimensional model of the catalytic core of mature monomer CC molecule consisting of heavy and light chains was constructed. The residual pro-part chain that is an integral part of mature CC monomer was omitted because of the lack of any known homologous structure that could be used as a structural template. The modeling with the program InsightII was based on the crystal structure of cathepsin B (Musil et al. 1991), the closest homolog of CC with a primary structure similarity of 41% (Rawlings and Barrett 2000). Root-mean-square deviation to the framework (cα) was less than 1.5 Å in the energy-optimized model, indicating correctly folded structure.
The overall structure of the bsCC catalytic core displays structural features typical for the papain family (Fig. 4A ▶). The disc-shaped molecule is organized into two distinct domains whose interdomain interface is partially opened to the V-shaped active site cleft. To adopt the nomenclature introduced for the papain family, the domains are named L and R (left and right hand, respectively) in orientation depicted in Figure 4A ▶ having the cleft on "top" of the molecule. The core possesses three disulfide bridges (D-3 to D-5), all confined to the L domain. Disulfide D-5, typical for CC and cathepsin B molecules, binds a base of the hairpin that is further protruded as the large "occluding loop" in cathepsin B (Musil et al. 1991). Absence of this loop is the major difference between the CC model and the cathepsin B structure. On the other hand, the multiple-turn loop segment on the "top" of R domain is enlarged by an insertion in the CC structure. It acquired a new function as the processing site between the heavy and light chain (LR370) during CC maturation. The CC processing site is located on the opposite site of the structure compared to the processing site of the cathepsin B molecule (loop at the "bottom" of L domain). CC molecule, on the contrary, lacks this cathepsin B processing site, and has in the topologically equivalent region an oligosaccharide moiety attached instead (N120).
Fig. 4.
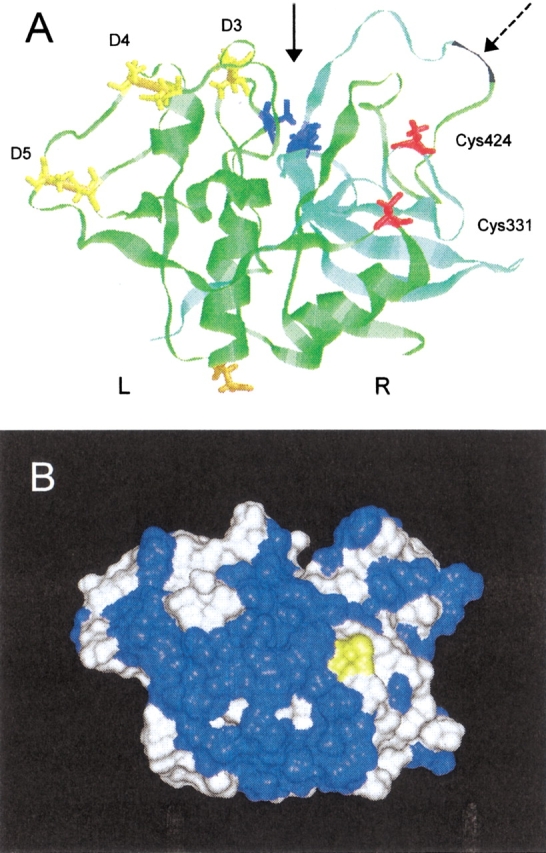
Model of the catalytic core of the bovine cathepsin C monomer. The cathepsin C tertiary structure was modeled using an X-ray structure of cathepsin B (Musil et al. 1991) as a template with the InsightII program. (A) Ribbon plot: proteolytic processing between the heavy (green) and light (bluegreen) chain ( ) results in Leu-Arg370 dipeptide (black) removal. The active site cleft (
) results in Leu-Arg370 dipeptide (black) removal. The active site cleft ( ) contains catalytic residues His381 and Cys234 (blue). Two other cysteines Cys331 and Cys424 are depicted in red, and disulfides D3-D5 in yellow. N-glycosylation residue Asn252 is in orange. L and R domains (nomenclature adopted from papain structure) of cathepsin C core are appropriately marked. (B) Molecular surface in the same orientation as above: the Cys331 residue critical for dimer–dimer interaction in the cathepsin C tetramer is in yellow and the surrounding cluster of hydrophobic residues in blue.
) contains catalytic residues His381 and Cys234 (blue). Two other cysteines Cys331 and Cys424 are depicted in red, and disulfides D3-D5 in yellow. N-glycosylation residue Asn252 is in orange. L and R domains (nomenclature adopted from papain structure) of cathepsin C core are appropriately marked. (B) Molecular surface in the same orientation as above: the Cys331 residue critical for dimer–dimer interaction in the cathepsin C tetramer is in yellow and the surrounding cluster of hydrophobic residues in blue.
Topology of cysteine residues
Cysteine residues C424 and C331 are located in the "front" of the R domain of the CC molecule with a mutual distance of 15 Å (Fig. 4A ▶). The buried C424 can easily be compared to its analog in the cathepsin B molecule. It is located in a depression framed by the side chain rings of several aromatic residues having the thiol group effectively removed from the surface and oriented to the interior.
On the contrary, C331 has a character of surface residue. Two other important structural features of C331 and its surroundings were discovered. First, the residues topologically equivalent to C331 were investigated on available tertiary structures of cysteine protease zymogens and were found to be in a close proximity (or with a potential to form interaction) to the C-terminal portion of the propeptide. This propeptide region, also called activation peptide, folds over the "front" of the main body of the molecule and its active site, and is removed during proteolytic activation of the zymogens and their conversion to mature enzymes (Coulombe et al. 1996; Cygler et al. 1996). Second, the surface of the "front" of the CC molecule, formed by a continuous and extended cluster of apolar and aromatic residues (Fig. 4B ▶), has a significantly pronounced hydrophobic character. This hydrophobic cluster is clearly unique to CC, and makes this region very different from the corresponding region of other cysteine proteinases of the papain family (in both mature and zymogen form), among which the hydrophobic residues are discontinuously scattered over the surface. The cluster originates from substitutions by hydrophobic residues as demonstrated in alignment of the CC sequence with the primary structures of other cysteine proteases (Berti and Storer 1995), and thus cannot be attributed to an improperly folded model. We propose that the cluster is directly involved in extended protein–protein interactions of the CC molecular core during CC oligomer formation. In this respect, we found experimentally through C331 selective modification and by model building that the "front" site of CC molecule forms the interface for dimer–dimer assembly.
The active site cleft contains cysteine C234 as a catalytic residue. Architecture of the cleft in the mature CC core resembles general structural features of the active site of papain-like cysteine proteases acting in the endopeptidase mode. Particularly, there is no extra peptide segment available that could sterically partially block the cleft to disable endopeptidase mode and provide for the exopeptidase mode of dipeptidyl peptidase activity of CC. Such obstructing structural entity must be (1) located in the "front" of active site to disable access of the substrate to the S3 binding subsite of CC, and (2) sufficiently large to prevent binding of polypeptides in the endopeptidase mode to the remaining restricted cleft. This type of mechanism can be found in the molecule of aminopeptidase cathepsin H containing a fragment of activation peptide ("minichain") blocking the S2 subsite (Baudyš et al. 1991; Guncar et al. 1998). This fragment is covalently attached through a disulfide bridge to the core of the cathepsin H molecule in the region coinciding with the location of the hydrophobic cluster on the CC molecule. We conclude that the obstruction of the active site cleft on the "front" site of the CC molecule originates from the dimer–dimer association into tetramer.
Activity of dimer
The dimer prepared using thiol-modifying agents (DTNB or pCMB) cannot be tested for activity, as the catalytic cysteine C234 is blocked together with the C331 residue critical for dimer–dimer interaction. However, the bsCC dimer with preserved active-site cysteine can be prepared using a three-step procedure. In the first step, the dimer was treated with DTNB (or pCMB) as described above (see Disassembly of tetramer). Next, the lysine residues were selectively modified with sulfo-NHS acetate. Finally, the dimer was dialyzed against β-mercaptoethanol to restore the original cysteine thiol groups. The dimer structure of this material was preserved (dimerLys), in contrast to reversible tetramerization that took place during dialysis if the lysine modification step was omitted (see Disassembly of tetramer). The one-step modification of native tetrameric bsCC with sulfo-NHS acetate did not result in any change of oligomerization degree (tetramerLys) due to an insufficient hydrophobicity of this reagent to penetrate the intact dimer–dimer interface. We conclude that a particular lysine residue(s) located on the dimer–dimer interface becomes accessible after disassembly into dimers, and its modification by sulfo-NHS acetate results in stabilization of the dimer structure. According to the 3D model, the candidate target residue is K285, located in the "front" of the L domain. Also, it should be noted that no lysine residue is located in the active site cleft, where modification could directly influence catalytic or substrate-binding function.
The enzymatic activities of dimerLys and tetramerLys were then compared. First, the activity of tetramerLys was found corresponding to that of the wild-type bsCC, while dimerLys was completely inactive. An absence of substrate cleavage was demonstrated with the standard dipeptidyl peptidase substrate Gly-Phe-AMC as well as with the endopeptidase substrate Z-Phe-Arg-AMC to test a possible change in activity mode (Kuribayashi et al. 1993). Second, the activity of tetramerLys can be fully inhibited by cystatin. The interaction of cystatin with inactive dimerLys was demonstrated by gel chromatography yielding a compound of a 125-kD mass. This material showed the bands corresponding to bsCC and cystatin on SDS-PAGE. Finally, the cysteine thiol groups in dimerLys were identified by labeling with IAEDANS under denaturing conditions. The marker was incorporated into both heavy and light bsCC chains as demonstrated on SDS-PAGE, indicating the original native status of cysteine residues.
CC from two bovine tissues
The data on posttranslational processing of bovine spleen CC (bsCC) were compared to that of bovine CC isolated from liver tissue (blCC). The results show no difference in the chain-size pattern on SDS-PAGE. A minor variation in proteolytic processing compared to bsCC is the more pronounced secondary fragmentation at G34 of the residual pro-part. This produces the clipped pro-part with a conversion yield of 10% of the total blCC pro-part (compare with a value of 5% for bsCC), as revealed by intact molecule sequencing. Because the specific activities of spleen and liver CC were the same, the minor fragmentation does not influence the enzyme function, and is possibly related to the aging of the cathepsin C molecule (Horst and Hasilik 1991).
Discussion
The primary goal of this investigation was to analyze structure–function relationships in the CC molecule. First, the mature bsCC molecule was studied with a focus on covalent structure, more specifically pairing of cysteines, glycosylation, and proteolytic processing. The three-chain composition of the bovine CC molecule is generally analogous to that found in other mammalian CCs (Nikawa et al. 1992; Dolenc et al. 1995), and has several important features. The N-terminal amino acid sequences of the chains suggest that they are evolutionarily perfected to resist autoproteolytic degradation. The dipeptidyl peptidase activity of CC is ineffective with substrates having proline residue in P1 or P1` subsite, which coincides with proline positions in the N-terminal sequences of residual pro-part (D1TPA . . . , Pro in P1` subsite), heavy (L207PTS . . . , Pro in P1 subsite), and light chain (D371PFN . . . , Pro in P1 subsite). The C-terminal sequence analysis of the chains revealed a trimming process on the heavy chain as a feature not found for human CC (Cigic et al. 1998). The removal of Leu-Arg dipeptide suggests that a carboxydipeptidase such as cathepsin B can be involved, as similar C-terminal trimming of Gly-Arg dipeptide in the bovine cathepsin B molecule was reported (Meloun et al. 1988). With respect to the residual pro-part, the C-terminal sequence is nine amino acids longer (containing N-glycosylated N120 residue) than that found in human CC (Cigic et al. 1998). This finding explains the reversal of SDS-PAGE band pattern of pro-part and heavy bsCC chains (pro-part mass is higher than the mass of heavy chain) compared to those for human and rat CC (Nikawa et al. 1992; Dolenc et al. 1995). Also, it indicates that the C-terminal region of the residual pro-part does not play any important role in the CC structure or function.
The bsCC sequence data and determined disulfide connectivities as well as identification of free-thiol cysteine residues served as a starting point for construction of the homology model of tertiary structure of the mature catalytic core (heavy and light chain) of bovine CC. The model later served as a basis for assigning function(s) to the uncovered structural determinants. First, the surface accessibility of three nonpaired cysteine residues identified in each monomer was studied. The catalytic residue C234 in the active site is freely available for modification, contrary to C424 having character of the buried residue similar to its homolog in cathepsin B (Baudyš et al. 1988). C331 represents a new CC-specific determinant that is conserved in all mammalian CCs while it is absent in other members of the papain family. According to the model, C331 is a typical surface residue that should be freely accessible from solvent. Experimentally, however, this residue behaves as partially obstructed, having reagent-selective accessibility. The modification of C331 is effective only with large hydrophobic thiol-specific reactants but not for hydrophilic and charged reagents. The specific role of C331 was unraveled during analysis of CC quaternary structure. After modification, the CC tetramer disassembled into the dimers, while this process was reversible upon removal of the thiol-label yielding back the functional tetramer (Table 1). We conclude that C331 is critical for the maintenance of the tetramer structure of the mature cathepsin C. Moreover, C331 and the adjacent surface of the molecule is very likely involved in the dimer–dimer interaction, which is further strongly supported by the 3D model. The large hydrophobic cluster, a specific structural feature of CC among the papain family members, is found on the surface of the molecule surrounding C331 residue (see Fig. 4B ▶). Such surface hydrophobic regions are usually involved in extensive protein–protein interactions, such as assembly of subunits into oligomers (Larsen et al. 1998) or domain–domain interactions (Jones et al. 2000). In this particular case, the hydrophobic part of the CC monomer surface takes direct part in the dimer–dimer assembly into the tetramer. Interestingly, two missense mutations (Y280 and Y323) in the cluster were found associated with Papillon-Lefévre syndrome (Hart et al. 2000). Most likely, these mutations interfere with CC tetramer formation, leading to accumulation of the inactive CC dimer. The involvement of the hydrophobic cluster in the dimer–dimer interaction is further supported by an unaltered stability of bsCC tetramer in solutions with high ionic strength such as 2.5 M NaCl (data not shown). The cysteine modification-dependent disassembly is a specific event because the dimers cannot be produced from mature CC tetramer by other means, such as an increased concentration of guanidine•HCl in gel filtration medium that rather leads to instant dissociation of the CC tetramer into its three constituent polypeptide chains (Cigic et al. 2000).
The results obtained provide new clues with regard to the structural events accompanying the maturation process of the procathepsin C (proCC) molecule. The proCC is a dimer (Muno et al. 1993; Dahl et al. 2001), which indicates that the proCC processing plays a direct role in formation of the mature tetramer. The N- and C-terminal parts of the full-length propeptide of proCC display strikingly different characteristics. The C-terminal portion is removed during processing as the so-called activation peptide (Cigic et al. 1998; this work). According to homology with other cysteine procathepsins (Coulombe et al. 1996; Cygler et al. 1996;Hola-Jarmiska et al. 1998), this portion folds over the "front" site of the catalytic core of CC. Here, it crosses over and partially occupies the proposed dimer–dimer interface, making potential contact with free C331. Moreover, Dahl et al. (2001) demonstrated that C-terminal part of the full-length proCC propart is accessible to the processing enzymes. Taken together, we propose that the removal of the C-terminal portion of the propeptide triggers the association of two dimers into a tetramer through the newly exposed surface on the dimer molecule. Thus, the mature CC tetrameric structure is not composed of four spatially equal monomers (centered around the fourfold axis) but of two dimers organized according to lower symmetry, for example, related by a twofold symmetry element. In addition, the internal organization of monomers within a dimer seems to be identical in the proCC dimer as well as the "mature dimer" produced by C331 modification.
The N-terminal portion of the propeptide (residual pro-part), which is an integral part of mature CC, is more likely involved in monomer–monomer interaction, and thus responsible for dimer formation/stability. We found that residual pro-part of bovine CC carries two intrachain disulfides, and is heavily glycosylated. These findings are in agreement with the recent work on its refolding that demonstrated that it forms an independent stable globular domain stabilizing human proCC (Cigic et al. 2000).
The 3D model of bovine CC shows high degree of structural homology in architecture of the core of the CC monomer, including the active site, with other representatives of the papain family. The X-ray studies demonstrated that the general endopeptidase active site cleft of cysteine proteinases of the papain family is preserved, and is sterically hindered in those members that function in the exopeptidase mode (Musil et al. 1991; Guncar et al. 1998). As an obstructive structural element specifically blocking part of the endopeptidase-like type binding cleft, the "occluding" loop and the disulfide bound "minichain" were identified for cathepsin B (carboxydipeptidase) and cathepsin H (aminopeptidase), respectively (Musil et al. 1991; Guncar et al. 1998). Neither of these mechanisms can be applied for CC based on our data. To ensure that the dipeptidyl peptidase activity of CC is dominant, an obstruction at the S3 substrate binding subsite is required. This can be achieved with a bulky structural segment localized in the "front" site of the binding cleft. Such an obstruction is very likely realized when two CC dimers come together to form a tetramer.
To address this question, we prepared a CC dimer stabilized through specific lysine modification (very likely K285) in the "front" site of the CC monomer having preserved the catalytic C234 residue (Table 1). The dimer is enzymatically inactive, while it still binds inhibitor cystatin. This suggests that the general architecture of papain-like proteinases' active site is preserved, so the dimer can specifically recognize the large proteinaceous inhibitor. However, to acquire the precise catalytic function, the CC exopeptidase active site cleft must be "finished up" with the help of the second interacting dimer.
In summary, the exhausting characterization of posttranslational modifications found in bovine CC molecule has been presented. Functional significance of these modifications was analyzed with focus on the role of cysteine residues in CC oligomerization. The model of CC structure is proposed that localizes the dimer–dimer interface formed during proCC processing, and that explains exopeptidase activity of CC by partial spatial obstruction of the endopeptidase-like cleft of the CC monomer realized through the dimer–dimer interaction within the mature tetramer.
Materials and methods
Materials
Cathepsin C was purified to homogeneity from bovine spleen and bovine liver as described previously (Mycek 1970). All buffers used during isolation contained 1 mM EDTA, and were free of exogenous thiol compounds. The enzyme exhibited exclusively the exopeptidase activity. Substrates Gly-Phe-AMC and Z-Phe-Arg-AMC were from Bachem. Enzymes TPCK-trypsin and α-chymotrypsin were from Serva, and endoproteinase Asp-N, carboxypeptidase Y, and endoglycosidase F/ N-Glycosidase F from Boehringer. Modification agents trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane (E-64), iodoacetic acid (IAA), N-iodoacetyl-N`-(5-sulfo-1-naphtyl)ethylenediamine (IAEDANS), 5,5`-dithiobis(2-nitrobenzoic acid) (DTNB), p-chloromercuribenzoic acid (pCMB) were from Sigma, 4-(dimethylamino)azobenzene-4`-iodoacetamide (DABIA) from Aldrich, and sulfo-N-hydroxysuccinimide acetate (sulfo-NHS acetate) from Pierce. Chicken cystatin was isolated as described by Nicklin and Barrett (1984). All other chemicals were analytical grade.
Separation of chains
Under denaturing conditions, bsCC was reduced by DTT and subsequently alkylated with IAA essentially as described previously (Meloun et al. 1988). This material (RCM-bsCC) was desalted on a Sephadex G-25 column equilibrated in 50 mM NH4HCO3, and the chains were separated by a two-step procedure. First, the RCM-bsCC was subjected to chromatography on a Sephadex G-100 column equilibrated in 6 M guanidine•HCl, 50 mM NH4HCO3 yielding the pro-part/heavy-chain fraction and the light-chain fraction. The pro-part/heavy chain fraction, desalted as above, was further fractionated by RP-HPLC (see below) producing the pro-part and heavy chain peaks, respectively.
Peptide fragmentation
For tryptic digestion, the 0.3% material (separated RCM-bsCC chains or nonreduced, carboxymethylated bsCC [CM-bsCC] [see Identification of disulfides]) was dissolved in 50 mM NH4HCO3, 0.1 mM CaCl2 and cleaved by TPCK-trypsin at a final enzyme to a substrate ratio of 1:80 (w/w). The cleavage proceeded 2.5 h at 37°C. The disulfide-clustered material (heavy chain) from CM-bsCC was treated with α-chymotrypsin at an enzyme-to-substrate ratio of 1:50 (w/w) for 5 h at 37°C. Cleavage of RCM-bsCC heavy chain by cyanogen bromide (2.5 mg/mg protein) was performed in 70% formic acid for 24 h at 26°C. Endoproteinase Asp-N was used for fragmentation (5 h, 37°C) of RCM-bsCC pro-part chain at an enzyme-to-substrate ratio of 1:500 (w/w) in 50 mM Na-phosphate, pH 6.5. The kinetics of digestion of particular RCM-bsCC chains with carboxypeptidase Y was monitored using amino acid analyzer (Hayashi 1976).
RP-HPLC purification
The mixtures of peptides were fractionated on a reverse-phase Vydac C18 218TP104 column. The Vydac C4 214TP54 column was used for separation of RCM-bsCC chains. The HPLC system (Hitachi LaChrom L7100) was equilibrated in 0.1% trifluoroacetic acid, and the material was eluted with a linear gradient of 90% acetonitrile, 0.1% trifluoroacetic acid.
Identification of disulfides
IAA modified bsCC (CM-bsCC) was prepared without prior reduction of disulfides under denaturing conditions as described previously (Baudyš et al. 1990). This CM-bsCC material was desalted, fragmented with trypsin, and group-separated on a Sephadex G-50 column equilibrated in 50 mM NH4HCO3. The separated disulfide cluster (containing disulfides D3-D5) was additionally fragmented with α-chymotrypsin. All digests were finally chromatographed on RP-HPLC. The disulfide-containing peptides were identified by a colorimetric reaction procedure (Henschen 1978). The purified disulfide-bound peptides were disconnected by DTT reduction and treated with 2-iodacetamide (Baudyš et al. 1990). The liberated peptides were separated by RP-HLPC, and characterized by amino acid sequencing, quantitative amino acid analysis, and mass spectrometry.
Analytical procedures
N-terminal amino acid sequencing was carried out in a Procise Protein Sequencer 491 (Applied Biosystems). The cysteine residues were monitored as alkylated derivatives; the glycosylated Asn residues were associated with a missing signal. The amino acid analysis was performed on a Biochrom 20 Analyzer (Pharmacia). MALDI-TOF mass spectrometry was done on a Lasermat 2000 instrument (Finnigan) using an α-cyano-4-hydroxycinnamic acid matrix.
Modification of cysteines and lysines
The cysteine residue content was determined spectrophotometrically by reaction with DTNB in 0.1 M Na-phosphate buffer, pH 7.2, 1 mM EDTA (buffer A), or the same buffer containing 6 M guanidine•HCl (buffer B) using the protocol of Riddles et al. (1983). Alkylation of cysteine residues with IAA (50- to 250-fold molar excess over cysteine thiols) was performed in buffer A for 30–240 min analogously as described in Baudyš et al. (1988). Selective modification with DTNB or pCMB was done under the same conditions (50- to 250-fold molar excess over cysteine thiols, 30–240 min). Active site cysteine was modified with 0.5 mM E-64 inhibitor during 8 h at 4°C, which produced enzyme having less than 1% of the original activity. The labeling with IAEDANS (20-fold molar excess over cysteine thiols) was carried out in buffer A containing 8 M urea as described by Seifried et al. (1988), and the analogous conditions were used for DABIA labeling (Baudyš et al. 1988). Acylation of the ɛ-amino groups of lysine residues was performed with sulfo-NHS acetate in buffer A under conditions described by Cuozzo and Sahagian (1994). All the modified protein materials were finally dialyzed against 0.1 M Na-acetate, pH 5.5, 1 mM EDTA, 0.2 M NaCl (for native proteins) or 0.1 M NH4HCO3 (for denatured proteins). The former solution, containing 1 mM β-mercaptoethanol, was used for reversible removal of DTNB or pCMB label during dialysis (8 h at 4°C).
Molecular mass determination
The oligomerization degree of bsCC was determined by gel filtration chromatography on an analytical Superdex 200 HR10/30 column operated with ÄKTAexplorer (Amersham Pharmacia Biotech). The measurement was performed in 0.1 M Na-acetate buffer, pH 5.5, 1 mM EDTA, 0.2 M NaCl. Molecular mass of deglycosylated bsCC was determined after treatment of bsCC with endoglycosidase F/ N-glycosidase F (2 U/100 μg bsCC, incubated at 37°C for 48 h in the buffer as described above). The obtained data are based on calibration with a set of proteins in a mass range of 13 to 440 kD (Gel Filtration Calibration Kit, Pharmacia). Mass determination by ultracentrifugation was carried out on a Beckman Spinco Model E with an AnH-Ti rotor. The experiments, done in the same buffer as above, were analyzed at sedimentation equilibrium formed after 7 h at 10,589 RPM and 20°C.
Activity measurement
Activity of bsCC was assayed with Gly-Phe-AMC substrate essentially as described previously (Horn et al. 2000). The assay was performed in a Perkin-Elmer LS-3B Fluorescence Spectrophotometer. Chicken cystatin was used for titration of bsCC by monitoring the loss of activity in the same activity test. The residual inhibited activity was measured for various cystatin concentrations, and the binding stoichiometry was determined according to Dolenc et al. (1996). Endosubstrate Z-Phe-Arg-AMC hydrolysis was tested analogously as that for Gly-Phe-AMC (Kuribayashi et al. 1993).
3D modeling
The core of the bsCC molecule (without residual pro-part) was modeled with the template of the tertiary structure of cathepsin B (PDB code 1HUC) using program modules under InsightII (Molecular Simulations Inc.). In short, the insertion/deletion regions were identified using sequence similarity between bsCC and cathepsin B on the basis of the structure-based alignment of the papain family members (Berti and Storer 1995). Molecular replacement of the structurally conserved regions and rebuilding of the variable regions was done with a Biopolymer module. The optimization of molecular geometry has been carried out by the Discover program using an Amber force field (Cornell et al. 1995). The steepest descent and conjugate gradient methods have been applied until the maximum difference was less than 0.05 kcal•mol−1.
Electronic supplemental material
Additional experimental data are presented for sequencing of bsCC molecule and peptide fragmentation (Fig. 5) (fig5.tif), for structure of isolated disulfide-containing peptides (Table 2) (tab2.doc), and for separation of bsCC chains (Fig. 6) (fig6.doc).
Acknowledgments
This work was supported by the Grant agency of the Czech Academy of Sciences Grant No. A4055006, the Grant agency of the Czech Republic Grant Nos. 522/00/1553 and GP203/01/D008, the Center for Complex Molecular Systems and Biomolecules (project LN00A032 MSMTCR), and by the research project Z4 055 905. The authors thank J. Neumann for analytical ultracentrifugation and Dr. K. Andriano (MacroMed, Inc., Salt Lake City, UT) for critical reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AMC, 7-amido-4-methylcoumarin
CC, cathepsin C
bsCC, bovine spleen cathepsin C
blCC, bovine liver cathepsin C
CM, S-carboxymethylated, RCM, reduced and S-carboxymethylated
DABIA, 4-(dimethylamino)azobenzene-4`-iodoacetamide
DTNB, 5,5`
-dithiobis(2-nitrobenzoic acid)
IAA, iodoacetic acid
IAEDANS, N-iodoacetyl-N`-(5-sulfo-1-naphtyl)ethylenediamine
pCMB, p-chloromercuribenzoic acid
sulfo-NHS acetate, sulfo-N-hydroxysuccinimide acetate
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.2910102.
References
- Barrett, A.J., Kembhavi, A.A., Brown, M.A., Kirschke, H., Knight, C.G., Tamai, M., and Hanada, K. 1982. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 201 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudyš, M., Meloun, B., Gan-Erdene, T., Fusek, M., Mareš, M., Kostka, V., Pohl, J., and Blake, C.C.F. 1991. S-S bridges of cathepsin B and H from bovine spleen: A basis for cathepsin B model building and possible functional implications for discrimination between exo- and endopeptidase activities among cathepsins B, H and L. Biomed. Biochem. Acta 50 569–577. [PubMed] [Google Scholar]
- Baudyš, M., Meloun, B., Gan-Erdene, T., Pohl, J., and Kostka, V. 1990. Disulfide bridges of bovine spleen cathepsin B. Biol. Chem. Hoppe-Seylers 371 485–491. [DOI] [PubMed] [Google Scholar]
- Baudyš, M., Meloun, B., Pohl, J., and Kostka, V. 1988. Identification of the second (buried) cysteine residue and of the C-terminal disulfide bridge of bovine spleen cathepsin B. Biol. Chem. Hoppe-Seylers 369 169–174. [PubMed] [Google Scholar]
- Berti, P.J. and Storer, A.C. 1995. Alignment/phylogeny of the papain superfamily of cysteine proteases. J. Mol. Biol. 246 273–283. [DOI] [PubMed] [Google Scholar]
- Cigic, B., Dahl, S.W., and Pain, R.H. 2000. The residual pro-part of cathepsin C fulfills the criteria required for an intramolecular chaperone in folding and stabilizing the human proenzyme. Biochemistry 39 12382–12390. [DOI] [PubMed] [Google Scholar]
- Cigic, B., Krizaj, I., Kralj, B., Turk, V., and Pain, R.H. 1998. Stoichiometry and heterogeneity of the pro-region chain in tetrameric human cathepsin C. Biochim. Biophys. Acta 1382 143–150. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I, Gould, I.R., Merz, K.M., Jr., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Coulombe, R., Grochulski, P., Sivaraman, J., Menard, R., Mort, J. S., and Cygler, M. 1996. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 15 5492–5503. [PMC free article] [PubMed] [Google Scholar]
- Cuozzo, J.W. and Sahagian, G.G. 1994. Lysine is a common determinant for mannose phosphorylation of lysosomal proteins. J. Biol. Chem. 269 14490–14496. [PubMed] [Google Scholar]
- Cygler, M., Sivaraman, J., Grochulski, P., Coulombe, R., Storer, A.C., and Mort, J.S. 1996. Structure of rat procathepsin B: Model for inhibition of cysteine protease activity by the proregion. Structure 4 405–416. [DOI] [PubMed] [Google Scholar]
- Dahl, S.W., Halkier, T., Lauritzen, C., Dolenc, I., Pedersen, J., Turk, V., and Turk, B. 2001. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 40 1671–1678. [DOI] [PubMed] [Google Scholar]
- Dolenc, I., Turk, B., Kos, J., and Turk, V. 1996. Interaction of human cathepsin C with chicken cystatin. FEBS Lett. 392 277–280. [DOI] [PubMed] [Google Scholar]
- Dolenc, I., Turk, B., Pungercic, G., Ritonja, A., and Turk, V. 1995. Oligomeric structure and substrate induced inhibition of human cathepsin C. J. Biol. Chem. 270 21626–21631. [DOI] [PubMed] [Google Scholar]
- Frye, C.C., Hershberger, C.L., and Zhang, H. 2000. Bovine dipeptidylaminopeptidase 1. U.S. Patent no. 6027911.
- Green, G.D.J., and Shaw, E. 1981. Peptidyl diazomethyl ketones are specific inactivators of thiol proteinases. J. Biol. Chem. 256 1923–1928. [PubMed] [Google Scholar]
- Guncar, G., Podobnik, M., Pungercar, J., Strukelj, B., Turk, V., Turk, D. 1998. Crystal structure of porcine cathepsin H determined at 2.1 A resolution: Location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 6 51–61. [DOI] [PubMed] [Google Scholar]
- Hart, T.C., Hart, P.S., Bowden, D.W., Michalec, M.D., Callison, S.A., Walker, S.J., Zhang, Y., Firatli, E. 1999. Mutations of the cathepsin C gene are responsible for Papillon-Lefevre syndrome. J. Med. Genet. 36 881–887. [PMC free article] [PubMed] [Google Scholar]
- Hart, P.S., Zhang, Y., Firatli, E., Uygur, C., Lotfazar, M., Michalec, M.D., Marks, J.J., Lu, X., Coates, B.J., Seow, W.K., Marshall, R., Williams, D., Reed, J.B., Wright, J.T., and Hart, T.C. 2000. Identification of cathepsin C mutations in ethnically diverse papillon-Lefevre syndrome patients. J. Med. Genet. 37 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, R. 1976. Carboxypeptidse Y. Methods Enzymol. 45 568–587. [DOI] [PubMed] [Google Scholar]
- Henschen, A. 1978. Disulfide bridges in the middle part of human fibrinogen. Hoppe-Seylers Z. Physiol. Chem. 359 1757–1770. [DOI] [PubMed] [Google Scholar]
- Hola-Jamriska, L., Tort, J.F., Dalton, J.P., Day, S.R., Fan, J., Aaskov, J., and Brindley, P.J. 1998. Cathepsin C from Schistosoma japonicum—cDNA encoding the preproenzyme and its phylogenetic relationships. Eur. J. Biochem. 255 527–534. [DOI] [PubMed] [Google Scholar]
- Horn, M., Pavlík, M., Dolečková, L., Baudyš, M., and Mareš, M. 2000. Arginine-based structures are specific inhibitors of cathepsin C. Application of peptide combinatorial libraries. Eur. J. Biochem. 267 3330–3336. [DOI] [PubMed] [Google Scholar]
- Horst, M. and Hasilik, A. 1991. Expression and maturation of human cathepsin D in baby-hamster kidney cells. Biochem. J. 273 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishidoh, K., Muno, D., Sato, N., and Kominami, E. 1991. Molecular cloning of cDNA for rat cathepsin C. Cathepsin C, a cysteine proteinase with an extremely long propeptide. J. Biol. Chem. 266 16312–16317. [PubMed] [Google Scholar]
- Jones, S., Marin, A., and Thornton, J.M. 2000. Protein domain interfaces: Characterization and comparison with oligomeric protein interfaces. Protein Eng. 13 77–82. [DOI] [PubMed] [Google Scholar]
- Kummer, J.A., Kamp, A.M., Citarella, F., Horrevoets, A.J., and Hack, C.E. 1996. Expression of human recombinant granzyme A zymogen and its activation by the cysteine proteinase cathepsin C. J. Biol. Chem. 271 9281–9286. [DOI] [PubMed] [Google Scholar]
- Kuribayashi, M., Yamada, H., Ohmori, T., Yanai, M., and Imoto, T. 1993. Endopeptidase activity of cathepsin C, dipeptidyl aminopeptidase I, from bovine spleen. J. Biochem. 113 441–449. [DOI] [PubMed] [Google Scholar]
- Larsen, T.A., Olson, A.J., and Goodsell, D.S. 1998. Morphology of protein–protein interfaces. Structure 6 421–427. [DOI] [PubMed] [Google Scholar]
- Lindley, H. 1972. The specificity of dipeptidyl aminopeptidase I (cathepsin C) and its use in peptide sequence studies. Biochem. J. 126 683–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J.K. and Schwabe, C. 1977. Intracellular exopeptidases. In Proteinases in mammalian cells tissues (ed. J.J. Barrett), pp. 311–391. North Holland Publishing, Amsterdam.
- McDonald, J.K., Zeitman, B.B., Reilly, and T.J., Ellis, S. 1969. New observations on the substrate specificity of cathepsin C (dipeptidyl aminopeptidase I). Including the degradation of beta-corticotropin and other peptide hormones. J. Biol. Chem. 244 2693–709. [PubMed] [Google Scholar]
- McEuen, A.R., Ashworth, D.M., and Walls, A.F. 1998. The conversion of recombinant human mast cell prochymase to enzymatically active chymase by dipeptidyl peptidase I is inhibited by heparin and histamine. Eur. J. Biochem. 253 300–308. [DOI] [PubMed] [Google Scholar]
- McGuire, M.J., Lipsky, P.E., and Thiele, D.L. 1992. Purification and characterization of dipeptidyl peptidase I from human spleen. Arch. Biochem. Biophys. 295 280–288. [DOI] [PubMed] [Google Scholar]
- McGuire, M.J., Lipsky, P.E., and Thiele, D.L. 1993. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J. Biol. Chem. 268 2458–2467. [PubMed] [Google Scholar]
- McGuire, M.J., Lipsky, P.E., and Thiele, D.L. 1997. Cloning and characterization of the cDNA encoding mouse dipeptidyl peptidase I (cathepsin C). Biochim. Biophys. Acta 1351 267–273. [DOI] [PubMed] [Google Scholar]
- Meloun, B., Baudyš, M., Pohl, J., Pavlík, M., and Kostka, V. 1988. Amino acid sequence of bovine spleen cathepsin B. J. Biol. Chem. 263 9087–9093. [PubMed] [Google Scholar]
- Metrione, R.M., Neves, A.G., and Fruton, J.S. 1966. Purification and properties of dipeptidyl transferase (cathepsin C). Biochemistry 5 1597–1604. [DOI] [PubMed] [Google Scholar]
- Metrione, R.M., Okuda, Y., and Fairclough, G.F. 1970. Subunit structure of dipeptidyl transferase. Biochemistry 9 2427–2432. [DOI] [PubMed] [Google Scholar]
- Muno, D., Ishidoh, K., Ueno, T., and Kominami, E. 1993. Processing and transport of the precursor of cathepsin C during its transfer into lysosomes. Arch. Biochem. Biophys. 306 103–110. [DOI] [PubMed] [Google Scholar]
- Murakami, M., Karnik, S.S., and Husain, A. 1995. Human prochymase activation. A novel role for heparin in zymogen processing. J. Biol. Chem. 270 2218–2223. [PubMed] [Google Scholar]
- Musil, D., Zucic, D., Turk, D., Engh, R.A., Mayr, I., Huber, R., Popovic, T., Turk, V., Towatari, T., Katunuma, N., and Bode, W. 1991. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: The structural basis for its specificity. EMBO J. 10 2321–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycek, M.J. 1970. Cathepsin C (dipeptidyl transferase). Methods Enzymol. 19 285–315. [Google Scholar]
- Nicklin, M.J. and Barrett, A.J. 1984. Inhibition of cysteine proteinases and dipeptidyl peptidase I by egg-white cystatin. Biochem. J. 223 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikawa, T., Towatari, T., and Katunuma, N. 1992. Purification and characterization of cathepsin J from rat liver. Eur. J. Biochem. 204 381–393. [DOI] [PubMed] [Google Scholar]
- Paris, A., Strukelj, B., Pungercar, J., Renko, M., Dolenc, I., and Turk, V. 1995. Molecular cloning and sequence analysis of human preprocathepsin C. FEBS Lett. 369 326–330. [DOI] [PubMed] [Google Scholar]
- Pham, C.T. and Ley, T.J. 1999. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc. Natl. Acad. Sci. 96 8627–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, C.T., Armstrong, R.J., Zimonjic, D.B., Popescu, N.C., Payan, D.G., and Ley, T.J. 1997. Molecular cloning, chromosomal localization, and expression of murine dipeptidyl peptidase I. J. Biol. Chem. 272 10695–10703. [DOI] [PubMed] [Google Scholar]
- Rao, N.V., Rao, G.V., Hoidal, J.R. 1997. Human dipeptidyl-peptidase I. Gene characterization, localization, and expression. J. Biol. Chem. 272 10260–10265. [DOI] [PubMed] [Google Scholar]
- Rawlings, N.D. and Barrett, A.J. 2000. MEROPS: The peptidase database. Nucleic Acids Res. 28 323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddles, P.W., Blakely, R.L., and Zerner, B. 1983. Reassessment of Ellman's reagent. Methods Enzymol. 91 49–60. [DOI] [PubMed] [Google Scholar]
- Sakai, K., Ren, S., and Schwartz, L.B. 1996. A novel heparin-dependent processing pathway for human tryptase. Autocatalysis followed by activation with dipeptidyl peptidase I. J. Clin. Invest. 97 988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifried, S.E., Wang, Y., and von Hippel, P.H. 1988. Fluorescent modification of the cysteine 202 residue of Escherichia coli transcription termination factor rho. J. Biol. Chem. 263 13511–13514 [PubMed] [Google Scholar]
- Smyth, M.J., McGuire, M.J., and Thia, K.Y. 1995. Expression of recombinant human granzyme B. A processing and activation role for dipeptidyl peptidase I. J. Immunol. 154 6299–6305. [PubMed] [Google Scholar]
- Toomes, C., James, J., Wood, A.J., Wu, C.L., McCormick, D., Lench, N., Hewitt, C., Moynihan, L., Roberts, E., Woods, C.G., Markham, A., Wong, M., Widmer, R., Ghaffar, K.A., Pemberton, M., Hussein, I.R., Temtamy, S.A., Davies, R., Read, A.P., Sloan, P., Dixon, M.J., and Thakker, N.S. 1999. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat. Genet. 23 421–424. [DOI] [PubMed] [Google Scholar]
- Veronese, F.M., Boccu, E., and Fontana, A. 1976. Isolation and properties of 6-phosphogluconate dehydrogenase from Escherichia coli. Some comparisons with the thermophilic enzyme from Bacillus stearothermophilus. Biochemistry 15 4026–4033. [DOI] [PubMed] [Google Scholar]
- Wilharm, E., Parry, M.A., Friebel, R., Tschesche, H., Matschiner, G., Sommerhoff, C.P., and Jenne, D.E. 1999. Generation of catalytically active granzyme K from Escherichia coli inclusion bodies and identification of efficient granzyme K inhibitors in human plasma. J. Biol. Chem. 274 27331–27337. [DOI] [PubMed] [Google Scholar]
- Wolters, P.J., Laig-Webster, M., and Caughey, G.H. 2000. Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways. Am. J. Respir. Cell. Mol. Biol. 22 183–190. [DOI] [PubMed] [Google Scholar]
- Wolters, P.J., Raymond, W.W., Blount, J.L, and Caughey, G.H. 1998. Regulated expression, processing, and secretion of dog mast cell dipeptidyl peptidase I. J. Biol. Chem. 273 15514–1520. [DOI] [PubMed] [Google Scholar]