

Abstract
Synpolydactyly (SPD) is a dominantly inherited congenital limb malformation. Typical cases have 3/4 finger and 4/5 toe syndactyly, with a duplicated digit in the syndactylous web, but incomplete penetrance and variable expressivity are common. The condition has recently been shown to be caused by expansions of an imperfect trinucleotide repeat sequence encoding a 15-residue polyalanine tract in HOXD13. We have studied 16 new and 4 previously published SPD families, with between 7 and 14 extra residues in the tract, to analyze the molecular basis for the observed variation in phenotype. Although there is no evidence of change in expansion size within families, even over six generations, there is a highly significant increase in the penetrance and severity of phenotype with increasing expansion size, affecting both hands (P = 0.012) and feet (P < 0.00005). Affected individuals from a family with a 14-alanine expansion, the largest so far reported, all have a strikingly similar and unusually severe limb phenotype, involving the first digits and distal carpals. Affected males from this family also have hypospadias, not previously described in SPD, but consistent with HOXD13 expression in the developing genital tubercle. The remarkable correlation between phenotype and expansion size suggests that expansion of the tract leads to a specific gain of function in the mutant HOXD13 protein, and has interesting implications for the role of polyalanine tracts in the control of transcription.
Dominantly inherited disorders frequently display incomplete penetrance (a normal phenotype in some mutation carriers) and variable expressivity (different degrees of phenotypic severity in affected individuals), the molecular basis for which is generally not understood. One such disorder is the rare dominantly inherited congenital limb malformation, synpolydactyly (SPD; OMIM No. 186000). Mutations in the first exon of HOXD13 have recently been found in three American SPD families (1), expanding a 15-residue polyalanine tract encoded by an imperfect trinucleotide repeat sequence by 7, 8, and 10 additional residues, respectively. Similar 9-residue expansions subsequently have been reported in two Turkish SPD families (2). SPD typically consists of 3/4 finger and 4/5 toe syndactyly, with a duplicated digit in the syndactylous web (3). Incomplete penetrance and variable expressivity both between and within affected families are common (4–6). From one to four limbs can be involved, and the severity of involvement ranges from partial skin syndactyly to complete reduplication of a digit, extending as far proximally as the metacarpals/tarsals. Associated distal limb abnormalities include fifth-finger clinodactyly, camptodactyly, or brachydactyly; variable syndactyly of the second to fifth toes; and middle phalanx hypoplasia/aplasia.
To investigate the molecular basis for this incomplete penetrance and variable expressivity, we analyzed the genotype and phenotype of 16 new SPD pedigrees, including one with an expansion that almost doubles the length of the polyalanine tract, as well as reviewed 4 previously published pedigrees. We found no evidence for instability of the expansion. There is, however, a clear correlation between expansion size and completeness of penetrance. The number of limbs affected and the severity of limb involvement increase progressively with increasing expansion size, and the family with the largest expansion has a consistent, unusually severe limb phenotype. Affected males in this family also have hypospadias, a genital malformation not previously associated with SPD. These findings have intriguing implications for the role of polyalanine tracts in the regulation of transcription.
MATERIALS AND METHODS
Subjects.
The majority of the new families were ascertained following referral for surgery and/or genetic counseling to the Beth Israel and Children’s Hospital, Boston [pedigrees C, E, F, K, L, and N (J.U.)], or to Great Ormond Street Hospital, London [pedigrees A, I, J, and Q (W.R. and R.W.)]. The remaining families were ascertained following a call for patients among European clinical geneticists [pedigrees B and H (F.M.); pedigree D (E.L.); pedigree G (J.M.); pedigree M (C.M.); and pedigree O (D.D.)]. One previously reported Italian family, pedigree P (7), and three previously reported American families, pedigrees 1, 2, and 3 (1), were also clinically reviewed (M.L.G.-U. and J.U.).
Mutation Analysis.
To screen for mutations, PCR was performed on genomic DNA extracted from whole venous blood, using primers designed to amplify a 172-bp fragment of exon 1 of HOXD13, which contains the trinucleotide repeat sequence encoding the polyalanine tract. Primers and reaction conditions were as previously described (1). PCR products were analyzed on a 4% agarose gel to detect the presence of an expanded allele. Products containing the expansions were subcloned into pCRII (Invitrogen) or pCRScript (Stratagene) and cycle sequenced (Applied Biosystems Prism Dye Terminator Kit).
Sequence Analysis.
To examine the complete HOXD13 coding sequence, normal and selected mutant alleles were amplified in three segments using sense and antisense primer pairs 5′-AAGGAGAGGAGGGAGGAGG-3′/5′-GACATACGGCAGCTGTAGTAGC-3′; 5′-CTACTACAGCTGCCGTATGTCG-3′/5′-TCGGTCCCTTTTCTCCATC-3′; and 5′-AGCTAGGTGCTCCGAATATCC-3′/5′-AAGCTGTCTGTGGCCAACC-3′, which yielded products of 505 bp, 406 bp, and 335 bp, respectively, starting 107 bases before the first coding methionine, 438 bases after it, and 56 bases upstream of the start of the second exon. Amplification reactions contained 1× Pfu buffer (Stratagene), 0.25 mM of each dNTP, 10% glycerol, 2% formamide, 1 μM of each primer, and 0.6 units of cloned Pfu DNA polymerase (Stratagene) in a final volume of 25 μl. Initial denaturation was at 97.5°C for 2 min, followed by 30 cycles of denaturation at 97.5°C for 30 sec, annealing for 30 sec, and extension at 72°C for 45 sec. Annealing temperatures were 61°C, 58°C, and 55.5°C, respectively. Amplified fragments were sequenced directly or subcloned into pCRScript as before. The complete nucleotide sequence, which corrects various minor errors in previously published work, has been deposited in the GenBank database (accession nos. AF005219 and AF005220).
Statistical Analysis.
The program cat mod (8) was used to fit an ordinal regression model to quantify the effect of expansion size on extent of limb involvement.
RESULTS
Pedigrees.
Sixteen new SPD pedigrees were ascertained in this study: seven British (pedigrees A, G, I, J, M, O, and Q), six American (pedigrees C, E, F, K, L, and N), two German (pedigrees B and H), and one Italian (pedigree D) (Fig. 1). In nine of these families the disorder could be traced back over three or more generations. The largest, pedigree A, was a 6-generation family with 24 living affected members spanning 4 generations. With the inclusion of the 4 previously reported families (pedigrees 1, 2, 3, and P) reassessed in the study, 99 affected individuals were examined. A further 44 affected individuals (33 deceased, 11 unavailable for examination) were identified, and for 36 of them, information about phenotype could be obtained from family members or old medical records.
Figure 1.

Pedigrees of 16 new SPD families. Solid symbols represent individuals with SPD. Dotted symbols represent phenotypically normal mutation carriers.
Genotypes.
Expansions in the first exon of HOXD13 have already been described in pedigrees 1, 2, and 3 (1). Mutation screening showed similar expansions in the remaining 17 pedigrees, segregating with the disease phenotype (results not shown). The expansions encode 7 additional alanine residues in 8 families (pedigrees A–G and 2); 8 in 3 families (pedigrees H, I, and 1); 9 in 7 families (pedigrees J–P); 10 in 1 family (pedigree 3); and 14 in 1 family (pedigree Q) (Fig. 2).
Figure 2.
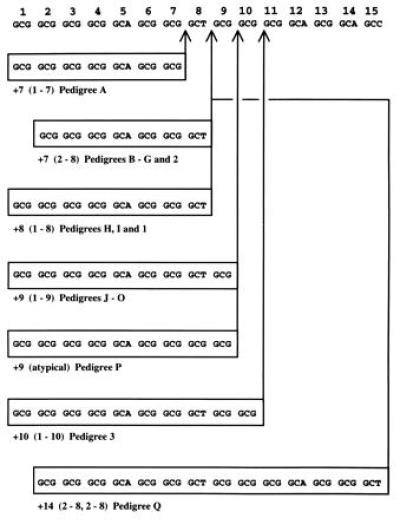
Polyalanine tract sequence in 20 SPD pedigrees. Across the top is a normal 15-residue sequence. Below the sequence are expansions (boxed) in SPD pedigrees. Probable sites of insertion of duplicated regions are indicated by vertical arrows. Note that many of these expansions could be interpreted as insertions of the same-sized duplications at a different site. Alanine 12 is encoded by GCG in pedigrees B, G–J, M, O, and P.
Affected individuals separated by three generations (I.1 and III.5 in pedigree I; III.4 and V.1 in pedigree P) and four generations (III.5 and VI.1 in pedigree A) had identical expansions, as did affected individuals from different branches of two large families (pedigrees A and 1), suggesting that the expansion had been stable over at least six generations. Similar stability over at least seven generations has recently been shown in another large SPD family (2).
Most expansions appear to be straightforward reduplications of short segments of the imperfect trinucleotide repeat sequence. The 9-alanine expansion in pedigree P, however, can only be interpreted as a reduplication of alanines 1–9 if alanines 8 and 12, normally encoded by GCT and GCA, respectively, were in this case encoded by GCG. Such a polymorphism in codon usage occurs at alanine 12 in 5/20 normal and 8/20 expanded alleles sequenced, but so far has not been found at alanine 8. The 14-alanine expansion in pedigree Q is also atypical: rather than a single duplication of alanines 1–14, there appears to have been a double duplication of alanines 2–8. This double duplication is likely to have arisen de novo in individual I.2, because her parents, who were both clinically normal (as was her one sibling), showed no evidence of an expansion in DNA extracted from peripheral blood lymphocytes (results not shown; paternity confirmed by analysis of polymorphic microsatellite repeats).
To exclude the possibility that polymorphisms or other mutations in HOXD13 cosegregating with the polyalanine tract expansions might be influencing the observed phenotypes, the entire mutant allele was sequenced for one individual (III.1) from pedigree D (7-alanine expansion, unusually low penetrance) and one individual (II.1) from pedigree Q (14-alanine expansion, unusually severe phenotype). In both cases, the sequence was normal apart from that coding for the polyalanine tract. Moreover, in 20 normal alleles sequenced, no polymorphisms were identified in the coding sequence of the gene, other than that at residue 12 of the polyalanine tract mentioned above.
Phenotypes.
Incomplete penetrance and variable expressivity were noted in many of these families, as in most of those previously reported. Neither penetrance nor severity of phenotype increased in successive generations in any one family (genetic anticipation), which is consistent with the observed stability of the expansions. We therefore examined whether penetrance and severity of phenotype was increased in patients with increasingly large expansions.
Seventeen nonpenetrant gene carriers were identified. Seven (IV.25, pedigree A; II.1, II.3, II.5, III.2, and III.3, pedigree D; and II.3, pedigree G) were completely normal on clinical examination but carried the same expanded allele seen in other family members (results not shown). The remaining 10 (II.6, III.8, III.14, and IV.15, pedigree A; I.1, pedigree D; I.2, pedigree E; I.3 and II.1, pedigree F; I.1, pedigree G; and the father of individual 8, pedigree 2), all obligate carriers on pedigree analysis, were deceased or unavailable for examination. All these individuals were from families with a 7-alanine expansion. One individual with a 10-alanine expansion (individual 14 in pedigree 3), previously described as unaffected, actually had mild bilateral hand involvement. Thus, in our series, nonpenetrance was observed only in families with the smallest expansion. Penetrance in the large, fully ascertained pedigree A, with a 7-alanine expansion, was only 86%, as opposed to the figure of 97% reported for a very large, fully ascertained family with a 9-alanine expansion (2).
The ideal method of assessing SPD phenotype is radiological examination. Most adult patients in this study had undergone corrective surgery in childhood, however, and it was not always possible to trace preoperative x-rays to establish the original extent of their malformation. Moreover, many affected individuals were either deceased or unavailable for radiological examination. To avoid biasing the analysis, we therefore elected to use the number of limbs involved as a measure of phenotypic severity, allowing us to include almost all the individuals identified (71, 9, 32, 18, and 5 individuals with 7-, 8-, 9-, 10-, and 14-alanine expansions, respectively). Because hands were affected more commonly than feet, we analyzed hand and foot involvement separately. There is a clear, progressive trend toward increasing involvement of both the hands and the feet with increasing expansion size (Fig. 3). This trend was statistically highly significant (P = 0.012 for hands; P < 0.00005 for feet). The slight exception seen for foot involvement in individuals with 8-alanine expansions is probably due to their small number. The figures for individuals with 9-alanine expansions are closely comparable to those of Sayli et al. (6). The individuals with 10- and 14-alanine expansions were all from pedigrees 3 and Q, respectively. The increased limb involvement in these individuals is probably due to the large expansion rather than the genetic background in these two pedigrees, because marked phenotypic variation between families with same-sized expansions was not observed for families with 7- to 9-alanine expansions. There was no evidence of statistically significant asymmetry of involvement in individuals with expansions of any size.
Figure 3.
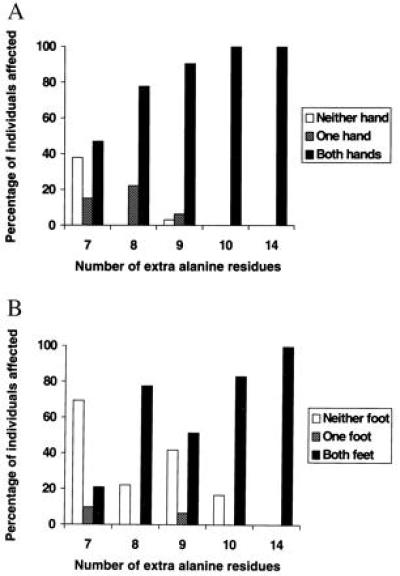
Bar charts showing correlation between expansion size and number of hands (A) and feet (B) affected in SPD patients.
Further information on severity of involvement was available for 156 affected hands. Three/four finger syndactyly, without digit duplication, occurred in approximately one-third of affected hands of 7- and 8-alanine expansion carriers (19/53 and 4/13 hands, respectively). In contrast, there was a duplicated digit in the syndactylous web in virtually all affected hands of 9-, 10-, and 14-alanine expansion carriers (44/44, 32/36, and 10/10 hands, respectively). Information on the level of branching (the most proximal point at which duplication was apparent) was also available for 84 hands with a duplicated digit (23, 9, 20, 22, and 10 hands for carriers of 7-, 8-, 9-, 10-, and 14-alanine expansions, respectively). Despite these relatively small numbers, there was a strong overall trend toward a more proximal level of branching with increased expansion size (Fig. 4).
Figure 4.
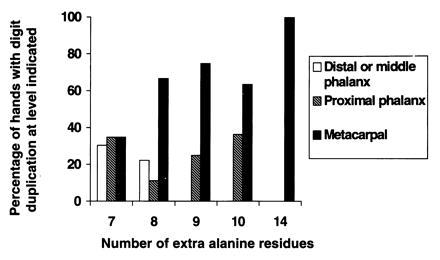
Bar chart showing correlation between expansion size and level of branching in SPD hands with digit duplication.
Nineteen affected individuals displayed an extremely mild foot phenotype—2/3 toe syndactyly, fifth-toe brachydactyly, or an overriding fourth or fifth toe—not typical of SPD, and therefore not included as affected in the foregoing analysis. Of these, 16 carried a 7-alanine expansion, 1 carried a 9-alanine expansion, and 2 carried a 10-alanine expansion. In 10 of the 7-alanine expansion carriers, the minor foot abnormality was the sole phenotypic manifestation and had generally gone unremarked by the patient. Similarly, two individuals with a 7-alanine expansion displayed an extremely mild hand phenotype—isolated fourth-finger brachydactyly—which is not typical of SPD. In these cases, our mutation screen was of practical value in determining carrier status.
The increasing phenotypic severity seen with increasing expansion size culminated in pedigree Q, with the 14-alanine expansion. All five affected members of this family had a virtually identical limb phenotype (Fig. 5). Typical SPD was present in all four limbs, in its most severe form. Both hands had a well formed additional finger that originated at the level of the third metacarpals, which were bifid, and was accompanied by marked fifth-finger camptodactyly and middle phalanx hypoplasia of all fingers. Both feet had an additional toe that originated at the level of the fourth/fifth metatarsals, accompanied by middle phalanx hypoplasia of all toes. In addition, however, there was involvement of the first digits and the distal carpals. Both thumbs were broad, radially deviated, and proximally placed, with short first metacarpals, and both halluces were unusually broad, with pseudoarthroses at the bases of the first and second metatarsals. II.1 also required surgery for a delta phalanx of the left thumb. In the wrist, the capitate bones (third distal carpals) were abnormally enlarged. The two affected males in this family (but no unaffected males on either side for at least the past three generations) were also born with mid-penile hypospadias, associated with marked chordee. Neither involvement of the first digit and distal carpals nor hypospadias has previously been reported in SPD heterozygotes.
Figure 5.
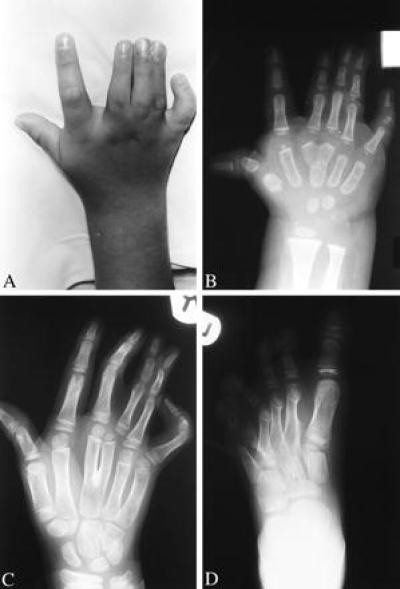
The limb phenotype in pedigree Q. Photograph (A) and x-ray (B) of right hand of II.1 at age 5 years and 18 months, respectively, showing severe SPD (duplicated third finger with soft tissue syndactyly as far as distal phalanx, bifid third metacarpal, fifth finger camptodactyly, and middle phalanx hypoplasia of all fingers), as well as broad, radially deviated thumb with short first metacarpal. (C) X-ray of right hand of II.2 at age 8 years, showing similar severe SPD, with soft tissue syndactyly as far as proximal phalanx, as well as an enlarged capitate bone. (D) X-ray of left foot of II.4 at age 6 years, showing severe SPD (additional toe between fourth and fifth toes; additional metatarsal between fourth and fifth metatarsals; middle phalanx hypoplasia of toes 2–5; and absent middle phalanx of toe 6, with bracket epiphysis of proximal phalanx), as well as broad hallux, with pseudoarthrosis at base of first and second metatarsals.
DISCUSSION
Nature of Expansions.
Of the 20 families with SPD described here, 18 have 7–9 extra alanine residues, as do the remaining two SPD families known to carry an expansion (3). Expansions of 6 residues or less either do not occur, or, more probably, produce effects so slight that they do not attract medical attention. The more severe phenotype caused by larger expansions is unlikely to be overlooked, however, so mutations producing large expansions must be rare.
The trinucleotide repeat in HOXD13 is normally short (15 repeats) and nonpolymorphic, and the expansions that cause SPD are also short (maximum length, 29 repeats) and stable. They thus differ sharply from the classic, long, unstable, disease-causing trinucleotide expansions found in coding sequence in, for example, Huntington’s disease (normal range, 10–35 repeats; disease-causing alleles, 36–121 repeats) (9). In HOXD13, moreover, the trinucleotide repeat is imperfect (Fig. 2), making replication slippage unlikely. The expansions are thus most probably caused by nonreciprocal recombination or misalignment during replication, mediated by different segments of the repeat. Interestingly, the 50-bp deletion in Hoxa13 recently found in the Hypodactyly mouse (10) can also be explained by nonreciprocal recombination in a similar trinucleotide repeat. If the de novo 14-alanine expansion described here was so produced, however, two successive such events must have occurred in the same germ line.
Range of Phenotype.
Abnormalities in SPD heterozygotes previously have been reported only in the distal limb, extending proximally as far as the metacarpals/tarsals and anteriorly as far as the third digit. The findings in our series of families considerably expand this range. At the mild end of the phenotypic spectrum, we have found that isolated fourth-finger brachydactyly, 2/3 toe syndactyly, overriding fourth or fifth toes, and fifth-toe brachydactyly can be minor manifestations of SPD. At the severe end, we have found limb abnormalities extending as far proximally as the distal carpals and as far anteriorly as the first digit. A similar distribution of abnormalities has been reported in SPD homozygotes (1, 11). These severe phenotypes cover the same area as the second phase of Hoxd13 expression in the developing limbs of chicks and mice (12), which occurs during specification of the autopod. During this phase, Hoxd13 has a larger expression domain than any other 5′ Hoxd gene and is also the first to be expressed. Hoxd13 is believed to play a dominant role in this phase, perhaps by controlling mesenchymal proliferation (13, 14), and it is this that appears to be perturbed in SPD.
In mice, Hoxd13 is also expressed in the developing genital tubercle, mainly in mesenchyme, but also in the most distal epithelium of the urogenital sinus, which subsequently produces the urogenital folds (15). It is therefore interesting that the two affected males in the pedigree with the largest expansion (pedigree Q) were born with hypospadias. Hypospadias affects 1 in 1,000 males, with a 10% recurrence risk in the brothers and sons of those affected (16). In pedigree Q, however, it was confined to the two males with SPD and may well be associated with the mutation. The condition occurs when the urogenital folds fail to fuse and form the penile portion of the urethra, producing an external urethral orifice on the ventral surface of the penis, and the mutation may interfere with the normal proliferation and fusion of these folds. It is interesting that a mutation in the paralogous human gene HOXA13 has recently been shown to cause a similar combination of developmental anomalies in a family with hand-foot-genital syndrome (17).
Relationship Between Genotype and Phenotype.
We have found that the number of limbs affected and the degree of limb involvement in SPD heterozygotes increase progressively with increasing expansion size. The level of branching in duplicated digits becomes more proximal in patients with larger expansions, and the proximal and anterior extent of the limb abnormalities is greatest in the family with the largest expansion. SPD homozygotes exhibit the same trend: the limb phenotype of seven patients homozygous for a 9-alanine expansion (11) is markedly more extensive than that of one patient homozygous for a 7-alanine expansion (1). Moreover, abnormalities outside the distal limb are associated only with the largest expansions.
These abnormalities differ from those seen in mice lacking Hoxd13. Only 36% of mice heterozygous for disruption of the Hoxd13 homeobox are affected, and those that are only minor carpal defects and an additional bony element postaxial to the fifth digit in the forelimb (18). All homozygotes have shortened digits, especially 2 and 5, and about half also have a rudimentary extra digit postaxial to digit 5 (13, 18). In addition, males are infertile, with a malformed penian bone, and both sexes have abnormalities of the lowest sacral vertebra (13). The difference between these phenotypes and those seen in SPD, together with the correlation between phenotypic severity and expansion size in SPD, suggests that expansions in the polyalanine tract confer a progressive gain of function on HOXD13.
Although differences in expansion size can account for interfamilial variation in SPD phenotype, they cannot explain intrafamilial variation, because the expansion is stable in any given family. Intrafamilial variation is most marked in families with the smallest expansions, perhaps reflecting the greater relative importance of other genetic and environmental factors. But there is also striking variation within affected individuals, because one hand or foot can be typically affected, while the other is completely normal. Here, variable expressivity is occurring against a uniform genetic and environmental background. Involvement of a given limb thus must reflect a stochastic event.
Effect of Expansion on Transcriptional Regulation.
The expansion responsible for SPD thus has several unusual properties, which must be accounted for in any explanation of its role. First, it apparently confers a gain of function. Second, this gain of function is confined to expansions with a threshold total length of 22 alanine residues. Third, increases beyond this threshold progressively enhance the effect. And fourth, the effect is manifested in a stochastic manner, increasing in frequency with increasing expansion size. These properties have interesting implications for the role of polyalanine tracts in regulating transcription.
Polyalanine tracts are found in many other homeodomain proteins, including Drosophila Krüppel, Engrailed, and Even-skipped (19–21), and mouse Hoxa3 (22), Hoxa11 (23), Hoxa13 (10), Evx 1, Evx 2 (24), Gsh-1 (25), Gsh-2 (26), Msx-1, and Msx-2 (27), as well as in other transcription factors, such as Drosophila AEF-1 (28) and human MAZ (29). Such tracts have been proposed to repress transcription directly, and the minimal repressor domains of Krüppel, Engrailed, and Even-skipped are indeed all strikingly alanine-rich (19–21, 30). The expanded tracts in SPD are unlikely to be acting in this way, however, because neither the role of the normal polyalanine tract in the wild-type protein nor the threshold effect seen at expansions totaling 22 residues is explained by such a model.
It has also been proposed that polyalanine tracts act as flexible spacer elements between functional domains (31), and lengthening such a spacer could readily perturb the interactions between these domains and their targets, either by affecting their orientation or by steric hindrance. This in itself, however, would be unlikely to produce the documented progressive gain of function.
The most plausible explanation of the phenotypes we observe, therefore, is that expansions of 7 or more residues confer on the mutant HOXD13 the ability to bind another protein, and that the binding affinity increases thereafter with increasing expansion size. Binding of such a protein could interfere not only with the action of the remaining wild-type HOXD13 but also with that of other HOX proteins. Such a “super” dominant negative effect might reflect binding either of a transcriptional corepressor or of another protein required for the action of other HOX proteins, thereby causing it to be sequestered or even degraded at an increased rate.
This hypothesis is consistent with the findings of three recent studies. First, mice homozygous for a deletion of Hoxd11, Hoxd12, and Hoxd13 have phenotypes similar to SPD heterozygotes (extra central digit fused to digit 3 or 4) and SPD homozygotes (reduced paw size and digit length, 1–2 extra digits, interdigital webbing and bony fusions, and small malformed carpal bones) (32). Second, mice that are both heterozygous for a targeted disruption of Hoxa13 and heterozygous or homozygous for a targeted disruption of Hoxd13 show some phenotypic resemblances to SPD heterozygotes (webbing between the central digits in the former and forelimb polydactyly with secondary bony fusions, resulting in central “synpolydactyly” in the latter) (33). Third, misexpression experiments in chicken limb buds suggest that ectopic Hoxd13 may interfere with the normal function of Hoxd11 and Hoxa11 (34). If this hypothesis is correct, it will be interesting to identify the interaction partner involved and analyze both its role in the control of transcription by HOX proteins and its perturbation in SPD.
Acknowledgments
We thank Dr. Christine Hall (Great Ormond Street Hospital, London) for help with assessment of x-rays and Dr. Angela Wade (Institute of Child Health, London) for help with the statistical analysis. This work was supported by the Medical Research Council (Clinical Training Fellowship to F.R.G.); the Deutsche Forschungsgemeinschaft (grant to S.M.); and the National Institutes of Health (Grants AR36819 and AR36820 to B.R.O.).
ABBREVIATION
- SPD
synpolydactyly
Footnotes
References
- 1.Muragaki Y, Mundlos S, Upton J, Olsen B. Science. 1996;272:548–551. doi: 10.1126/science.272.5261.548. [DOI] [PubMed] [Google Scholar]
- 2.Akarsu A, Stoilov I, Yilmaz E, Sayli B, Sarfarazi M. Hum Mol Genet. 1996;5:945–952. doi: 10.1093/hmg/5.7.945. [DOI] [PubMed] [Google Scholar]
- 3.Temtamy S, McKusick V. Birth Defects: Original Article Series. Vol. 14. New York: Liss; 1978. pp. 301–322. [PubMed] [Google Scholar]
- 4.Thomsen O. Acta Med Scand. 1927;65:609–644. [Google Scholar]
- 5.Cross H, Lerberg D, McKusick V. Am J Hum Genet. 1968;20:368–380. [PMC free article] [PubMed] [Google Scholar]
- 6.Sayli B, Akarsu A, Sayli U, Akhan O, Ceylaner S, Sarfarazi M. J Med Genet. 1995;32:421–434. doi: 10.1136/jmg.32.6.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chessa-Ricotti G, Lapi E, De Bernardi A, Corti P. Pediatr Med Chir. 1990;12:259–264. [PubMed] [Google Scholar]
- 8.SAS Institute. SAS/STAT User’s Guide. Cary, NC: SAS Institute; 1989. , Version 6, 4th Ed., Vol. 1, pp. 405–517. [Google Scholar]
- 9.Ashley C, Warren S. Annu Rev Genet. 1995;29:703–728. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- 10.Mortlock D, Post L, Innis J. Nat Genet. 1996;13:284–289. doi: 10.1038/ng0796-284. [DOI] [PubMed] [Google Scholar]
- 11.Akarsu A, Akhan O, Sayli B, Sayli U, Baskaya G, Sarfarazi M. J Med Genet. 1995;32:435–441. doi: 10.1136/jmg.32.6.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson C, Morgan B, Burke A, Laufer E, DiMambro E, Murtaugh C, Gonzales E, Tessarollo L, Parada L, Tabin C. Development (Cambridge, UK) 1996;122:1449–1466. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- 13.Dollé P, Dierich A, LeMeur M, Schimmang T, Schuhbaur B, Chambon P, Duboule D. Cell. 1993;75:431–441. doi: 10.1016/0092-8674(93)90378-4. [DOI] [PubMed] [Google Scholar]
- 14.Duboule D. Science. 1994;266:575–576. doi: 10.1126/science.7939709. [DOI] [PubMed] [Google Scholar]
- 15.Dollé P, Izpisùa-Belmonte J-C, Brown J, Tickle C, Duboule D. Genes Dev. 1991;5:1767–1776. doi: 10.1101/gad.5.10.1767. [DOI] [PubMed] [Google Scholar]
- 16.Harper P. Practical Genetic Counselling. 4th Ed. Oxford: Butterworth–Heinemann; 1993. pp. 268–269. [Google Scholar]
- 17.Mortlock D, Innis J. Nat Genet. 1997;15:179–180. doi: 10.1038/ng0297-179. [DOI] [PubMed] [Google Scholar]
- 18.Davis A, Capecchi M. Development (Cambridge, UK) 1996;122:1175–1185. doi: 10.1242/dev.122.4.1175. [DOI] [PubMed] [Google Scholar]
- 19.Licht J, Grossel M, Figge J, Hansen U. Nature (London) 1990;346:76–79. doi: 10.1038/346076a0. [DOI] [PubMed] [Google Scholar]
- 20.Han K, Manley J. EMBO J. 1993;12:2723–2733. doi: 10.1002/j.1460-2075.1993.tb05934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han K, Manley J. Genes Dev. 1993;7:491–503. doi: 10.1101/gad.7.3.491. [DOI] [PubMed] [Google Scholar]
- 22.Izpisùa-Belmonte J-C, Dollé P, Renucci A, Zappavigna V, Falkenstein H, Duboule D. Development (Cambridge, UK) 1990;110:733–745. [PubMed] [Google Scholar]
- 23.Tan D-P, Ferrante J, Nazarali A, Shao X, Kozak C, Guo V, Nirenberg M. Proc Natl Acad Sci USA. 1992;89:6280–6284. doi: 10.1073/pnas.89.14.6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bastian H, Gruss P. EMBO J. 1990;9:1839–1852. doi: 10.1002/j.1460-2075.1990.tb08309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valerius M, Li H, Stock J, Weinstein M, Kaur S, Singh G, Potter S. Dev Dyn. 1995;203:337–351. doi: 10.1002/aja.1002030306. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh-Li H, Witte D, Szucsik J, Weinstein M, Li H, Potter S. Mech Dev. 1995;50:177–186. doi: 10.1016/0925-4773(94)00334-j. [DOI] [PubMed] [Google Scholar]
- 27.Catron K, Wang H, Hu G, Shen M, Abate-Shen C. Mech Dev. 1996;55:185–199. doi: 10.1016/0925-4773(96)00503-5. [DOI] [PubMed] [Google Scholar]
- 28.Falb D, Maniatis T. Mol Cell Biol. 1992;12:4093–4103. doi: 10.1128/mcb.12.9.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossone S, Asselini C, Patel A, Marcu K. Proc Natl Acad Sci USA. 1992;89:7452–7456. doi: 10.1073/pnas.89.16.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Licht J, Hanna-Rose W, Reddy J, English M, Ro M, Grossel M, Shaknovich R, Hansen U. Mol Cell Biol. 1994;14:4057–4066. doi: 10.1128/mcb.14.6.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlin S, Burge C. Proc Natl Acad Sci USA. 1996;93:1560–1565. doi: 10.1073/pnas.93.4.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zákány J, Duboule D. Nature (London) 1996;384:69–71. doi: 10.1038/384069a0. [DOI] [PubMed] [Google Scholar]
- 33.Fromental-Ramain C, Warot X, Messadecq N, LeMeur M, Dollé P, Chambon P. Development (Cambridge, UK) 1996;122:2997–3011. doi: 10.1242/dev.122.10.2997. [DOI] [PubMed] [Google Scholar]
- 34.Goff D, Tabin C. Development (Cambridge, UK) 1997;124:627–636. doi: 10.1242/dev.124.3.627. [DOI] [PubMed] [Google Scholar]